
Characterization of the Human Blood Virome in Iranian Multiple Transfused Patients
 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and Ethics Statement
2.2. Plasma Virome Sequencing
2.3. Read Processing and Taxonomical Annotation
2.4. Phage Host Prediction
2.5. Phylogenetic Analysis
2.6. Quantitative PCR
2.7. Statistical Analysis
3. Results
3.1. Population Characteristics
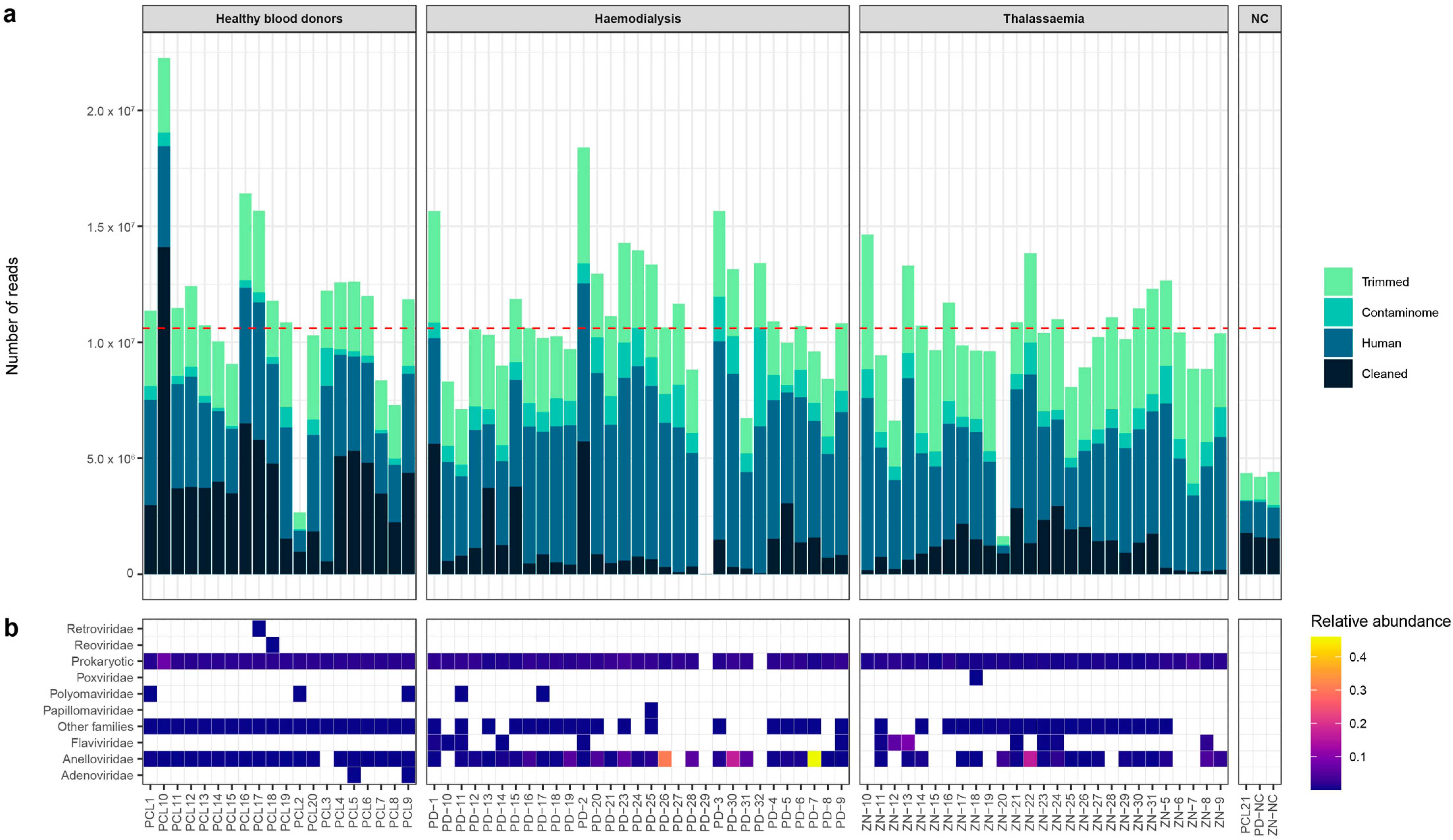
3.2. Sequencing Data
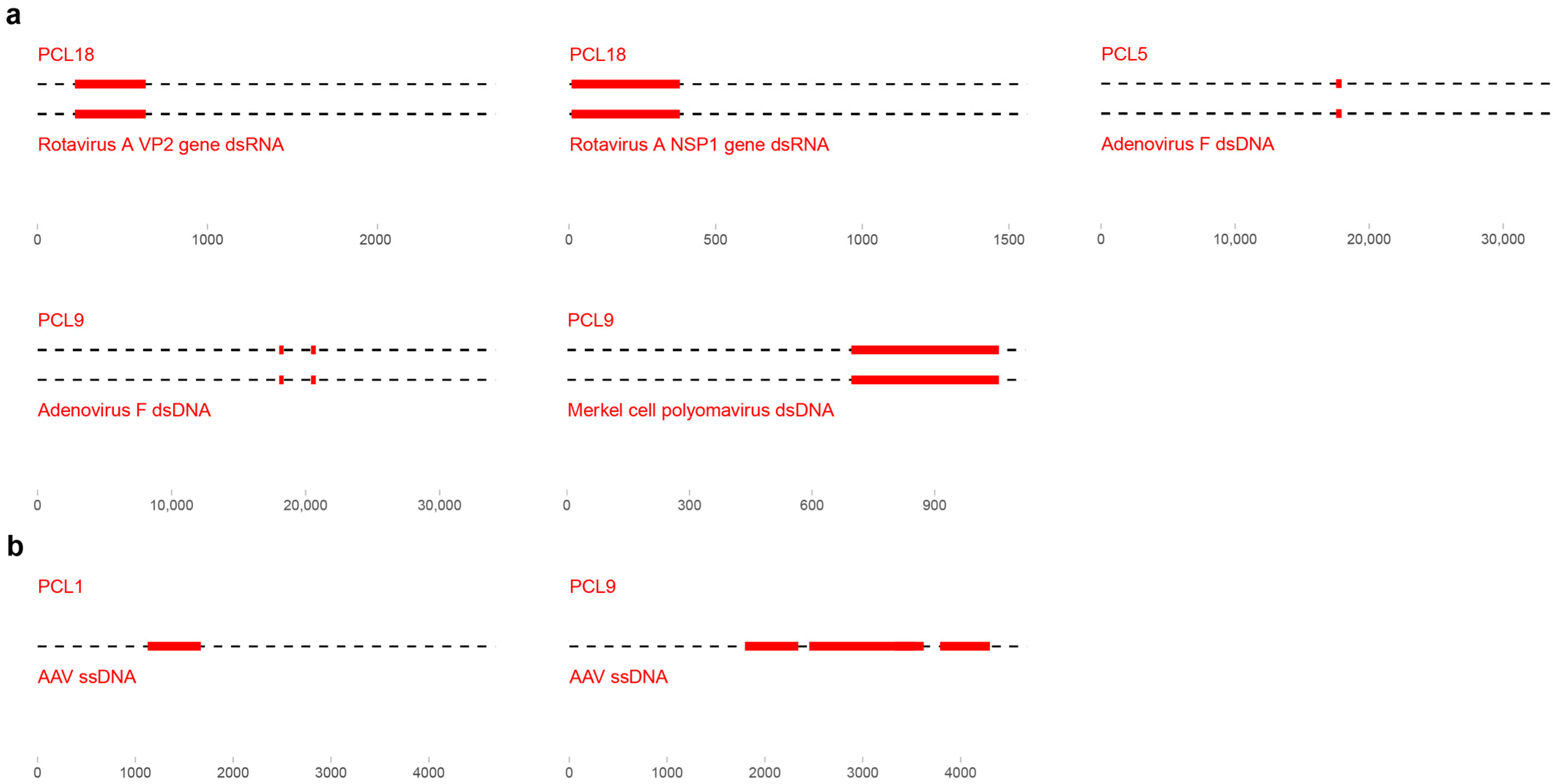
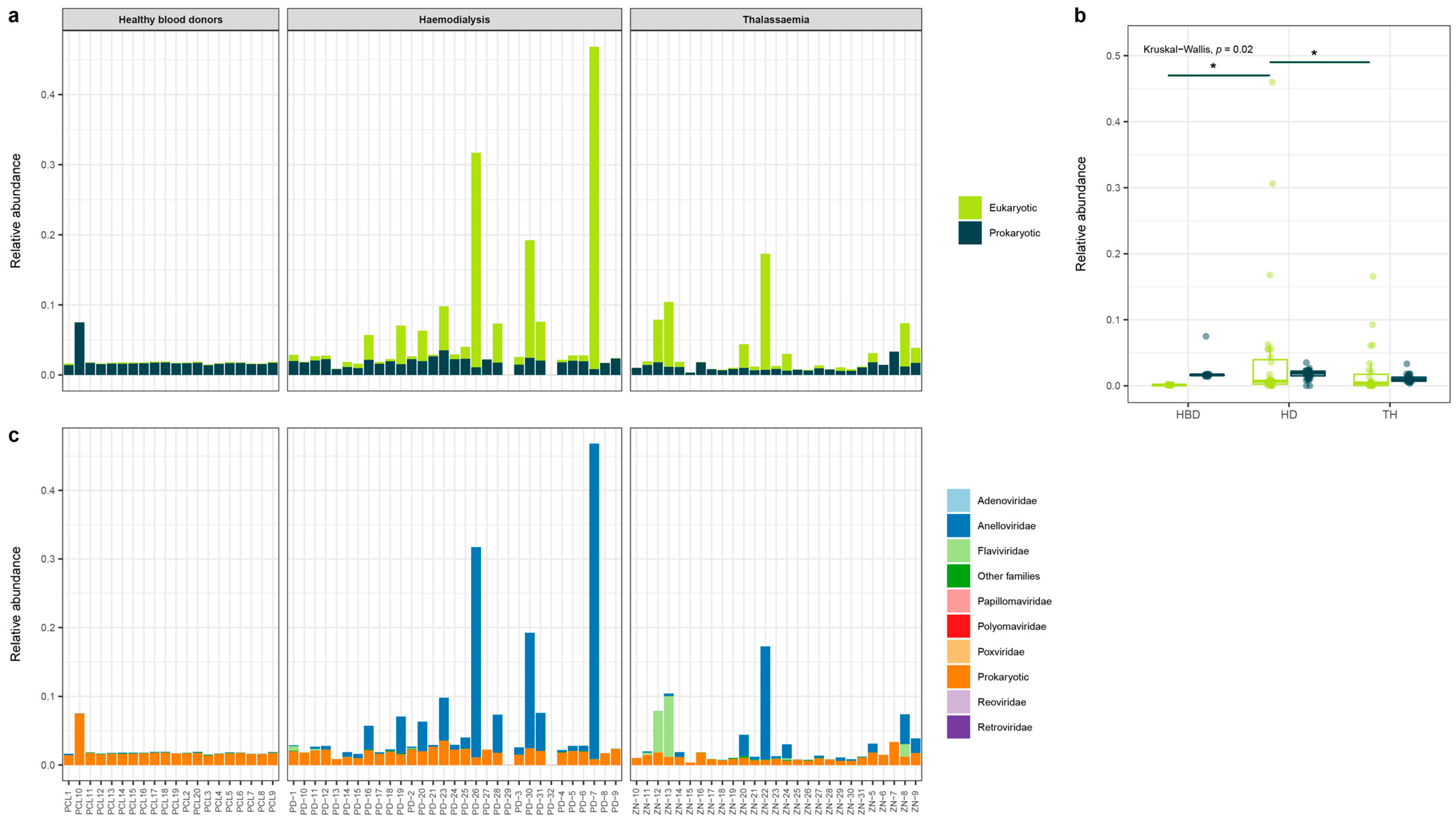
3.3. Eukaryotic and Prokaryotic Virome
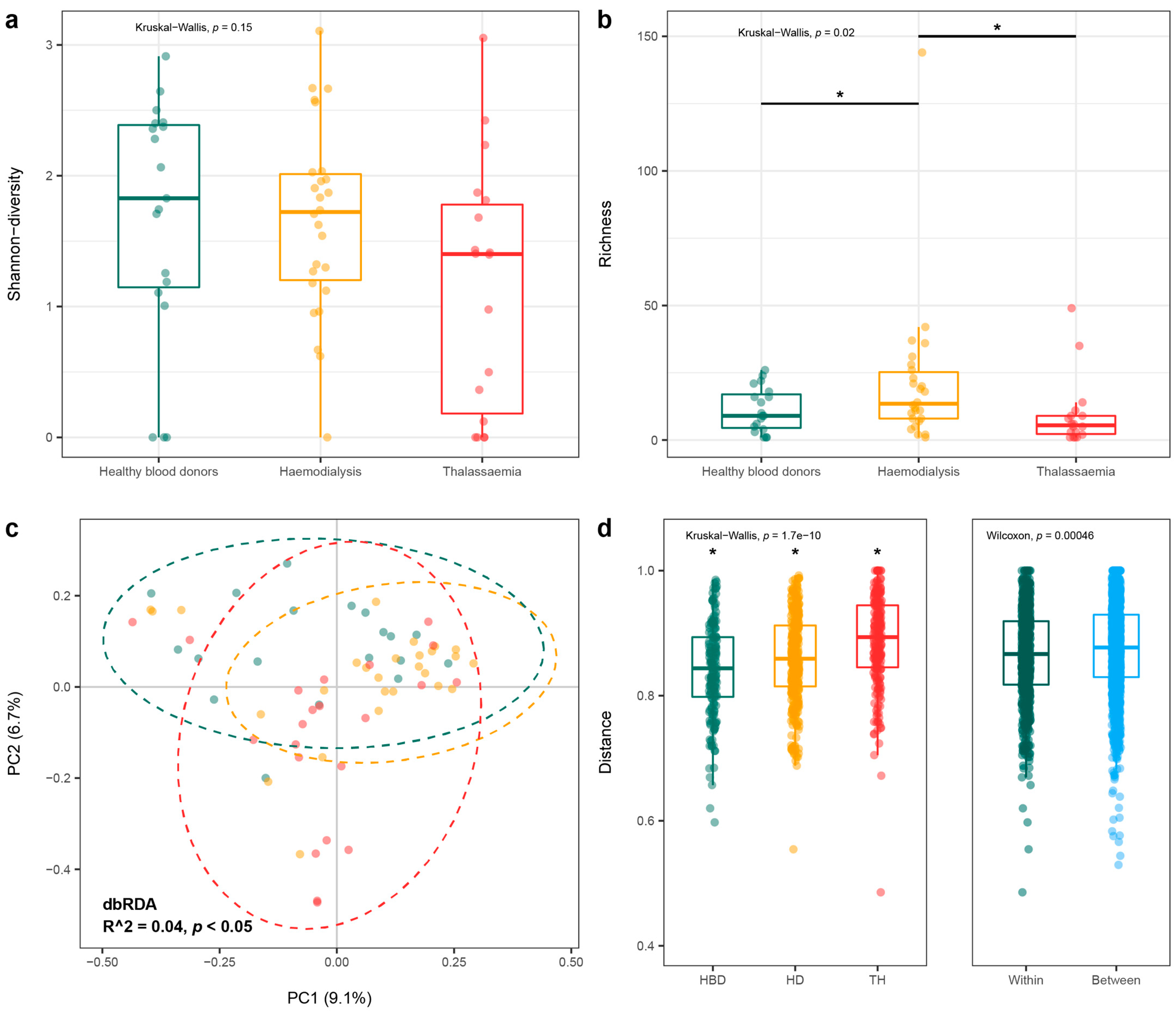
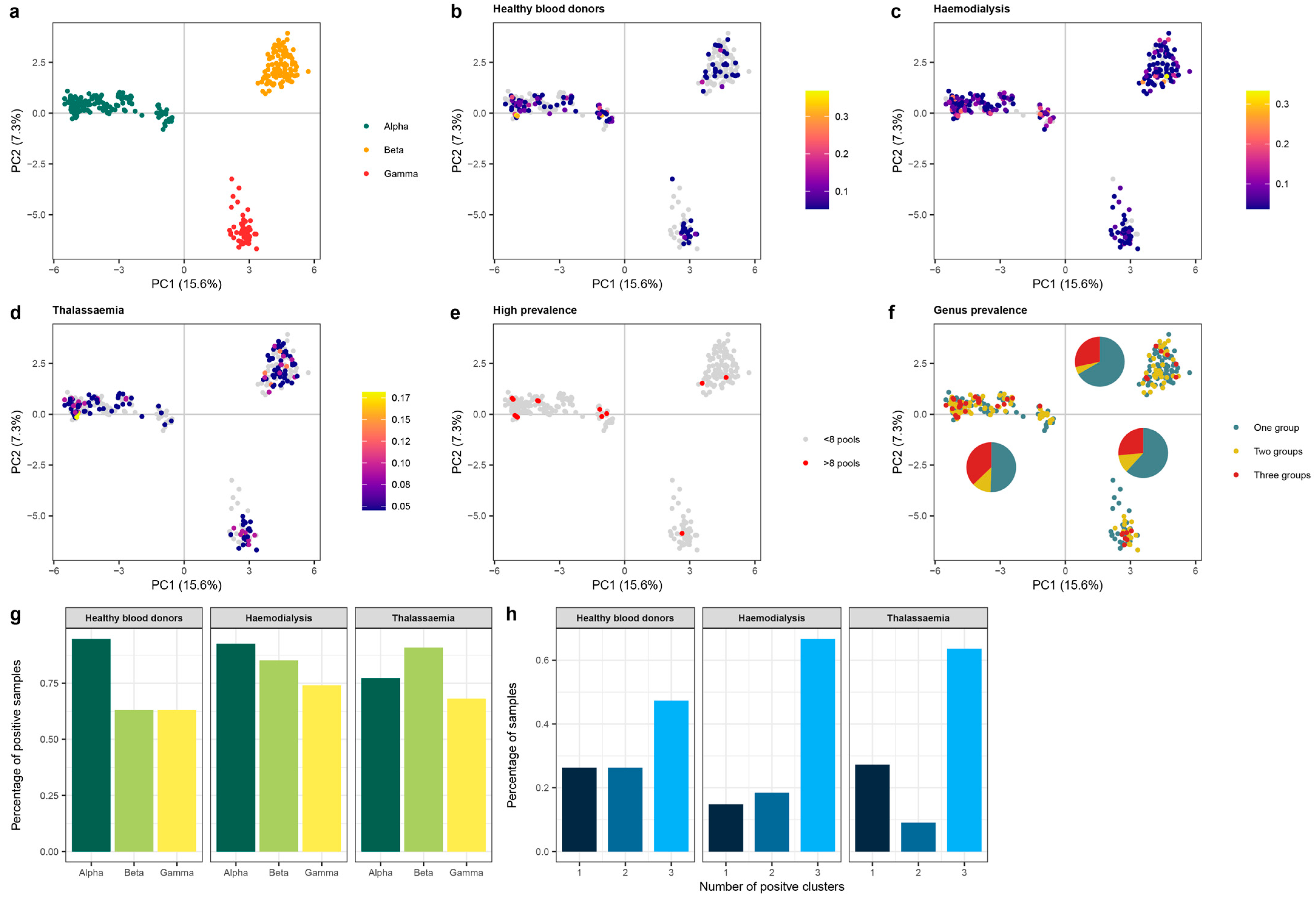
3.4. Anelloviridae Family
3.4.1. Number of Reads VS Abundance of Anellovirus
3.4.2. Anellovirus Diversity
3.4.3. Anelloviridae Genera
3.5. Pegivirus
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thijssen, M.; Devos, T.; Ejtahed, H.-S.; Amini-Bavil-Olyaee, S.; Pourfathollah, A.A.; Pourkarim, M.R. Convalescent plasma against COVID-19: A broad-spectrum therapeutic approach for emerging infectious diseases. Microorganisms 2020, 8, 1733. [Google Scholar] [CrossRef]
- He, M.; Wang, J.; Chen, L.; Liu, J.; Zeng, P. The Impact of Emerging Infectious Diseases on Chinese Blood Safety. Transfus. Med. Rev. 2017, 31, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Brunson, D.C.; Belanger, G.A.; Sussmann, H.; Fine, A.M.; Pandey, S.; Pham, T.D. Factors associated with f irst-time and repeat blood donation: Adverse reactions and effects on donor behavior. Transfusion 2022, 62, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Pierik, R.; Verweij, M.; van de Laar, T.; Zaaijer, H. Facing difficult but unavoidable choices: Donor blood safety and the deferral of men who have sex with men. Bioethics 2022, 36, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, K.Y.; Yan, K.; Ou, G.; Li, W.; Wang, J.; Song, N.; Tian, L.; Ji, X.; Chen, Y.; et al. The History and Challenges of Blood Donor Screening in China. Transfus. Med. Rev. 2017, 31, 89–93. [Google Scholar] [CrossRef]
- Stramer, S.L.; Hollinger, F.B.; Katz, L.M.; Kleinman, S.; Metzel, P.S.; Gregory, K.R.; Dodd, R.Y. Emerging infectious disease agents and their potential threat to transfusion safety. Transfusion 2009, 49 (Suppl. 2), 1S–29S. [Google Scholar] [CrossRef]
- Kapoor, A.; Kumar, A.; Simmonds, P.; Bhuva, N.; Singh Chauhan, L.; Lee, B.; Sall, A.A.; Jin, Z.; Morse, S.S.; Shaz, B.; et al. Virome Analysis of Transfusion Recipients Reveals a Novel Human Virus That Shares Genomic Features with Hepaciviruses and Pegiviruses. MBio 2015, 6, e01466-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buisson, Y. COVID-19, an unfinished story. La Presse Médicale 2022, 51, 104131. [Google Scholar] [CrossRef]
- Yu, H.; Peng, C.; Zhang, C.; Stoian, A.M.; Tazi, L.; Brennan, G.; Rothenburg, S. Maladaptation after a virus host switch leads to increased activation of the pro-inflammatory NF-κB pathway. Proc. Natl. Acad. Sci. USA 2022, 119, e2115354119. [Google Scholar] [CrossRef]
- Lipkin, W.I. The changing face of pathogen discovery and surveillance. Nat. Rev. Microbiol. 2013, 11, 133–141. [Google Scholar] [CrossRef]
- Sauvage, V.; Eloit, M. Viral metagenomics and blood safety. Transfus. Clin. Biol. 2016, 23, 28–38. [Google Scholar] [CrossRef]
- Thijssen, M.; Tacke, F.; Beller, L.; Deboutte, W.; Yinda, K.C.; Nevens, F.; Laleman, W.; Van Ranst, M.; Pourkarim, M.R. Clinical relevance of plasma virome dynamics in liver transplant recipients. EBioMedicine 2020, 60, 103009. [Google Scholar] [CrossRef] [PubMed]
- Ainley, L.I.; Hewitt, P.E. Haematology patients and the risk of transfusion transmitted infection. Br. J. Haematol. 2018, 180, 473–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedayati-Moghaddam, M.R.; Soltanian, H.; Ahmadi-Ghezeldasht, S. Occult hepatitis C virus infection in the Middle East and Eastern Mediterranean countries: A systematic review and meta-analysis. World J. Hepatol. 2021, 13, 242. [Google Scholar] [CrossRef]
- Pourkarim, M.; Khamisipour, G.; Hajiani, G.; Tahmasebi, R.; Ardeshirdavani, N. Seroepidemiological investigation of HTLV I, II infection among Busherian multi-transfused patients in 2003. Sci. J. Iran Blood Transfus Organ 2005, 2, 99–104. [Google Scholar]
- Poukarim, M.R.; Khamisipour, G.; Zandi, K.; Roustaee, M.H. Prevalence of anti-HTLV-I & anti-HTLV-II antibodies in blood donors in Bushehr province. ISMJ 2004, 6, 161–164. [Google Scholar]
- Dalvand, N.; Dalvand, A.; Sharifi, Z.; Hosseini, S.M. Prevalence of hepatitis E virus in thalassemia patients with hepatitis C in Tehran, Iran. Iran. J. Microbiol. 2019, 11, 535. [Google Scholar] [CrossRef]
- Lau, P.; Cordey, S.; Brito, F.; Tirefort, D.; Petty, T.J.; Turin, L.; Guichebaron, A.; Docquier, M.; Zdobnov, E.M.; Waldvogel-Abramowski, S.; et al. Metagenomics analysis of red blood cell and fresh-frozen plasma units. Transfusion 2017, 57, 1787–1800. [Google Scholar] [CrossRef] [Green Version]
- Furuta, R.A.; Sakamoto, H.; Kuroishi, A.; Yasiui, K.; Matsukura, H.; Hirayama, F. Metagenomic profiling of the viromes of plasma collected from blood donors with elevated serum alanine aminotransferase levels. Transfusion 2015, 55, 1889–1899. [Google Scholar] [CrossRef]
- Conceicao-Neto, N.; Zeller, M.; Lefrere, H.; De Bruyn, P.; Beller, L.; Deboutte, W.; Yinda, C.K.; Lavigne, R.; Maes, P.; Van Ranst, M.; et al. Modular approach to customise sample preparation procedures for viral metagenomics: A reproducible protocol for virome analysis. Sci. Rep. 2015, 5, 16532. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [Green Version]
- Simon, R.; Ben, B. ClusterGenomes. Available online: https://bitbucket.org/MAVERICLab/stampede-clustergenomes/src/ (accessed on 22 April 2022).
- Benjamin, B.; Simon, R. Clustering Viral Genomes in iVirus. Available online: https://www.protocols.io/view/clustering-viral-genomes-in-ivirus-rm7vzkx4vx1w/v1 (accessed on 24 May 2023).
- Vasimuddin, M.; Misra, S.; Li, H.; Aluru, S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. In Proceedings of the 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS), Rio de Janeiro, Brazil, 20–24 May 2019; pp. 314–324. [Google Scholar]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitúa, M.C.; Vik, D.; Sullivan, M.B. VirSorter2: A multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 2021, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2021, 39, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Bland, C.; Ramsey, T.L.; Sabree, F.; Lowe, M.; Brown, K.; Kyrpides, N.C.; Hugenholtz, P. CRISPR recognition tool (CRT): A tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinform. 2007, 8, 209. [Google Scholar] [CrossRef] [Green Version]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. The RAxML v8. 2. X Manual. Heidleberg Institute for Theoretical Studies. 2016. Available online: https://cme.h-its.org/exelixis/resource/download/NewManual.pdf (accessed on 10 June 2022).
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Toppinen, M.; Pratas, D.; Väisänen, E.; Söderlund-Venermo, M.; Hedman, K.; Perdomo, M.F.; Sajantila, A. The landscape of persistent human DNA viruses in femoral bone. Forensic Sci. Int. Genet. 2020, 48, 102353. [Google Scholar] [CrossRef]
- R Core Team. R: A language and environment for statistical computing. Vienna Austria, 2013. Available online: https://www.R-project.org/ (accessed on 3 January 2022).
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Stevens, M.H.H.; Oksanen, M.J.; Suggests, M. The vegan package. Community Ecol. Package 2007, 10, 719. [Google Scholar]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelé, J.; Bécu, J.-M.; Abdi, H.; Chabbert, M. Bios2mds: An R package for comparing orthologous protein families by metric multidimensional scaling. BMC Bioinform. 2012, 13, 133. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, R.A.M.; Chen, Z.J. ggplot2: Elegant Graphics for Data Analysis; Taylor & Francis: Oxfordshire, UK, 2019. [Google Scholar]
- Moustafa, A.; Xie, C.; Kirkness, E.; Biggs, W.; Wong, E.; Turpaz, Y.; Bloom, K.; Delwart, E.; Nelson, K.E.; Venter, J.C. The blood DNA virome in 8000 humans. PLoS Pathog. 2017, 13, e1006292. [Google Scholar]
- Davies, S.I.; Barrett, J.; Wong, S.; Chang, M.J.; Muranski, P.J.; Brownell, I. Robust production of merkel cell polyomavirus oncogene specific T cells from healthy donors for adoptive transfer. Front. Immunol. 2020, 11, 592721. [Google Scholar] [CrossRef] [PubMed]
- Kamminga, S.; van der Meijden, E.; de Brouwer, C.; Feltkamp, M.; Zaaijer, H. Prevalence of DNA of fourteen human polyomaviruses determined in blood donors. Transfusion 2019, 59, 3689–3697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blutt, S.E.; Kirkwood, C.D.; Parreño, V.; Warfield, K.L.; Ciarlet, M.; Estes, M.K.; Bok, K.; Bishop, R.F.; Conner, M.E. Rotavirus antigenaemia and viraemia: A common event? Lancet 2003, 362, 1445–1449. [Google Scholar] [CrossRef] [PubMed]
- Lynch III, J.P.; Kajon, A.E. Adenovirus: Epidemiology, global spread of novel serotypes, and advances in treatment and prevention. Seminars in Respiratory and Critical Care Medicine 2016, 37, 586–602. [Google Scholar]
- Wollants, E.; Keyaerts, E.; Cuypers, L.; Bloemen, M.; Thijssen, M.; Ombelet, S.; Raymenants, J.; Beuselinck, K.; Laenen, L.; Budts, L. Environmental circulation of adenovirus 40/41 and SARS-CoV-2 in the context of the emergence of acute hepatitis of unknown origin. medRxiv 2022. [Google Scholar]
- Pourkarim, M.R. Acute Hepatitis of Unknown Origin in Children; Lessons Learned from the COVID-19 Pandemic; Brieflands: North Brabant, The Netherlands, 2022; Volume 22. [Google Scholar]
- Hüser, D.; Khalid, D.; Lutter, T.; Hammer, E.-M.; Weger, S.; Heßler, M.; Kalus, U.; Tauchmann, Y.; Hensel-Wiegel, K.; Lassner, D. High prevalence of infectious adeno-associated virus (AAV) in human peripheral blood mononuclear cells indicative of T lymphocytes as sites of AAV persistence. J. Virol. 2017, 91, e02137-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, A.F.; Tobler, K.; Michaelsen, K.; Vogt, B.; Henckaerts, E.; Fraefel, C. Herpes Simplex Virus 1 Coinfection Modifies Adeno-associated Virus Genome End Recombination. J. Virol. 2021, 95, e00486-21. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, A.; Massy, Z.A.; Meijers, B.; Evenepoel, P.; Vanholder, R.; Raj, D.S. Role of the gut microbiome in uremia: A potential therapeutic target. Am. J. Kidney Dis. 2016, 67, 483–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delistefani, F.; Wallbach, M.; Müller, G.A.; Koziolek, M.J.; Grupp, C. Risk factors for catheter-related infections in patients receiving permanent dialysis catheter. BMC Nephrol. 2019, 20, 199. [Google Scholar] [CrossRef] [Green Version]
- Ducloux, D.; Legendre, M.; Bamoulid, J.; Rebibou, J.-M.; Saas, P.; Courivaud, C.; Crepin, T. ESRD-associated immune phenotype depends on dialysis modality and iron status: Clinical implications. Immun. Ageing 2018, 15, 16. [Google Scholar] [CrossRef] [PubMed]
- Taylo, L.J.; Keeler, E.L.; Bushman, F.D.; Collman, R.G. The enigmatic roles of Anelloviridae and Redondoviridae in humans. Curr. Opin. Virol. 2022, 55, 101248. [Google Scholar] [CrossRef]
- Arze, C.A.; Springer, S.; Dudas, G.; Patel, S.; Bhattacharyya, A.; Swaminathan, H.; Brugnara, C.; Delagrave, S.; Ong, T.; Kahvejian, A. Global genome analysis reveals a vast and dynamic anellovirus landscape within the human virome. Cell Host Microbe 2021, 29, 1305–1315.e1306. [Google Scholar] [CrossRef] [PubMed]
- Kaczorowska, J.; Deijs, M.; Klein, M.; Bakker, M.; Jebbink, M.F.; Sparreboom, M.; Kinsella, C.M.; Timmerman, A.L.; van der Hoek, L. Diversity and Long-Term Dynamics of Human Blood Anelloviruses. J. Virol. 2022, 96, e00109-22. [Google Scholar] [CrossRef]
- Brito, F.; Cordey, S.; Delwart, E.; Deng, X.; Tirefort, D.; Lemoine-Chaduc, C.; Zdobnov, E.; Lecompte, T.; Kaiser, L.; Waldvogel-Abramowski, S. Metagenomics analysis of the virome of 300 concentrates from a Swiss platelet bank. Vox Sang. 2018, 113, 601–604. [Google Scholar] [CrossRef] [Green Version]
- Thijssen, M.; Tacke, F.; Van Espen, L.; Cassiman, D.; Naser Aldine, M.; Nevens, F.; Van Ranst, M.; Matthijnssens, J.; Pourkarim, M. Plasma virome dynamics in chronic hepatitis B virus infected patients. Front. Microbiol. 2023, 14, 1426. [Google Scholar] [CrossRef]
- Wahid, N.M.; Israa, S. Torque Teno Virus (TTV) as a Risk Factor in Hemodialysis Process in Kirkuk. Indian J. Forensic Med. Toxicol 2019, 13, 1425–1431. [Google Scholar] [CrossRef]
- Spandole-Dinu, S.; Cimponeriu, D.G.; Crăciun, A.-M.; Radu, I.; Nica, S.; Toma, M.; Alexiu, O.A.; Iorga, C.S.; Berca, L.-M.; Nica, R. Prevalence of human anelloviruses in Romanian healthy subjects and patients with common pathologies. BMC Infect. Dis. 2018, 18, 334. [Google Scholar] [CrossRef]
- Jalali, H.; Mahdavi, M.R.; Zaeromali, N. Torque Teno Virus (TTV) among β-thalassemia and haemodialysis patients in Mazandaran Province (North of Iran). Int. J. Mol. Cell. Med. 2017, 6, 56. [Google Scholar]
- Kasraian, L.; Karimi, M.H. The Incidence rate of acute transfusion reactions in thalassemia patients referred to the Shiraz Thalassemia Centre, Shiraz, Iran, before and after the establishment of the hemovigilance system. Hemoglobin 2015, 39, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Fan, P.-C.; Kuo, G.; Lin, Y.-S.; Tsai, T.-Y.; Chang, S.-W.; Tian, Y.-C.; Lee, C.-C. Infection in advanced chronic kidney disease and subsequent adverse outcomes after dialysis initiation: A nationwide cohort study. Sci. Rep. 2020, 10, 2938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samadi, M.; Salimi, V.; Haghshenas, M.R.; Miri, S.M.; Mohebbi, S.R.; Ghaemi, A. Clinical and molecular aspects of human pegiviruses in the interaction host and infectious agent. Virol. J. 2022, 19, 41. [Google Scholar] [CrossRef]
- Wang, H.; Wan, Z.; Xu, R.; Guan, Y.; Zhu, N.; Li, J.; Xie, Z.; Lu, A.; Zhang, F.; Fu, Y. A novel human pegivirus, HPgV-2 (HHpgV-1), is tightly associated with hepatitis C virus (HCV) infection and HCV/human immunodeficiency virus type 1 coinfection. Clin. Infect. Dis. 2018, 66, 29–35. [Google Scholar] [CrossRef] [PubMed]
- N’Guessan, K.F.; Anderson, M.; Phinius, B.; Moyo, S.; Malick, A.; Mbangiwa, T.; Choga, W.T.; Makhema, J.; Marlink, R.; Essex, M. The impact of human pegivirus on CD4 cell count in HIV-positive persons in Botswana. In Proceedings of Open Forum Infectious Diseases; Oxford University Press: Oxford, UK, 2017; p. ofx222. [Google Scholar]
- Tillmann, H.L.; Heiken, H.; Knapik-Botor, A.; Heringlake, S.; Ockenga, J.; Wilber, J.C.; Goergen, B.; Detmer, J.; McMorrow, M.; Stoll, M. Infection with GB virus C and reduced mortality among HIV-infected patients. N. Engl. J. Med. 2001, 345, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Lanteri, M.C.; Vahidnia, F.; Tan, S.; Stapleton, J.T.; Norris, P.J.; Heitman, J.; Deng, X.; Keating, S.M.; Brambilla, D.; Busch, M.P. Downregulation of cytokines and chemokines by GB virus C after transmission via blood transfusion in HIV-positive blood recipients. J. Infect. Dis. 2015, 211, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Kandathil, A.J.; Cox, A.L.; Page, K.; Mohr, D.; Razaghi, R.; Ghanem, K.G.; Tuddenham, S.A.; Hsieh, Y.-H.; Evans, J.L.; Coller, K.E. Plasma virome and the risk of blood-borne infection in persons with substance use disorder. Nat. Commun. 2021, 12, 6909. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wan, Z.; Wang, J.-H.; Yang, X.; Zhang, C. Review of human pegivirus: Prevalence, transmission, pathogenesis, and clinical implication. Virulence 2022, 13, 323–340. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Thalassemia (ZN) | Hemodialysis (PD) | Blood Donors (PCL) |
|---|---|---|---|
| Number of samples | 155 | 149 | 100 |
| Age (mean ± sd) | 24.7 ± 8.5 | 45 ± 10.4 1 | 41 ± 9.7 1 |
| Sex (male/female) | 79/21 | 58/41 | 90/10 |
| Transfusion/year (mean ± sd) | 19 ± 6 | 92 ± 55 2 | NA |
| Transfusion range | 6–40 | 24–264 | NA |
| Transfused product | Red blood cell | Red blood cell | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thijssen, M.; Khamisipour, G.; Maleki, M.; Devos, T.; Li, G.; Van Ranst, M.; Matthijnssens, J.; Pourkarim, M.R. Characterization of the Human Blood Virome in Iranian Multiple Transfused Patients. Viruses 2023, 15, 1425. https://doi.org/10.3390/v15071425
Thijssen M, Khamisipour G, Maleki M, Devos T, Li G, Van Ranst M, Matthijnssens J, Pourkarim MR. Characterization of the Human Blood Virome in Iranian Multiple Transfused Patients. Viruses. 2023; 15(7):1425. https://doi.org/10.3390/v15071425
Chicago/Turabian StyleThijssen, Marijn, Gholamreza Khamisipour, Mohammad Maleki, Timothy Devos, Guangdi Li, Marc Van Ranst, Jelle Matthijnssens, and Mahmoud Reza Pourkarim. 2023. "Characterization of the Human Blood Virome in Iranian Multiple Transfused Patients" Viruses 15, no. 7: 1425. https://doi.org/10.3390/v15071425