
Mechanisms of Action of the Host-Targeting Agent Cyclosporin A and Direct-Acting Antiviral Agents against Hepatitis C Virus
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Compounds and Antibodies
2.3. The In Vitro Transcription and Electroporation of HCV RNAs
2.4. Gaussia Luciferase Reporter Assay
2.5. Western Blot Analysis
2.6. Quantitative Reverse Transcription Quantitative PCR (RT-qPCR)
2.7. Strand-Specific RT-qPCR
2.8. Fluorescence Microscopy
2.9. Limited Dilution Assay (TCID50)
2.10. Extracellular and Intracellular Infectivity Assays
2.11. Statistical Analysis
3. Results
3.1. Time Course of HCV Inhibition by Antiviral Inhibitors
3.2. Kinetics of HCV Protein Suppression
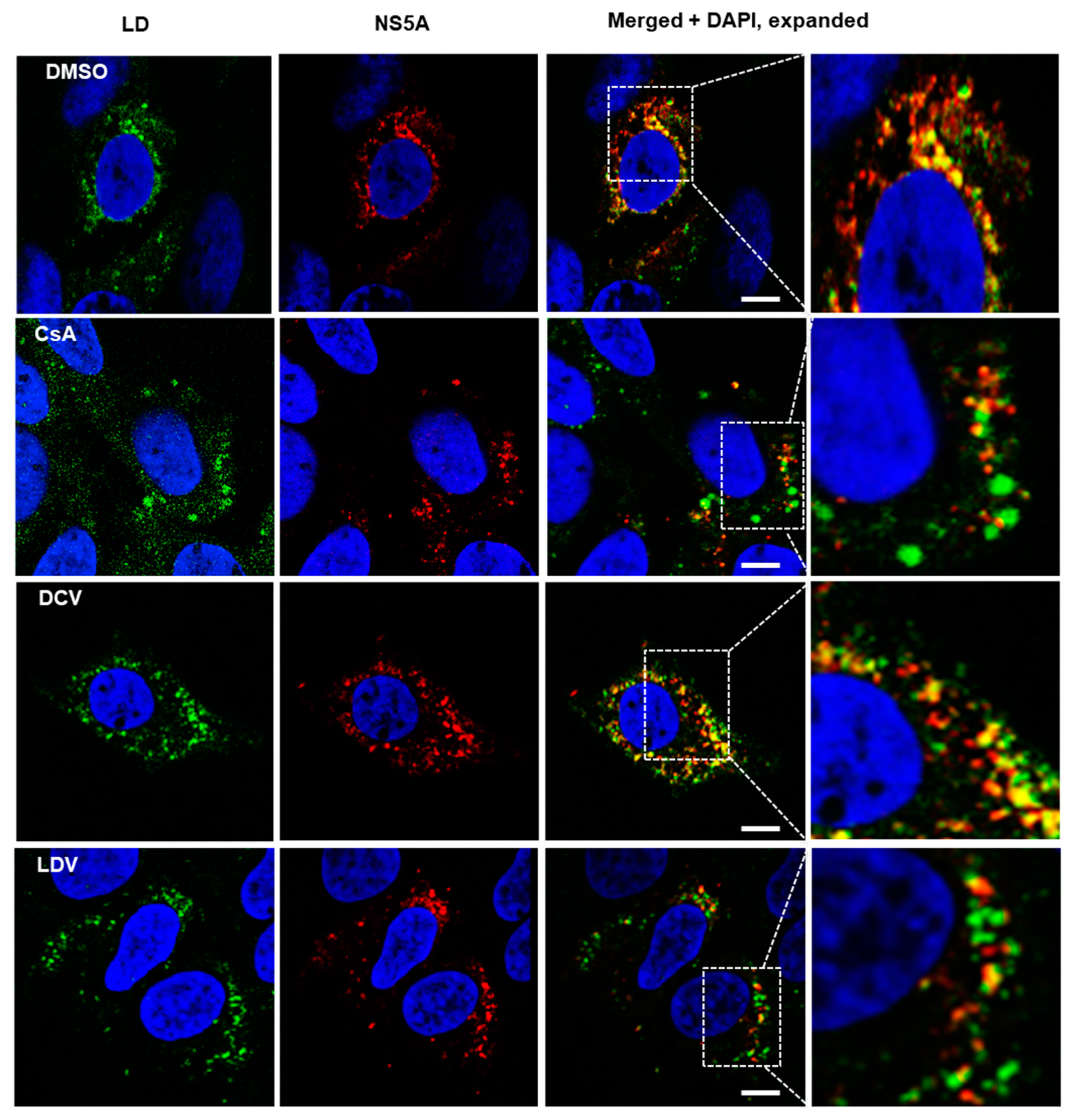
3.3. The Effect of CsA and NS5A Inhibitors on the Localization of NS5A at LDs
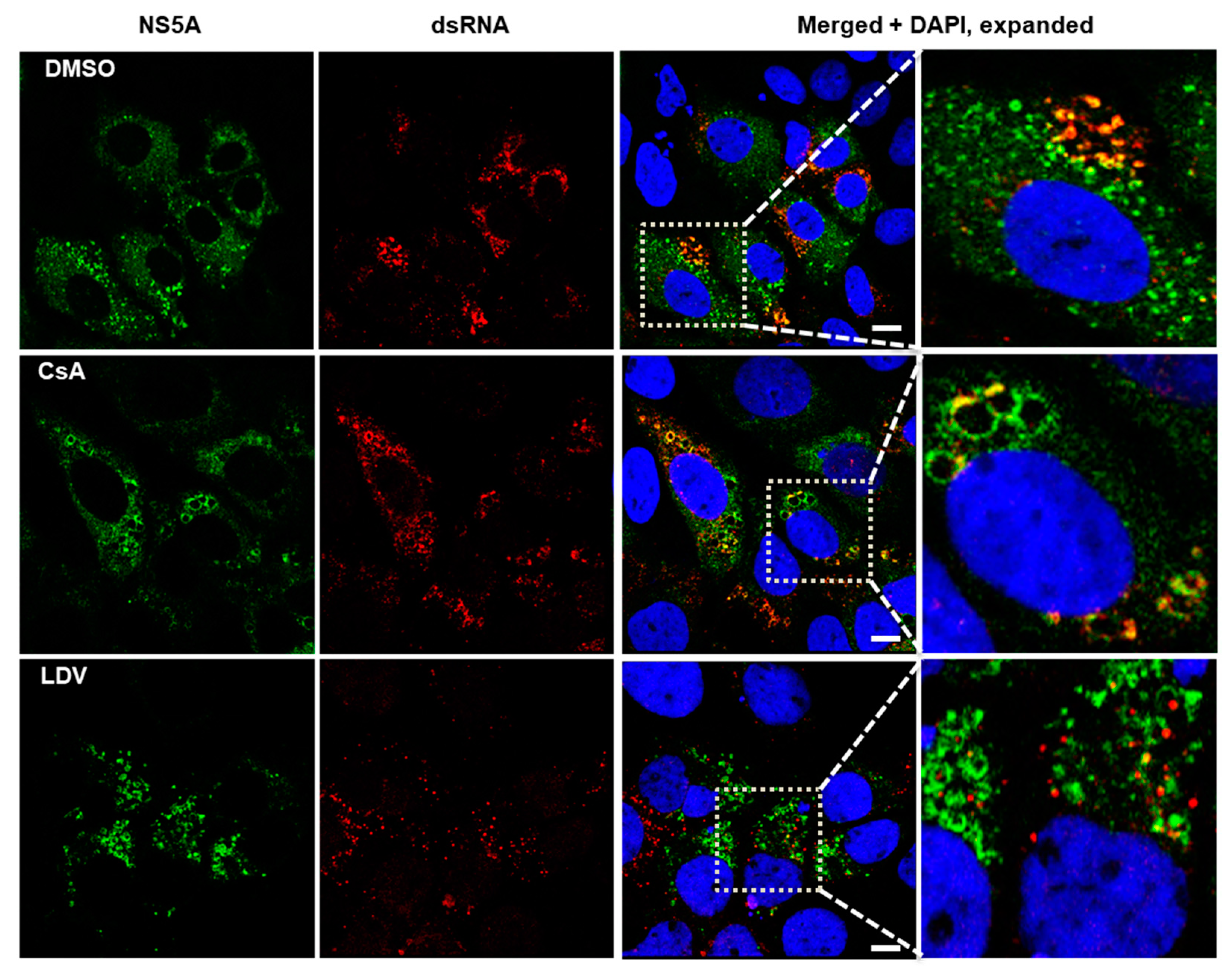
3.4. Interaction between NS5A and HCV Double-Stranded RNA
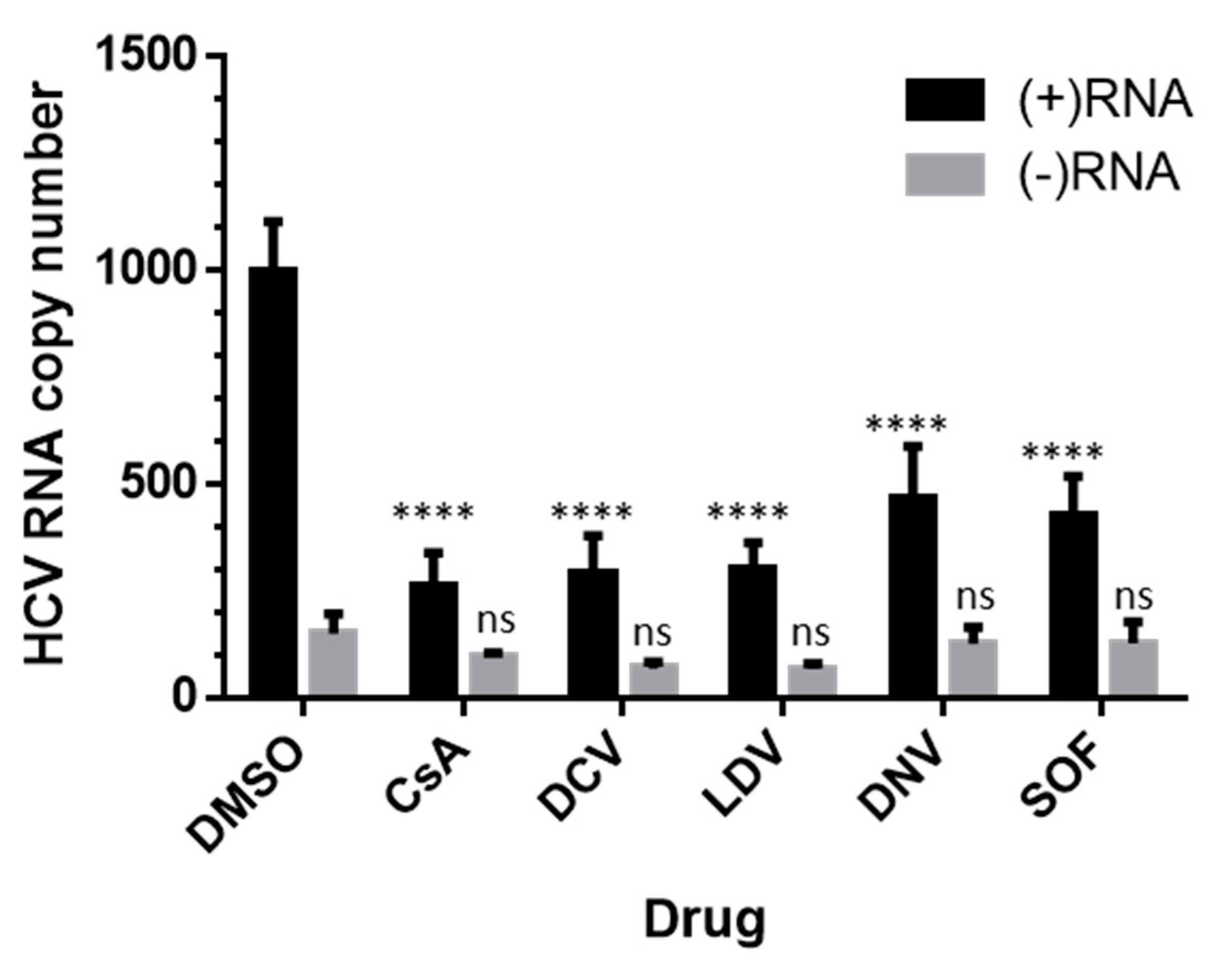
3.5. Effects on HCV RNA Assessed by Strand-Specific RT-qPCR
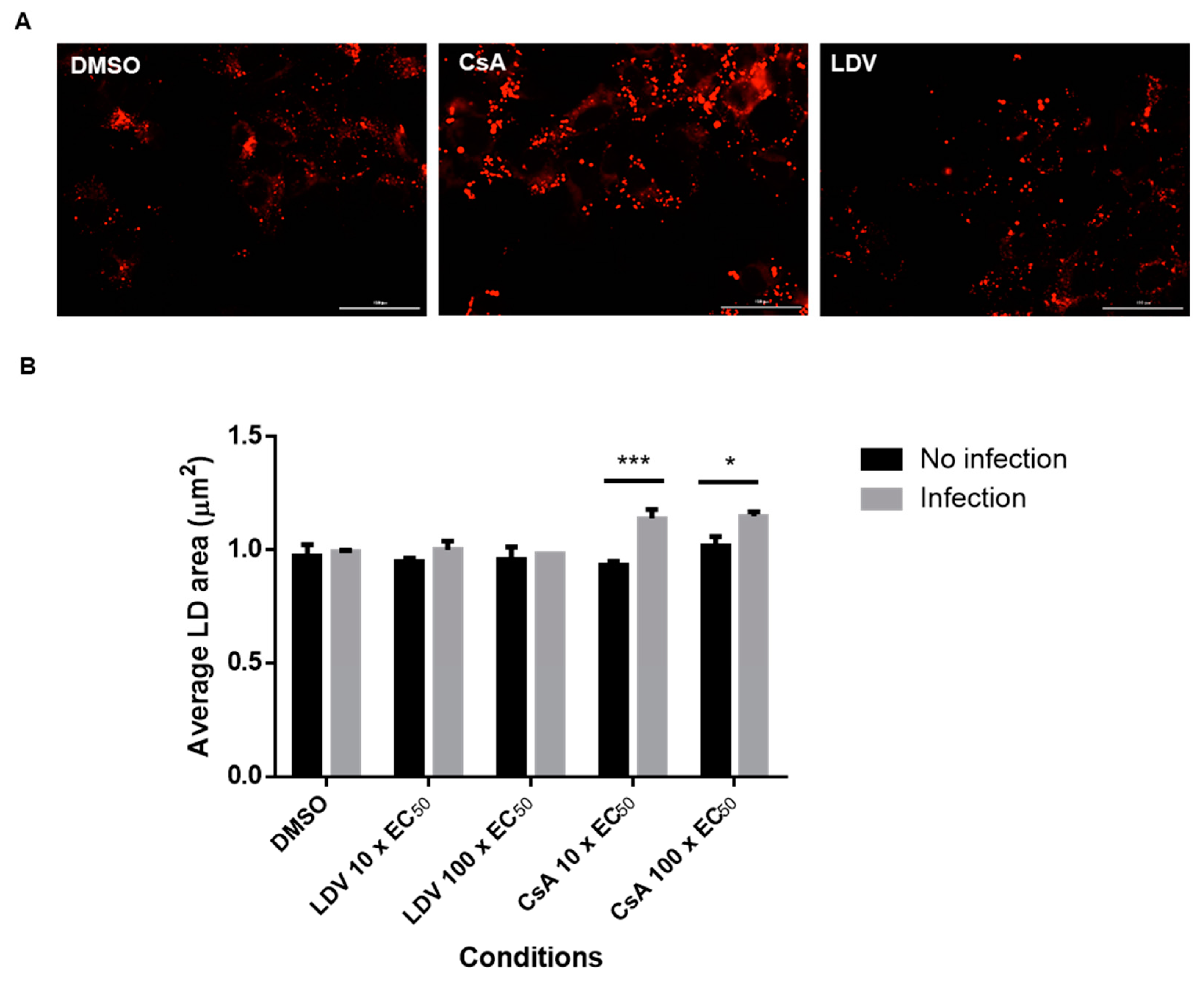
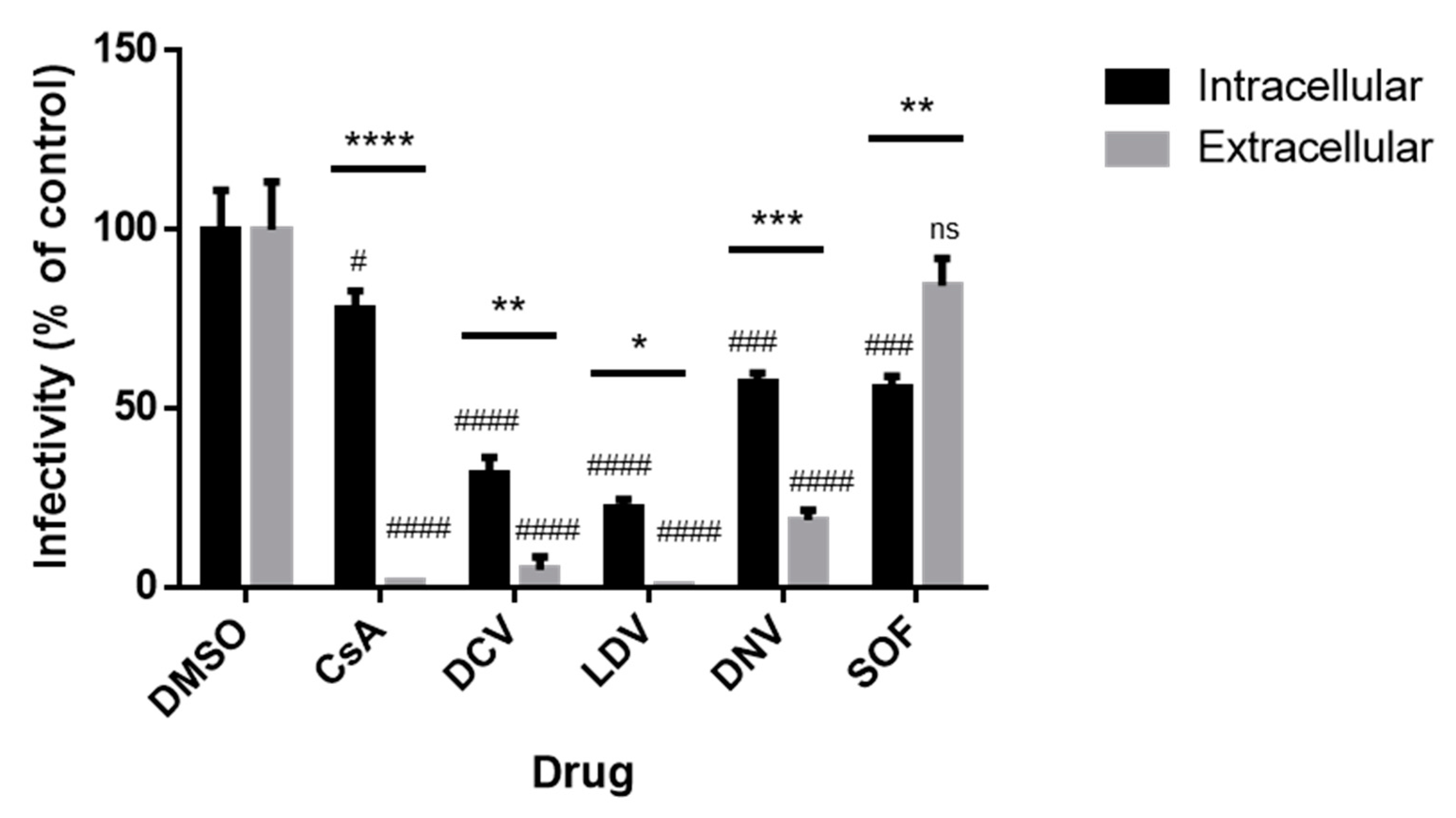
3.6. The Infectivity of Intracellular and Extracellular Viruses
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Hepatitis C. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-c (accessed on 16 June 2022).
- Manns, M.P.; Buti, M.; Gane, E.; Pawlotsky, J.M.; Razavi, H.; Terrault, N.; Younossi, Z. Hepatitis C virus infection. Nat. Rev. Dis. Prim. 2017, 3, 17006. [Google Scholar] [CrossRef]
- Feld, J.J.; Hoofnagle, J.H. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 2005, 436, 967–972. [Google Scholar] [CrossRef]
- Sulkowski, M.S.; Cooper, C.; Hunyady, B.; Jia, J.; Ogurtsov, P.; Peck-Radosavljevic, M.; Shiffman, M.L.; Yurdaydin, C.; Dalgard, O. Management of adverse effects of Peg-IFN and ribavirin therapy for hepatitis C. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Gotte, M.; Feld, J.J. Direct-acting antiviral agents for hepatitis C: Structural and mechanistic insights. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 338–351. [Google Scholar] [CrossRef]
- Schinazi, R.; Halfon, P.; Marcellin, P.; Asselah, T. HCV direct-acting antiviral agents: The best interferon-free combinations. Liver Int. 2014, 34 (Suppl. S1), 69–78. [Google Scholar] [CrossRef]
- Dietz, C.; Maasoumy, B. Direct-Acting Antiviral Agents for Hepatitis C Virus Infection-from Drug Discovery to Successful Implementation in Clinical Practice. Viruses 2022, 14, 1325. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Frese, M.; Pietschmann, T. Novel insights into hepatitis C virus replication and persistence. Adv. Virus. Res. 2004, 63, 71–180. [Google Scholar] [CrossRef]
- Moradpour, D.; Penin, F. Hepatitis C virus proteins: From structure to function. Curr. Top. Microbiol. Immunol. 2013, 369, 113–142. [Google Scholar] [CrossRef]
- Penin, F.; Brass, V.; Appel, N.; Ramboarina, S.; Montserret, R.; Ficheux, D.; Blum, H.E.; Bartenschlager, R.; Moradpour, D. Structure and function of the membrane anchor domain of hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 2004, 279, 40835–40843. [Google Scholar] [CrossRef]
- Lim, P.J.; Chatterji, U.; Cordek, D.; Sharma, S.D.; Garcia-Rivera, J.A.; Cameron, C.E.; Lin, K.; Targett-Adams, P.; Gallay, P.A. Correlation between NS5A dimerization and hepatitis C virus replication. J. Biol. Chem. 2012, 287, 30861–30873. [Google Scholar] [CrossRef]
- Tellinghuisen, T.L.; Marcotrigiano, J.; Rice, C.M. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 2005, 435, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, S.; Nichols, A.K.; Saravanabalaji, D.; Welsch, C.; Yi, M. HCV NS5A dimer interface residues regulate HCV replication by controlling its self-interaction, hyperphosphorylation, subcellular localization and interaction with cyclophilin A. PLoS Pathog. 2018, 14, e1007177. [Google Scholar] [CrossRef] [PubMed]
- Ross-Thriepland, D.; Harris, M. Hepatitis C virus NS5A: Enigmatic but still promiscuous 10 years on! J. Gen. Virol. 2015, 96, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Alvisi, G.; Madan, V.; Bartenschlager, R. Hepatitis C virus and host cell lipids: An intimate connection. RNA Biol. 2011, 8, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Camus, G.; Herker, E.; Modi, A.A.; Haas, J.T.; Ramage, H.R.; Farese, R.V., Jr.; Ott, M. Diacylglycerol acyltransferase-1 localizes hepatitis C virus NS5A protein to lipid droplets and enhances NS5A interaction with the viral capsid core. J. Biol. Chem. 2013, 288, 9915–9923. [Google Scholar] [CrossRef] [PubMed]
- Ross-Thriepland, D.; Amako, Y.; Harris, M. The C terminus of NS5A domain II is a key determinant of hepatitis C virus genome replication, but is not required for virion assembly and release. J. Gen. Virol. 2013, 94, 1009–1018. [Google Scholar] [CrossRef]
- Tellinghuisen, T.L.; Marcotrigiano, J.; Gorbalenya, A.E.; Rice, C.M. The NS5A protein of hepatitis C virus is a zinc metalloprotein. J. Biol. Chem. 2004, 279, 48576–48587. [Google Scholar] [CrossRef]
- Appel, N.; Zayas, M.; Miller, S.; Krijnse-Locker, J.; Schaller, T.; Friebe, P.; Kallis, S.; Engel, U.; Bartenschlager, R. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 2008, 4, e1000035. [Google Scholar] [CrossRef]
- Verdegem, D.; Badillo, A.; Wieruszeski, J.M.; Landrieu, I.; Leroy, A.; Bartenschlager, R.; Penin, F.; Lippens, G.; Hanoulle, X. Domain 3 of NS5A protein from the hepatitis C virus has intrinsic alpha-helical propensity and is a substrate of cyclophilin A. J. Biol. Chem. 2011, 286, 20441–20454. [Google Scholar] [CrossRef]
- Dujardin, M.; Madan, V.; Gandhi, N.S.; Cantrelle, F.X.; Launay, H.; Huvent, I.; Bartenschlager, R.; Lippens, G.; Hanoulle, X. Cyclophilin A allows the allosteric regulation of a structural motif in the disordered domain 2 of NS5A and thereby fine-tunes HCV RNA replication. J. Biol. Chem. 2019, 294, 13171–13185. [Google Scholar] [CrossRef]
- Foster, T.L.; Gallay, P.; Stonehouse, N.J.; Harris, M. Cyclophilin A interacts with domain II of hepatitis C virus NS5A and stimulates RNA binding in an isomerase-dependent manner. J. Virol. 2011, 85, 7460–7464. [Google Scholar] [CrossRef]
- Abe, T.; Minami, N.; Bawono, R.G.; Matsui, C.; Deng, L.; Fukuhara, T.; Matsuura, Y.; Shoji, I. ISGylation of Hepatitis C Virus NS5A Protein Promotes Viral RNA Replication via Recruitment of Cyclophilin A. J. Virol. 2020, 94, e00532-20. [Google Scholar] [CrossRef] [PubMed]
- Galat, A. Function-dependent clustering of orthologues and paralogues of cyclophilins. Proteins 2004, 56, 808–820. [Google Scholar] [CrossRef]
- Fernandes, F.; Ansari, I.U.; Striker, R. Cyclosporine inhibits a direct interaction between cyclophilins and hepatitis C NS5A. PLoS ONE 2010, 5, e9815. [Google Scholar] [CrossRef]
- Yang, F.; Robotham, J.M.; Nelson, H.B.; Irsigler, A.; Kenworthy, R.; Tang, H. Cyclophilin A is an essential cofactor for hepatitis C virus infection and the principal mediator of cyclosporine resistance in vitro. J. Virol. 2008, 82, 5269–5278. [Google Scholar] [CrossRef]
- Kaul, A.; Stauffer, S.; Berger, C.; Pertel, T.; Schmitt, J.; Kallis, S.; Zayas, M.; Lohmann, V.; Luban, J.; Bartenschlager, R. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog. 2009, 5, e1000546. [Google Scholar] [CrossRef]
- Jesudian, A.B.; Gambarin-Gelwan, M.; Jacobson, I.M. Advances in the treatment of hepatitis C virus infection. Gastroenterol. Hepatol. 2012, 8, 91–101. [Google Scholar]
- Gaska, J.M.; Balev, M.; Ding, Q.; Heller, B.; Ploss, A. Differences across cyclophilin A orthologs contribute to the host range restriction of hepatitis C virus. Elife 2019, 8, e44436. [Google Scholar] [CrossRef]
- Liu, J.; Farmer, J.D., Jr.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66, 807–815. [Google Scholar] [CrossRef]
- Ma, S.; Boerner, J.E.; TiongYip, C.; Weidmann, B.; Ryder, N.S.; Cooreman, M.P.; Lin, K. NIM811, a cyclophilin inhibitor, exhibits potent in vitro activity against hepatitis C virus alone or in combination with alpha interferon. Antimicrob. Agents. Chemother. 2006, 50, 2976–2982. [Google Scholar] [CrossRef]
- Paeshuyse, J.; Kaul, A.; De Clercq, E.; Rosenwirth, B.; Dumont, J.M.; Scalfaro, P.; Bartenschlager, R.; Neyts, J. The non-immunosuppressive cyclosporin DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology 2006, 43, 761–770. [Google Scholar] [CrossRef]
- Ciesek, S.; Steinmann, E.; Wedemeyer, H.; Manns, M.P.; Neyts, J.; Tautz, N.; Madan, V.; Bartenschlager, R.; von Hahn, T.; Pietschmann, T. Cyclosporine A inhibits hepatitis C virus nonstructural protein 2 through cyclophilin A. Hepatology 2009, 50, 1638–1645. [Google Scholar] [CrossRef]
- Liu, Z.; Robida, J.M.; Chinnaswamy, S.; Yi, G.; Robotham, J.M.; Nelson, H.B.; Irsigler, A.; Kao, C.C.; Tang, H. Mutations in the hepatitis C virus polymerase that increase RNA binding can confer resistance to cyclosporine A. Hepatology 2009, 50, 25–33. [Google Scholar] [CrossRef]
- Robida, J.M.; Nelson, H.B.; Liu, Z.; Tang, H. Characterization of hepatitis C virus subgenomic replicon resistance to cyclosporine in vitro. J. Virol. 2007, 81, 5829–5840. [Google Scholar] [CrossRef]
- Yang, F.; Robotham, J.M.; Grise, H.; Frausto, S.; Madan, V.; Zayas, M.; Bartenschlager, R.; Robinson, M.; Greenstein, A.E.; Nag, A.; et al. A major determinant of cyclophilin dependence and cyclosporine susceptibility of hepatitis C virus identified by a genetic approach. PLoS. Pathog. 2010, 6, e1001118. [Google Scholar] [CrossRef]
- Chatel-Chaix, L.; Baril, M.; Lamarre, D. Hepatitis C Virus NS3/4A Protease Inhibitors: A Light at the End of the Tunnel. Viruses 2010, 2, 1752–1765. [Google Scholar] [CrossRef]
- Marks, K.M.; Jacobson, I.M. The first wave: HCV NS3 protease inhibitors telaprevir and boceprevir. Antivir. Ther. 2012, 17, 1119–1131. [Google Scholar] [CrossRef]
- Miao, M.; Jing, X.; De Clercq, E.; Li, G. Danoprevir for the Treatment of Hepatitis C Virus Infection: Design, Development, and Place in Therapy. Drug. Des. Devel. Ther. 2020, 14, 2759–2774. [Google Scholar] [CrossRef]
- Gao, M.; Nettles, R.E.; Belema, M.; Snyder, L.B.; Nguyen, V.N.; Fridell, R.A.; Serrano-Wu, M.H.; Langley, D.R.; Sun, J.H.; O’Boyle, D.R., 2nd; et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 2010, 465, 96–100. [Google Scholar] [CrossRef]
- Afdhal, N.; Zeuzem, S.; Kwo, P.; Chojkier, M.; Gitlin, N.; Puoti, M.; Romero-Gomez, M.; Zarski, J.P.; Agarwal, K.; Buggisch, P.; et al. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N. Engl. J. Med. 2014, 370, 1889–1898. [Google Scholar] [CrossRef] [PubMed]
- Afdhal, N.; Reddy, K.R.; Nelson, D.R.; Lawitz, E.; Gordon, S.C.; Schiff, E.; Nahass, R.; Ghalib, R.; Gitlin, N.; Herring, R.; et al. Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N. Engl. J. Med. 2014, 370, 1483–1493. [Google Scholar] [CrossRef]
- Gamal, N.; Gitto, S.; Andreone, P. Efficacy and Safety of Daclatasvir in Hepatitis C: An Overview. J. Clin. Transl. Hepatol. 2016, 4, 336–344. [Google Scholar] [CrossRef]
- Fridell, R.A.; Wang, C.; Sun, J.H.; O’Boyle, D.R., 2nd; Nower, P.; Valera, L.; Qiu, D.; Roberts, S.; Huang, X.; Kienzle, B.; et al. Genotypic and phenotypic analysis of variants resistant to hepatitis C virus nonstructural protein 5A replication complex inhibitor BMS-790052 in humans: In vitro and in vivo correlations. Hepatology 2011, 54, 1924–1935. [Google Scholar] [CrossRef]
- Pawlotsky, J.M. NS5A inhibitors in the treatment of hepatitis C. J. Hepatol. 2013, 59, 375–382. [Google Scholar] [CrossRef]
- Targett-Adams, P.; Graham, E.J.; Middleton, J.; Palmer, A.; Shaw, S.M.; Lavender, H.; Brain, P.; Tran, T.D.; Jones, L.H.; Wakenhut, F.; et al. Small molecules targeting hepatitis C virus-encoded NS5A cause subcellular redistribution of their target: Insights into compound modes of action. J. Virol. 2011, 85, 6353–6368. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Peng, B.; Corsa, A.; Yu, M.; Nash, M.; Lee, Y.-J.; Xu, Y.; Kirschberg, T.; Tian, Y.; Taylor, J.; et al. 1172 Antiviral Activity and Resistance Profile of the Novel Hcv Ns5a Inhibitor GS-5885. J. Hepatol. 2012, 56, S464. [Google Scholar] [CrossRef]
- Zeuzem, S.; Foster, G.R.; Wang, S.; Asatryan, A.; Gane, E.; Feld, J.J.; Asselah, T.; Bourliere, M.; Ruane, P.J.; Wedemeyer, H.; et al. Glecaprevir-Pibrentasvir for 8 or 12 Weeks in HCV Genotype 1 or 3 Infection. N. Engl. J. Med. 2018, 378, 354–369. [Google Scholar] [CrossRef]
- Feld, J.J.; Jacobson, I.M.; Hezode, C.; Asselah, T.; Ruane, P.J.; Gruener, N.; Abergel, A.; Mangia, A.; Lai, C.L.; Chan, H.L.; et al. Sofosbuvir and Velpatasvir for HCV Genotype 1, 2, 4, 5, and 6 Infection. N. Engl. J. Med. 2015, 373, 2599–2607. [Google Scholar] [CrossRef]
- Curry, M.P.; O’Leary, J.G.; Bzowej, N.; Muir, A.J.; Korenblat, K.M.; Fenkel, J.M.; Reddy, K.R.; Lawitz, E.; Flamm, S.L.; Schiano, T.; et al. Sofosbuvir and Velpatasvir for HCV in Patients with Decompensated Cirrhosis. N. Engl. J. Med. 2015, 373, 2618–2628. [Google Scholar] [CrossRef]
- Foster, G.R.; Afdhal, N.; Roberts, S.K.; Brau, N.; Gane, E.J.; Pianko, S.; Lawitz, E.; Thompson, A.; Shiffman, M.L.; Cooper, C.; et al. Sofosbuvir and Velpatasvir for HCV Genotype 2 and 3 Infection. N. Engl. J. Med. 2015, 373, 2608–2617. [Google Scholar] [CrossRef]
- Llaneras, J.; Riveiro-Barciela, M.; Lens, S.; Diago, M.; Cachero, A.; Garcia-Samaniego, J.; Conde, I.; Arencibia, A.; Arenas, J.; Gea, F.; et al. Effectiveness and safety of sofosbuvir/velpatasvir/voxilaprevir in patients with chronic hepatitis C previously treated with DAAs. J. Hepatol. 2019, 71, 666–672. [Google Scholar] [CrossRef]
- Degasperi, E.; Spinetti, A.; Lombardi, A.; Landonio, S.; Rossi, M.C.; Pasulo, L.; Pozzoni, P.; Giorgini, A.; Fabris, P.; Romano, A.; et al. Real-life effectiveness and safety of sofosbuvir/velpatasvir/voxilaprevir in hepatitis C patients with previous DAA failure. J. Hepatol. 2019, 71, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Buti, M.; Flisiak, R.; Kao, J.H.; Chuang, W.L.; Streinu-Cercel, A.; Tabak, F.; Calistru, P.; Goeser, T.; Rasenack, J.; Horban, A.; et al. Alisporivir with peginterferon/ribavirin in patients with chronic hepatitis C genotype 1 infection who failed to respond to or relapsed after prior interferon-based therapy: FUNDAMENTAL, a Phase II trial. J. Viral. Hepat. 2015, 22, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Flisiak, R.; Jaroszewicz, J.; Flisiak, I.; Lapinski, T. Update on alisporivir in treatment of viral hepatitis C. Expert. Opin. Investig. Drugs. 2012, 21, 375–382. [Google Scholar] [CrossRef]
- Pawlotsky, J.M.; Flisiak, R.; Sarin, S.K.; Rasenack, J.; Piratvisuth, T.; Chuang, W.L.; Peng, C.Y.; Foster, G.R.; Shah, S.; Wedemeyer, H.; et al. Alisporivir plus ribavirin, interferon free or in combination with pegylated interferon, for hepatitis C virus genotype 2 or 3 infection. Hepatology. 2015, 62, 1013–1023. [Google Scholar] [CrossRef]
- Liu, D.; Ji, J.; Ndongwe, T.P.; Michailidis, E.; Rice, C.M.; Ralston, R.; Sarafianos, S.G. Fast hepatitis C virus RNA elimination and NS5A redistribution by NS5A inhibitors studied by a multiplex assay approach. Antimicrob. Agents Chemother. 2015, 59, 3482–3492. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 2005, 102, 9294–9299. [Google Scholar] [CrossRef] [PubMed]
- Marukian, S.; Jones, C.T.; Andrus, L.; Evans, M.J.; Ritola, K.D.; Charles, E.D.; Rice, C.M.; Dustin, L.B. Cell culture-produced hepatitis C virus does not infect peripheral blood mononuclear cells. Hepatology 2008, 48, 1843–1850. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; et al. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef]
- Komurian-Pradel, F.; Perret, M.; Deiman, B.; Sodoyer, M.; Lotteau, V.; Paranhos-Baccala, G.; Andre, P. Strand specific quantitative real-time PCR to study replication of hepatitis C virus genome. J. Virol. Methods. 2004, 116, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; Meunch, H.A. A Simple Method of Estimating Fifty per Cent Endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Dittmar, M.; Lee, J.S.; Whig, K.; Segrist, E.; Li, M.; Kamalia, B.; Castellana, L.; Ayyanathan, K.; Cardenas-Diaz, F.L.; Morrisey, E.E.; et al. Drug repurposing screens reveal cell-type-specific entry pathways and FDA-approved drugs active against SARS-CoV-2. Cell Rep. 2021, 35, 108959. [Google Scholar] [CrossRef]
- Targett-Adams, P.; Boulant, S.; McLauchlan, J. Visualization of double-stranded RNA in cells supporting hepatitis C virus RNA replication. J. Virol. 2008, 82, 2182–2195. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Tedbury, P.R.; Lan, S.; Huber, A.D.; Puray-Chavez, M.N.; Ji, J.; Michailidis, E.; Saeed, M.; Ndongwe, T.P.; Bassit, L.C.; et al. Visualization of Positive and Negative Sense Viral RNA for Probing the Mechanism of Direct-Acting Antivirals against Hepatitis C Virus. Viruses. 2019, 11, 1039. [Google Scholar] [CrossRef] [PubMed]
- Filipe, A.; McLauchlan, J. Hepatitis C virus and lipid droplets: Finding a niche. Trends. Mol. Med. 2015, 21, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Hueging, K.; Weller, R.; Doepke, M.; Vieyres, G.; Todt, D.; Wolk, B.; Vondran, F.W.; Geffers, R.; Lauber, C.; Kaderali, L.; et al. Several Human Liver Cell Expressed Apolipoproteins Complement HCV Virus Production with Varying Efficacy Conferring Differential Specific Infectivity to Released Viruses. PLoS ONE 2015, 10, e0134529. [Google Scholar] [CrossRef] [PubMed]
- Gastaminza, P.; Kapadia, S.B.; Chisari, F.V. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J. Virol. 2006, 80, 11074–11081. [Google Scholar] [CrossRef]
- Huang, H.; Sun, F.; Owen, D.M.; Li, W.; Chen, Y.; Gale, M., Jr.; Ye, J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. USA 2007, 104, 5848–5853. [Google Scholar] [CrossRef]
- Chang, K.S.; Jiang, J.; Cai, Z.; Luo, G. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 2007, 81, 13783–13793. [Google Scholar] [CrossRef] [PubMed]
- Ascher, D.B.; Wielens, J.; Nero, T.L.; Doughty, L.; Morton, C.J.; Parker, M.W. Potent hepatitis C inhibitors bind directly to NS5A and reduce its affinity for RNA. Sci. Rep. 2014, 4, 4765. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.R.; An, N.; Liu, Z.L.; Xu, F.W.; Raniga, K.; Li, Q.J.; Zhou, R.; Wang, J.; Zhang, Y.X.; Zhou, J.M.; et al. Human MxB Inhibits the Replication of Hepatitis C Virus. J. Virol. 2019, 93, e01285-18. [Google Scholar] [CrossRef] [PubMed]
- Waller, H.; Chatterji, U.; Gallay, P.; Parkinson, T.; Targett-Adams, P. The use of AlphaLISA technology to detect interaction between hepatitis C virus-encoded NS5A and cyclophilin A. J. Virol. Methods. 2010, 165, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Watashi, K.; Inoue, D.; Hijikata, M.; Shimotohno, K. Identification of cellular and viral factors related to anti-hepatitis C virus activity of cyclophilin inhibitor. Cancer. Sci. 2009, 100, 1943–1950. [Google Scholar] [CrossRef] [PubMed]
- von Hahn, T.; Schiene-Fischer, C.; Van, N.D.; Pfaender, S.; Karavul, B.; Steinmann, E.; Potthoff, A.; Strassburg, C.; Hamdi, N.; Abdelaziz, A.I.; et al. Hepatocytes that express variants of cyclophilin A are resistant to HCV infection and replication. Gastroenterology 2012, 143, 439–447. [Google Scholar] [CrossRef]
- Nag, A.; Robotham, J.M.; Tang, H. Suppression of viral RNA binding and the assembly of infectious hepatitis C virus particles in vitro by cyclophilin inhibitors. J. Virol. 2012, 86, 12616–12624. [Google Scholar] [CrossRef]
- Madan, V.; Paul, D.; Lohmann, V.; Bartenschlager, R. Inhibition of HCV replication by cyclophilin antagonists is linked to replication fitness and occurs by inhibition of membranous web formation. Gastroenterology 2014, 146, 1361–1372.e1–9. [Google Scholar] [CrossRef]
- Chatterji, U.; Bobardt, M.; Tai, A.; Wood, M.; Gallay, P.A. Cyclophilin and NS5A inhibitors, but not other anti-hepatitis C virus (HCV) agents, preclude HCV-mediated formation of double-membrane-vesicle viral factories. Antimicrob. Agents. Chemother. 2015, 59, 2496–2507. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, U.; Bobardt, M.; Schaffer, L.; Wood, M.; Gallay, P.A. Cyclophilin Inhibitors Remodel the Endoplasmic Reticulum of HCV-Infected Cells in a Unique Pattern Rendering Cells Impervious to a Reinfection. PLoS ONE 2016, 11, e0159511. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yang, F.; Robotham, J.M.; Tang, H. Critical role of cyclophilin A and its prolyl-peptidyl isomerase activity in the structure and function of the hepatitis C virus replication complex. J. Virol. 2009, 83, 6554–6565. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; An, N.; Yin, X.; Zhang, R.; Shao, H.; Yi, D.; Cen, S. MxB Disrupts Hepatitis C Virus NS5A-CypA Complex: Insights From a Combined Theoretical and Experimental Approach. Front. Microbiol. 2022, 13, 849084. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.J.; Lin, K.; Compton, T.; Wiedmann, B. Inhibition of cyclophilins alters lipid trafficking and blocks hepatitis C virus secretion. Virol. J. 2011, 8, 329. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Ma, Y.; Yates, J.; Lemon, S.M. Trans-complementation of an NS2 defect in a late step in hepatitis C virus (HCV) particle assembly and maturation. PLoS Pathog. 2009, 5, e1000403. [Google Scholar] [CrossRef]
- Badillo, A.; Receveur-Brechot, V.; Sarrazin, S.; Cantrelle, F.X.; Delolme, F.; Fogeron, M.L.; Molle, J.; Montserret, R.; Bockmann, A.; Bartenschlager, R.; et al. Overall Structural Model of NS5A Protein from Hepatitis C Virus and Modulation by Mutations Confering Resistance of Virus Replication to Cyclosporin A. Biochemistry 2017, 56, 3029–3048. [Google Scholar] [CrossRef] [PubMed]
- Ngure, M.; Issur, M.; Shkriabai, N.; Liu, H.W.; Cosa, G.; Kvaratskhelia, M.; Gotte, M. Interactions of the Disordered Domain II of Hepatitis C Virus NS5A with Cyclophilin A, NS5B, and Viral RNA Show Extensive Overlap. ACS Infect. Dis. 2016, 2, 839–851. [Google Scholar] [CrossRef]
- Jones, C.T.; Murray, C.L.; Eastman, D.K.; Tassello, J.; Rice, C.M. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 2007, 81, 8374–8383. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, D.; Ndongwe, T.P.; Ji, J.; Huber, A.D.; Michailidis, E.; Rice, C.M.; Ralston, R.; Tedbury, P.R.; Sarafianos, S.G. Mechanisms of Action of the Host-Targeting Agent Cyclosporin A and Direct-Acting Antiviral Agents against Hepatitis C Virus. Viruses 2023, 15, 981. https://doi.org/10.3390/v15040981
Liu D, Ndongwe TP, Ji J, Huber AD, Michailidis E, Rice CM, Ralston R, Tedbury PR, Sarafianos SG. Mechanisms of Action of the Host-Targeting Agent Cyclosporin A and Direct-Acting Antiviral Agents against Hepatitis C Virus. Viruses. 2023; 15(4):981. https://doi.org/10.3390/v15040981
Chicago/Turabian StyleLiu, Dandan, Tanya P. Ndongwe, Juan Ji, Andrew D. Huber, Eleftherios Michailidis, Charles M. Rice, Robert Ralston, Philip R. Tedbury, and Stefan G. Sarafianos. 2023. "Mechanisms of Action of the Host-Targeting Agent Cyclosporin A and Direct-Acting Antiviral Agents against Hepatitis C Virus" Viruses 15, no. 4: 981. https://doi.org/10.3390/v15040981