
Coronaviral Infection and Interferon Response: The Virus-Host Arms Race and COVID-19


Abstract
:1. Introduction
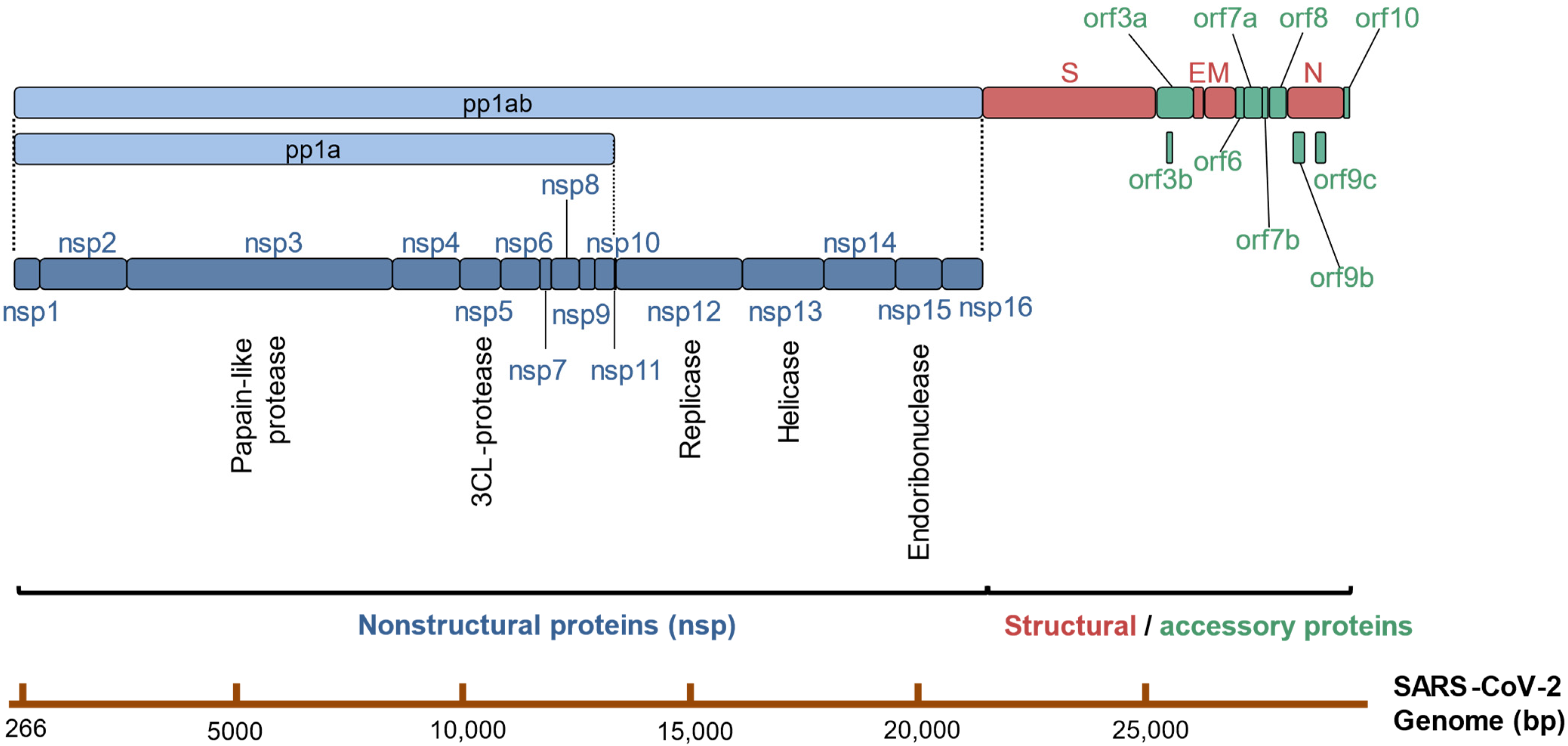
2. Overview of the SARS-CoV-2 Life Cycle
3. Pattern-Recognition Receptors and Interferons
3.1. Pattern-Recognition Receptors
3.2. Type I Interferon Pathway
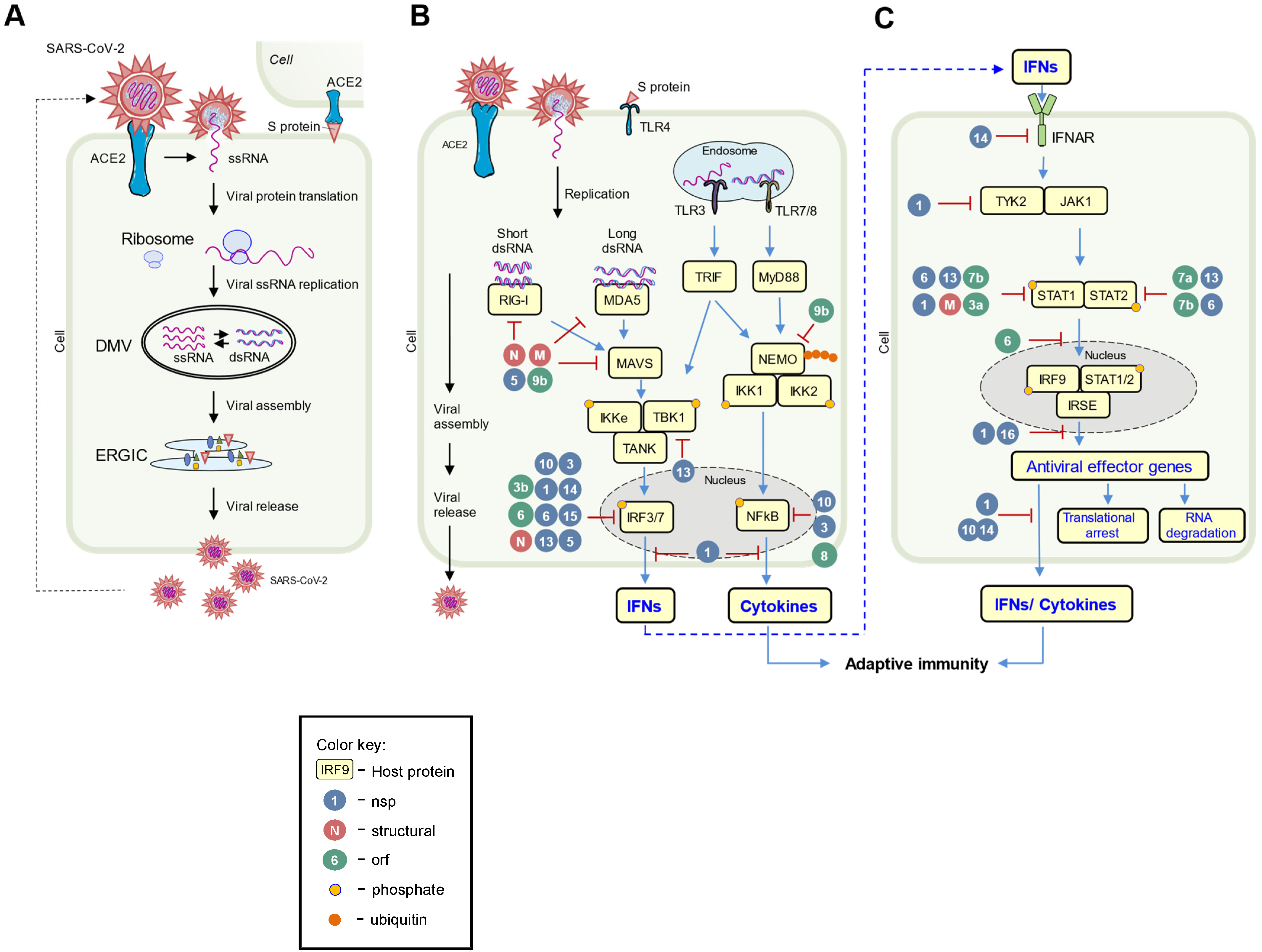
4. Host-Virus Arms Race
4.1. Arms Race in Pathogen Sensing and IFN Induction
4.2. Arms Race in IFN Signaling
4.3. Arms Race between Virus and IFN-Induced Effectors, and Cellular Events
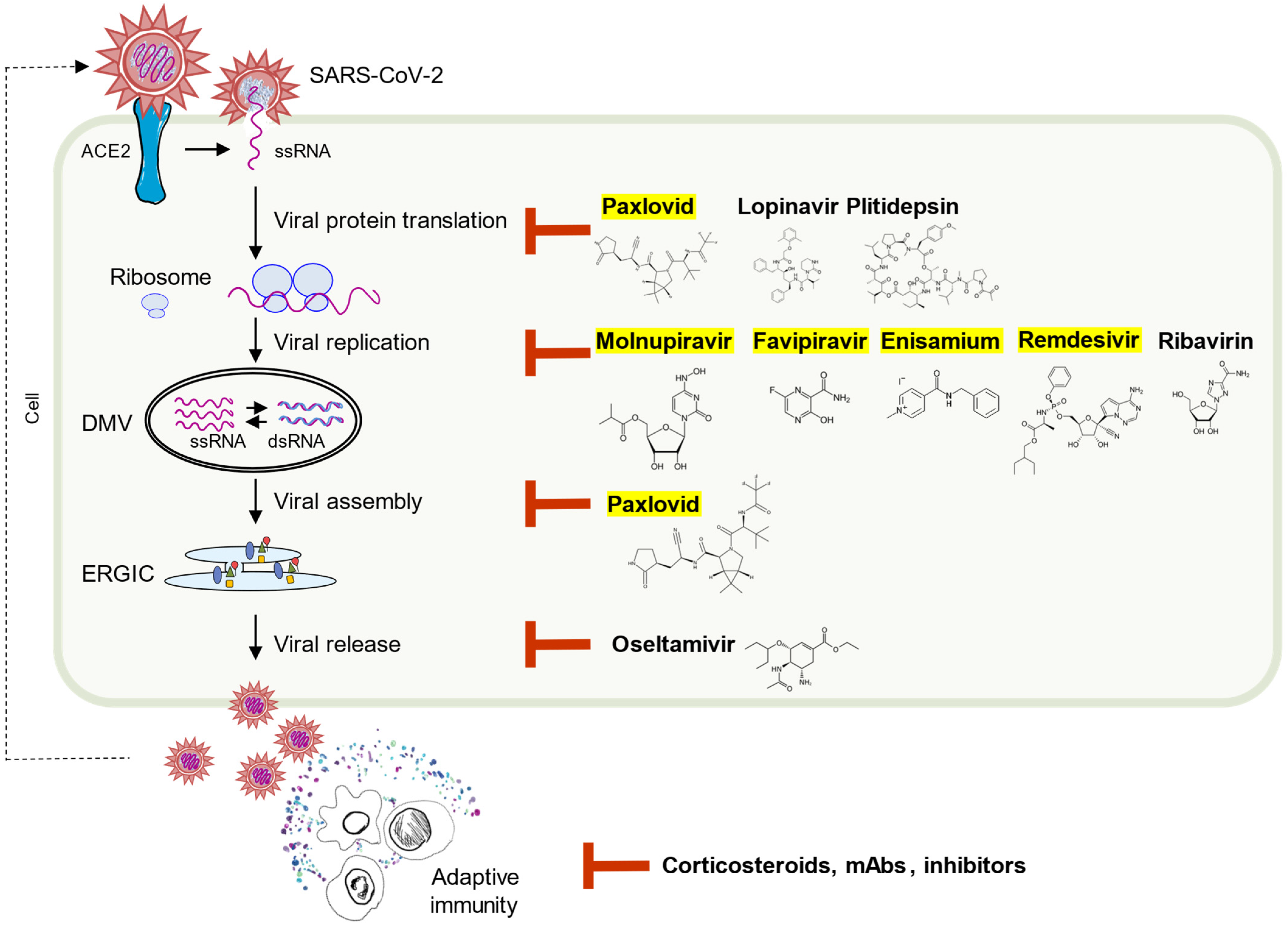
5. Beyond the Nature—Antiviral Pharmaceuticals
6. Conclusions and Future Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- WHO. Available online: https://covid19.who.int/ (accessed on 30 May 2022).
- Payne, S. Family Coronaviridae. In Viruses: From Understanding to Investigation; Elsevier: Amsterdam, The Netherlands, 2017; pp. 149–158. [Google Scholar]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Christie, M.J.; Irving, A.T.; Forster, S.C.; Marsland, B.J.; Hansbro, P.M.; Hertzog, P.J.; Nold-Petry, C.A.; Nold, M.F. Of bats and men: Immunomodulatory treatment options for COVID-19 guided by the immunopathology of SARS-CoV-2 infection. Sci. Immunol. 2021, 6, eabd0205. [Google Scholar] [CrossRef]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y.; et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat. Microbiol. 2021, 6, 899–909. [Google Scholar] [CrossRef]
- Romano, M.; Ruggiero, A.; Squeglia, F.; Maga, G.; Berisio, R. A Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping. Cells 2020, 9, 1267. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [Green Version]
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 2013, 4, e00524–e00613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snijder, E.J.; Limpens, R.; de Wilde, A.H.; de Jong, A.W.M.; Zevenhoven-Dobbe, J.C.; Maier, H.J.; Faas, F.; Koster, A.J.; Barcena, M. A unifying structural and functional model of the coronavirus replication organelle: Tracking down RNA synthesis. PLoS Biol. 2020, 18, e3000715. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Cortese, M.; Winter, S.L.; Wachsmuth-Melm, M.; Neufeldt, C.J.; Cerikan, B.; Stanifer, M.L.; Boulant, S.; Bartenschlager, R.; Chlanda, P. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nat. Commun. 2020, 11, 5885. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 1989, 54, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [Green Version]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate immune pattern recognition: A cell biological perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef] [Green Version]
- Herwald, H.; Egesten, A. On PAMPs and DAMPs. J. Innate Immun. 2016, 8, 427–428. [Google Scholar] [CrossRef]
- Zindel, J.; Kubes, P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu. Rev. Pathol. 2020, 15, 493–518. [Google Scholar] [CrossRef] [Green Version]
- Pandolfi, F.; Altamura, S.; Frosali, S.; Conti, P. Key Role of DAMP in Inflammation, Cancer, and Tissue Repair. Clin. Ther. 2016, 38, 1017–1028. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, G.; D’Osualdo, A.; Schubert, D.A.; Weber, A.; Bruscia, E.M.; Hartl, D. Cellular Innate Immunity: An Old Game with New Players. J. Innate Immun. 2017, 9, 111–125. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, B.N.; Hammad, H. The airway epithelium in asthma. Nat. Med. 2012, 18, 684–692. [Google Scholar] [CrossRef]
- Uehara, A.; Fujimoto, Y.; Fukase, K.; Takada, H. Various human epithelial cells express functional Toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol. Immunol. 2007, 44, 3100–3111. [Google Scholar] [CrossRef] [PubMed]
- Golebski, K.; Hoepel, W.; van Egmond, D.; de Groot, E.J.; Amatngalim, G.D.; Beekman, J.M.; Fokkens, W.J.; van Drunen, C.M.; den Dunnen, J. FcgammaRIII stimulation breaks the tolerance of human nasal epithelial cells to bacteria through cross-talk with TLR4. Mucosal Immunol. 2019, 12, 425–433. [Google Scholar] [CrossRef] [Green Version]
- Van Tongeren, J.; Roschmann, K.I.L.; Reinartz, S.M.; Luiten, S.; Fokkens, W.J.; de Jong, E.C.; van Drunen, C.M. Expression profiling and functional analysis of Toll-like receptors in primary healthy human nasal epithelial cells shows no correlation and a refractory LPS response. Clin. Transl. Allergy 2015, 5, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, K.; Ferrari, J.D.; Cao, Y.; Ramirez, M.I.; Jones, M.R.; Quinton, L.J.; Mizgerd, J.P. Type I alveolar epithelial cells mount innate immune responses during pneumococcal pneumonia. J. Immunol. 2012, 189, 2450–2459. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Cen, X.; Liu, S.; Cheng, K. The Role of Toll-Like Receptor in Inflammation and Tumor Immunity. Front. Pharmacol. 2018, 9, 878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [Green Version]
- Sonnenberg, G.F.; Hepworth, M.R. Functional interactions between innate lymphoid cells and adaptive immunity. Nat. Rev. Immunol. 2019, 19, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, J.A. The immune response of Drosophila. Nature 2003, 426, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature 2004, 430, 257–263. [Google Scholar] [CrossRef]
- Beutler, B. Innate immunity: An overview. Mol. Immunol. 2004, 40, 845–859. [Google Scholar] [CrossRef]
- Bowie, A.; O’Neill, L.A. The interleukin-1 receptor/Toll-like receptor superfamily: Signal generators for pro-inflammatory interleukins and microbial products. J. Leukoc. Biol. 2000, 67, 508–514. [Google Scholar] [CrossRef]
- Bowie, A.; Kiss-Toth, E.; Symons, J.A.; Smith, G.L.; Dower, S.K.; O’Neill, L.A. A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc. Natl. Acad. Sci. USA 2000, 97, 10162–10167. [Google Scholar] [CrossRef] [Green Version]
- Enkhbayar, P.; Kamiya, M.; Osaki, M.; Matsumoto, T.; Matsushima, N. Structural principles of leucine-rich repeat (LRR) proteins. Proteins 2004, 54, 394–403. [Google Scholar] [CrossRef]
- Kim, H.M.; Park, B.S.; Kim, J.I.; Kim, S.E.; Lee, J.; Oh, S.C.; Enkhbayar, P.; Matsushima, N.; Lee, H.; Yoo, O.J.; et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 2007, 130, 906–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, M.S.; Kim, S.E.; Heo, J.Y.; Lee, M.E.; Kim, H.M.; Paik, S.G.; Lee, H.; Lee, J.O. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 2007, 130, 1071–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, D.; Li, W. Structures and recognition modes of toll-like receptors. Proteins 2017, 85, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Carty, M.; Guy, C.; Bowie, A.G. Detection of Viral Infections by Innate Immunity. Biochem. Pharmacol. 2021, 183, 114316. [Google Scholar] [CrossRef]
- Onomoto, K.; Onoguchi, K.; Yoneyama, M. Regulation of RIG-I-like receptor-mediated signaling: Interaction between host and viral factors. Cell Mol. Immunol. 2021, 18, 539–555. [Google Scholar] [CrossRef]
- Gee, P.; Chua, P.K.; Gevorkyan, J.; Klumpp, K.; Najera, I.; Swinney, D.C.; Deval, J. Essential Role of the N-terminal Domain in the Regulation of RIG-I ATPase Activity. J. Biol. Chem. 2008, 283, 9488–9496. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Ramanathan, A.; Miller, M.T.; Tang, G.Q.; Gale, M., Jr.; Patel, S.S.; Marcotrigiano, J. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature 2011, 479, 423–427. [Google Scholar] [CrossRef]
- Luo, D.; Ding, S.C.; Vela, A.; Kohlway, A.; Lindenbach, B.D.; Pyle, A.M. Structural insights into RNA recognition by RIG-I. Cell 2011, 147, 409–422. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.X.; Wan, H.; Nie, L.; Shao, T.; Xiang, L.X.; Shao, J.Z. RIG-I: A multifunctional protein beyond a pattern recognition receptor. Protein Cell 2018, 9, 246–253. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.C.; Gopalkrishnan, R.V.; Wu, Q.; Jankowsky, E.; Pyle, A.M.; Fisher, P.B. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc. Natl. Acad. Sci. USA 2002, 99, 637–642. [Google Scholar] [CrossRef] [Green Version]
- Cui, S.; Eisenacher, K.; Kirchhofer, A.; Brzozka, K.; Lammens, A.; Lammens, K.; Fujita, T.; Conzelmann, K.K.; Krug, A.; Hopfner, K.P. The C-terminal regulatory domain is the RNA 5’-triphosphate sensor of RIG-I. Mol. Cell 2008, 29, 169–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalinski, E.; Lunardi, T.; McCarthy, A.A.; Louber, J.; Brunel, J.; Grigorov, B.; Gerlier, D.; Cusack, S. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 2011, 147, 423–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ludwig, J.; Schuberth, C.; Goldeck, M.; Schlee, M.; Li, H.; Juranek, S.; Sheng, G.; Micura, R.; Tuschl, T.; et al. Structural and functional insights into 5’-ppp RNA pattern recognition by the innate immune receptor RIG-I. Nat. Struct. Mol. Biol. 2010, 17, 781–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruns, A.M.; Leser, G.P.; Lamb, R.A.; Horvath, C.M. The innate immune sensor LGP2 activates antiviral signaling by regulating MDA5-RNA interaction and filament assembly. Mol. Cell 2014, 55, 771–781. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Shi, H.; Wu, J.; Zhang, X.; Sun, L.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 2013, 51, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Chiu, Y.H.; Chen, Z.J. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 2014, 54, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Slavik, K.M.; Morehouse, B.R.; Ragucci, A.E.; Zhou, W.; Ai, X.; Chen, Y.; Li, L.; Wei, Z.; Bahre, H.; Konig, M.; et al. cGAS-like receptors sense RNA and control 3′2′-cGAMP signalling in Drosophila. Nature 2021, 597, 109–113. [Google Scholar] [CrossRef]
- Holleufer, A.; Winther, K.G.; Gad, H.H.; Ai, X.; Chen, Y.; Li, L.; Wei, Z.; Deng, H.; Liu, J.; Frederiksen, N.A.; et al. Two cGAS-like receptors induce antiviral immunity in Drosophila. Nature 2021, 597, 114–118. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blander, J.M. A long-awaited merger of the pathways mediating host defence and programmed cell death. Nat. Rev. Immunol. 2014, 14, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe 2016, 19, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Beziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Park, S.; Jeong, H.W.; Ahn, J.Y.; Choi, S.J.; Lee, H.; Choi, B.; Nam, S.K.; Sa, M.; Kwon, J.S.; et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci. Immunol. 2020, 5, eabd1554. [Google Scholar] [CrossRef] [PubMed]
- Cheemarla, N.R.; Watkins, T.A.; Mihaylova, V.T.; Wang, B.; Zhao, D.; Wang, G.; Landry, M.L.; Foxman, E.F. Dynamic innate immune response determines susceptibility to SARS-CoV-2 infection and early replication kinetics. J. Exp. Med. 2021, 218, e20210583. [Google Scholar] [CrossRef]
- Van der Made, C.I.; Simons, A.; Schuurs-Hoeijmakers, J.; van den Heuvel, G.; Mantere, T.; Kersten, S.; van Deuren, R.C.; Steehouwer, M.; van Reijmersdal, S.V.; Jaeger, M.; et al. Presence of Genetic Variants Among Young Men With Severe COVID-19. JAMA 2020, 324, 663–673. [Google Scholar] [CrossRef]
- Mdkhana, B.; Saheb Sharif-Askari, N.; Ramakrishnan, R.K.; Goel, S.; Hamid, Q.; Halwani, R. Nucleic Acid-Sensing Pathways During SARS-CoV-2 Infection: Expectations versus Reality. J. Inflamm. Res. 2021, 14, 199–216. [Google Scholar] [CrossRef]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020, 92, 2105–2113. [Google Scholar] [CrossRef]
- Shirato, K.; Kizaki, T. SARS-CoV-2 spike protein S1 subunit induces pro-inflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon 2021, 7, e06187. [Google Scholar] [CrossRef]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Amor, S.; Fernandez Blanco, L.; Baker, D. Innate immunity during SARS-CoV-2: Evasion strategies and activation trigger hypoxia and vascular damage. Clin. Exp. Immunol. 2020, 202, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Renner, D.M.; Comar, C.E.; Whelan, J.N.; Reyes, H.M.; Cardenas-Diaz, F.L.; Truitt, R.; Tan, L.H.; Dong, B.; Alysandratos, K.D.; et al. SARS-CoV-2 induces double-stranded RNA-mediated innate immune responses in respiratory epithelial-derived cells and cardiomyocytes. Proc. Natl. Acad. Sci. USA 2021, 118, e2022643118. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.K.; Blanco, M.R.; Bruce, E.A.; Honson, D.D.; Chen, L.M.; Chow, A.; Bhat, P.; Ollikainen, N.; Quinodoz, S.A.; Loney, C.; et al. SARS-CoV-2 Disrupts Splicing, Translation, and Protein Trafficking to Suppress Host Defenses. Cell 2020, 183, 1325–1339. [Google Scholar] [CrossRef]
- Mendez, A.S.; Ly, M.; Gonzalez-Sanchez, A.M.; Hartenian, E.; Ingolia, N.T.; Cate, J.H.; Glaunsinger, B.A. The N-terminal domain of SARS-CoV-2 nsp1 plays key roles in suppression of cellular gene expression and preservation of viral gene expression. Cell Rep. 2021, 37, 109841. [Google Scholar] [CrossRef]
- Yuan, S.; Peng, L.; Park, J.J.; Hu, Y.; Devarkar, S.C.; Dong, M.B.; Shen, Q.; Wu, S.; Chen, S.; Lomakin, I.B.; et al. Nonstructural Protein 1 of SARS-CoV-2 Is a Potent Pathogenicity Factor Redirecting Host Protein Synthesis Machinery toward Viral RNA. Mol. Cell 2020, 80, 1055–1066. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Starokadomskyy, P.; Escala Perez-Reyes, A.; Burstein, E. Immune Dysfunction in Mendelian Disorders of POLA1 Deficiency. J. Clin. Immunol. 2021, 41, 285–293. [Google Scholar] [CrossRef]
- Burke, J.M.; St Clair, L.A.; Perera, R.; Parker, R. SARS-CoV-2 infection triggers widespread host mRNA decay leading to an mRNA export block. RNA 2021, 27, 1318–1329. [Google Scholar] [CrossRef]
- Zhang, K.; Miorin, L.; Makio, T.; Dehghan, I.; Gao, S.; Xie, Y.; Zhong, H.; Esparza, M.; Kehrer, T.; Kumar, A.; et al. Nsp1 protein of SARS-CoV-2 disrupts the mRNA export machinery to inhibit host gene expression. Sci. Adv. 2021, 7, eabe7386. [Google Scholar] [CrossRef]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.Y. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Ishida, R.; Strilets, T.; Cole, J.; Lopez-Orozco, J.; Fayad, N.; Felix-Lopez, A.; Elaish, M.; Evseev, D.; Magor, K.E.; et al. SARS-CoV-2 Nonstructural Protein 1 Inhibits the Interferon Response by Causing Depletion of Key Host Signaling Factors. J. Virol. 2021, 95, e0026621. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.W.; Tang, C.; Wei, H.C.; Du, B.; Chen, C.; Wang, M.; Zhou, Y.; Yu, M.X.; Cheng, L.; Kuivanen, S.; et al. Genomic monitoring of SARS-CoV-2 uncovers an Nsp1 deletion variant that modulates type I interferon response. Cell Host Microbe 2021, 29, 489–502. [Google Scholar] [CrossRef]
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; Mackens-Kiani, T.; Cheng, J.; et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 2020, 369, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Hayn, M.; Hirschenberger, M.; Koepke, L.; Nchioua, R.; Straub, J.H.; Klute, S.; Hunszinger, V.; Zech, F.; Prelli Bozzo, C.; Aftab, W.; et al. Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. 2021, 35, 109126. [Google Scholar] [CrossRef]
- Russo, L.C.; Tomasin, R.; Matos, I.A.; Manucci, A.C.; Sowa, S.T.; Dale, K.; Caldecott, K.W.; Lehtio, L.; Schechtman, D.; Meotti, F.C.; et al. The SARS-CoV-2 Nsp3 macrodomain reverses PARP9/DTX3L-dependent ADP-ribosylation induced by interferon signaling. J. Biol. Chem. 2021, 297, 101041. [Google Scholar] [CrossRef]
- Liu, Y.; Qin, C.; Rao, Y.; Ngo, C.; Feng, J.J.; Zhao, J.; Zhang, S.; Wang, T.Y.; Carriere, J.; Savas, A.C.; et al. SARS-CoV-2 Nsp5 Demonstrates Two Distinct Mechanisms Targeting RIG-I and MAVS To Evade the Innate Immune Response. mBio 2021, 12, e0233521. [Google Scholar] [CrossRef]
- Wu, Y.; Ma, L.; Zhuang, Z.; Cai, S.; Zhao, Z.; Zhou, L.; Zhang, J.; Wang, P.H.; Zhao, J.; Cui, J. Main protease of SARS-CoV-2 serves as a bifunctional molecule in restricting type I interferon antiviral signaling. Signal Transduct. Target. Ther. 2020, 5, 221. [Google Scholar] [CrossRef]
- Fung, S.Y.; Siu, K.L.; Lin, H.; Yeung, M.L.; Jin, D.Y. SARS-CoV-2 main protease suppresses type I interferon production by preventing nuclear translocation of phosphorylated IRF3. Int. J. Biol. Sci. 2021, 17, 1547–1554. [Google Scholar] [CrossRef]
- Yuen, C.K.; Lam, J.Y.; Wong, W.M.; Mak, L.F.; Wang, X.; Chu, H.; Cai, J.P.; Jin, D.Y.; To, K.K.; Chan, J.F.; et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg. Microbes Infect. 2020, 9, 1418–1428. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.C.; Laurent-Rolle, M.; Pawlak, J.B.; Wilen, C.B.; Cresswell, P. Translational shutdown and evasion of the innate immune response by SARS-CoV-2 NSP14 protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2101161118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zheng, Y.; Niu, Z.; Zhang, B.; Wang, C.; Yao, X.; Peng, H.; Franca, D.N.; Wang, Y.; Zhu, Y.; et al. SARS-CoV-2 spike protein dictates syncytium-mediated lymphocyte elimination. Cell Death Differ. 2021, 28, 2765–2777. [Google Scholar] [CrossRef] [PubMed]
- Buchrieser, J.; Dufloo, J.; Hubert, M.; Monel, B.; Planas, D.; Rajah, M.M.; Planchais, C.; Porrot, F.; Guivel-Benhassine, F.; Van der Werf, S.; et al. Syncytia formation by SARS-CoV-2-infected cells. EMBO J. 2020, 39, e106267. [Google Scholar] [CrossRef] [PubMed]
- Rajah, M.M.; Bernier, A.; Buchrieser, J.; Schwartz, O. The Mechanism and Consequences of SARS-CoV-2 Spike-Mediated Fusion and Syncytia Formation. J. Mol. Biol. 2021, 434, 167280. [Google Scholar] [CrossRef]
- Fu, Y.Z.; Wang, S.Y.; Zheng, Z.Q.; Yi, H.; Li, W.W.; Xu, Z.S.; Wang, Y.Y. SARS-CoV-2 membrane glycoprotein M antagonizes the MAVS-mediated innate antiviral response. Cell Mol. Immunol. 2021, 18, 613–620. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhuang, M.W.; Han, L.; Zhang, J.; Nan, M.L.; Zhan, P.; Kang, D.; Liu, X.; Gao, C.; Wang, P.H. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal Transduct. Target. Ther. 2020, 5, 299. [Google Scholar] [CrossRef]
- Chen, K.; Xiao, F.; Hu, D.; Ge, W.; Tian, M.; Wang, W.; Pan, P.; Wu, K.; Wu, J. SARS-CoV-2 Nucleocapsid Protein Interacts with RIG-I and Represses RIG-Mediated IFN-beta Production. Viruses 2020, 13, 47. [Google Scholar] [CrossRef]
- Oh, S.J.; Shin, O.S. SARS-CoV-2 Nucleocapsid Protein Targets RIG-I-Like Receptor Pathways to Inhibit the Induction of Interferon Response. Cells 2021, 10, 530. [Google Scholar] [CrossRef]
- Konno, Y.; Kimura, I.; Uriu, K.; Fukushi, M.; Irie, T.; Koyanagi, Y.; Sauter, D.; Gifford, R.J.; Consortium, U.-C.; Nakagawa, S.; et al. SARS-CoV-2 ORF3b Is a Potent Interferon Antagonist Whose Activity Is Increased by a Naturally Occurring Elongation Variant. Cell Rep. 2020, 32, 108185. [Google Scholar] [CrossRef]
- Miorin, L.; Kehrer, T.; Sanchez-Aparicio, M.T.; Zhang, K.; Cohen, P.; Patel, R.S.; Cupic, A.; Makio, T.; Mei, M.; Moreno, E.; et al. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 28344–28354. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Y.; Li, Y.; Huang, F.; Luo, B.; Yuan, Y.; Xia, B.; Ma, X.; Yang, T.; Yu, F.; et al. The ORF8 protein of SARS-CoV-2 mediates immune evasion through down-regulating MHC-Iota. Proc. Natl. Acad. Sci. USA 2021, 118, e2024202118. [Google Scholar] [CrossRef]
- Li, J.Y.; Liao, C.H.; Wang, Q.; Tan, Y.J.; Luo, R.; Qiu, Y.; Ge, X.Y. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. 2020, 286, 198074. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Shi, Y.; Pan, X.; Wu, S.; Hou, R.; Zhang, Y.; Zhong, T.; Tang, H.; Du, W.; Wang, L.; et al. SARS-CoV-2 ORF9b inhibits RIG-I-MAVS antiviral signaling by interrupting K63-linked ubiquitination of NEMO. Cell Rep 2021, 34, 108761. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Zhuang, M.W.; Deng, J.; Zheng, Y.; Zhang, J.; Nan, M.L.; Zhang, X.J.; Gao, C.; Wang, P.H. SARS-CoV-2 ORF9b antagonizes type I and III interferons by targeting multiple components of the RIG-I/MDA-5-MAVS, TLR3-TRIF, and cGAS-STING signaling pathways. J. Med. Virol. 2021, 93, 5376–5389. [Google Scholar] [CrossRef] [PubMed]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.Y.; Schneller, S.; Zust, R.; Dong, H.; et al. 2’-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010, 468, 452–456. [Google Scholar] [CrossRef]
- Hyde, J.L.; Diamond, M.S. Innate immune restriction and antagonism of viral RNA lacking 2-O methylation. Virology 2015, 479, 66–74. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhou, Z.; Xiao, X.; Tian, Z.; Dong, X.; Wang, C.; Li, L.; Ren, L.; Lei, X.; Xiang, Z.; et al. SARS-CoV-2 nsp12 attenuates type I interferon production by inhibiting IRF3 nuclear translocation. Cell Mol. Immunol. 2021, 18, 945–953. [Google Scholar] [CrossRef]
- Li, A.; Zhao, K.; Zhang, B.; Hua, R.; Fang, Y.; Jiang, W.; Zhang, J.; Hui, L.; Zheng, Y.; Li, Y.; et al. SARS-CoV-2 NSP12 Protein Is Not an Interferon-beta Antagonist. J. Virol. 2021, 95, e0074721. [Google Scholar] [CrossRef]
- Devarkar, S.C.; Wang, C.; Miller, M.T.; Ramanathan, A.; Jiang, F.; Khan, A.G.; Patel, S.S.; Marcotrigiano, J. Structural basis for m7G recognition and 2’-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc. Natl. Acad. Sci. USA 2016, 113, 596–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilamowski, M.; Sherrell, D.A.; Minasov, G.; Kim, Y.; Shuvalova, L.; Lavens, A.; Chard, R.; Maltseva, N.; Jedrzejczak, R.; Rosas-Lemus, M.; et al. 2′-O methylation of RNA cap in SARS-CoV-2 captured by serial crystallography. Proc. Natl. Acad. Sci. USA 2021, 118, e2100170118. [Google Scholar] [CrossRef] [PubMed]
- Cortese, M.; Lee, J.Y.; Cerikan, B.; Neufeldt, C.J.; Oorschot, V.M.J.; Kohrer, S.; Hennies, J.; Schieber, N.L.; Ronchi, P.; Mizzon, G.; et al. Integrative Imaging Reveals SARS-CoV-2-Induced Reshaping of Subcellular Morphologies. Cell Host Microbe 2020, 28, 853–866. [Google Scholar] [CrossRef] [PubMed]
- Fung, S.Y.; Siu, K.L.; Lin, H.; Chan, C.P.; Yeung, M.L.; Jin, D.Y. SARS-CoV-2 NSP13 helicase suppresses interferon signaling by perturbing JAK1 phosphorylation of STAT1. Cell Biosci. 2022, 12, 36. [Google Scholar] [CrossRef] [PubMed]
- Schubert, K.; Karousis, E.D.; Jomaa, A.; Scaiola, A.; Echeverria, B.; Gurzeler, L.A.; Leibundgut, M.; Thiel, V.; Muhlemann, O.; Ban, N. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. 2020, 27, 959–966. [Google Scholar] [CrossRef]
- Kilkenny, M.L.; Veale, C.E.; Guppy, A.; Hardwick, S.W.; Chirgadze, D.Y.; Rzechorzek, N.J.; Maman, J.D.; Pellegrini, L. Structural basis for the interaction of SARS-CoV-2 virulence factor nsp1 with DNA polymerase alpha-primase. Protein Sci. 2021, 31, 333–344. [Google Scholar] [CrossRef] [PubMed]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Ghosh, S.; Dellibovi-Ragheb, T.A.; Kerviel, A.; Pak, E.; Qiu, Q.; Fisher, M.; Takvorian, P.M.; Bleck, C.; Hsu, V.W.; Fehr, A.R.; et al. beta-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell 2020, 183, 1520–1535. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Consortium, C.-G.U.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Thorne, L.G.; Bouhaddou, M.; Reuschl, A.K.; Zuliani-Alvarez, L.; Polacco, B.; Pelin, A.; Batra, J.; Whelan, M.V.X.; Hosmillo, M.; Fossati, A.; et al. Evolution of enhanced innate immune evasion by the SARS-CoV-2. Nature 2022, 602, 487–495. [Google Scholar] [CrossRef]
- Choy, K.T.; Wong, A.Y.; Kaewpreedee, P.; Sia, S.F.; Chen, D.; Hui, K.P.Y.; Chu, D.K.W.; Chan, M.C.W.; Cheung, P.P.; Huang, X.; et al. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antivir. Res. 2020, 178, 104786. [Google Scholar] [CrossRef] [PubMed]
- Williamson, B.N.; Feldmann, F.; Schwarz, B.; Meade-White, K.; Porter, D.P.; Schulz, J.; van Doremalen, N.; Leighton, I.; Yinda, C.K.; Perez-Perez, L.; et al. Clinical benefit of remdesivir in rhesus macaques infected with SARS-CoV-2. Nature 2020, 585, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Mozaffari, E.; Chandak, A.; Zhang, Z.; Liang, S.; Thrun, M.; Gottlieb, R.L.; Kuritzkes, D.R.; Sax, P.E.; Wohl, D.A.; Casciano, R.; et al. Remdesivir treatment in hospitalized patients with COVID-19: A comparative analysis of in-hospital all-cause mortality in a large multi-center observational cohort. Clin. Infect. Dis. 2021, ciab875. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Echevarria, A.; Perez-Martinez, A.; Gonzalez Del Valle, L.; Ara, M.F.; Melendo, S.; Ruiz de Valbuena, M.; Vazquez-Martinez, J.L.; Morales-Martinez, A.; Remesal, A.; Sandor-Bajusz, K.A.; et al. Compassionate use of remdesivir in children with COVID-19. Eur. J. Pediatr. 2021, 180, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.H.; Lau, K.T.K.; Au, I.C.H.; Xiong, X.; Chung, M.S.H.; Lau, E.H.Y.; Cowling, B.J. Optimal timing of remdesivir initiation in hospitalized COVID-19 patients administered with dexamethasone. Clin. Infect. Dis. 2021, ciab728. [Google Scholar] [CrossRef]
- Benfield, T.; Bodilsen, J.; Brieghel, C.; Harboe, Z.B.; Helleberg, M.; Holm, C.; Israelsen, S.B.; Jensen, J.; Jensen, T.O.; Johansen, I.S.; et al. Improved Survival Among Hospitalized Patients With Coronavirus Disease 2019 (COVID-19) Treated With Remdesivir and Dexamethasone. A Nationwide Population-Based Cohort Study. Clin. Infect. Dis. 2021, 73, 2031–2036. [Google Scholar] [CrossRef]
- Gottlieb, R.L.; Vaca, C.E.; Paredes, R.; Mera, J.; Webb, B.J.; Perez, G.; Oguchi, G.; Ryan, P.; Nielsen, B.U.; Brown, M.; et al. Early Remdesivir to Prevent Progression to Severe COVID-19 in Outpatients. N. Engl. J. Med. 2022, 386, 305–315. [Google Scholar] [CrossRef]
- Kabinger, F.; Stiller, C.; Schmitzova, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Hobartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef]
- Jayk Bernal, A.; Gomes da Silva, M.M.; Musungaie, D.B.; Kovalchuk, E.; Gonzalez, A.; Delos Reyes, V.; Martin-Quiros, A.; Caraco, Y.; Williams-Diaz, A.; Brown, M.L.; et al. Molnupiravir for Oral Treatment of COVID-19 in Nonhospitalized Patients. N. Engl. J. Med. 2021, 386, 509–520. [Google Scholar] [CrossRef]
- Fischer, W.A., 2nd; Eron, J.J., Jr.; Holman, W.; Cohen, M.S.; Fang, L.; Szewczyk, L.J.; Sheahan, T.P.; Baric, R.; Mollan, K.R.; Wolfe, C.R.; et al. A phase 2a clinical trial of molnupiravir in patients with COVID-19 shows accelerated SARS-CoV-2 RNA clearance and elimination of infectious virus. Sci. Transl. Med. 2022, 14, eabl7430. [Google Scholar] [CrossRef]
- Mahase, E. COVID-19: UK becomes first country to authorise antiviral molnupiravir. BMJ 2021, 375, n2697. [Google Scholar] [CrossRef] [PubMed]
- Holman, W.; Holman, W.; McIntosh, S.; Painter, W.; Painter, G.; Bush, J.; Cohen, O. Accelerated first-in-human clinical trial of EIDD-2801/MK-4482 (molnupiravir), a ribonucleoside analog with potent antiviral activity against SARS-CoV-2. Trials 2021, 22, 561. [Google Scholar] [CrossRef]
- Zhou, S.; Hill, C.S.; Sarkar, S.; Tse, L.V.; Woodburn, B.M.D.; Schinazi, R.F.; Sheahan, T.P.; Baric, R.S.; Heise, M.T.; Swanstrom, R. beta-d-N4-hydroxycytidine Inhibits SARS-CoV-2 Through Lethal Mutagenesis But Is Also Mutagenic To Mammalian Cells. J. Infect. Dis. 2021, 224, 415–419. [Google Scholar] [CrossRef]
- Greasley, S.E.; Noell, S.; Plotnikova, O.; Ferre, R.A.; Liu, W.; Bolanos, B.; Fennell, K.; Nicki, J.; Craig, T.; Zhu, Y.; et al. Structural basis for the in vitro efficacy of Nirmatrelvir against of SARS-CoV-2 variant. J. Biol. Chem. 2022, 298, 101972. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.; Leister-Tebbe, H.; Gardner, A.; Abreu, P.; Bao, W.; Wisemandle, W.; Baniecki, M.; Hendrick, V.M.; Damle, B.; Simon-Campos, A.; et al. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with COVID-19. N. Engl. J. Med. 2022, 386, 1397–1408. [Google Scholar] [CrossRef]
- Mahase, E. COVID-19: Pfizer’s paxlovid is 89% effective in patients at risk of serious illness, company reports. BMJ 2021, 375, n2713. [Google Scholar] [CrossRef]
- Abdool Karim, S.S.; de Oliveira, T. New SARS-CoV-2 Variants-Clinical, Public Health, and Vaccine Implications. N. Engl. J. Med. 2021, 384, 1866–1868. [Google Scholar] [CrossRef]
- Mahase, E. Delta variant: What is happening with transmission, hospital admissions, and restrictions? BMJ 2021, 373, n1513. [Google Scholar] [CrossRef]
- Hoffmann, M.; Arora, P.; Gross, R.; Seidel, A.; Hornich, B.F.; Hahn, A.S.; Kruger, N.; Graichen, L.; Hofmann-Winkler, H.; Kempf, A.; et al. SARS-CoV-2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cell 2021, 184, 2384–2393. [Google Scholar] [CrossRef]
- Tian, D.; Sun, Y.; Zhou, J.; Ye, Q. The global epidemic of SARS-CoV-2 variants and their mutational immune escape. J. Med. Virol. 2022, 94, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef] [PubMed]
- WHO. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 30 May 2022).
- Ren, S.Y.; Wang, W.B.; Gao, R.D.; Zhou, A.M. Omicron variant (B.1.1.529) of SARS-CoV-2: Mutation, infectivity, transmission, and vaccine resistance. World. J. Clin. Cases 2022, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vangeel, L.; Chiu, W.; De Jonghe, S.; Maes, P.; Slechten, B.; Raymenants, J.; Andre, E.; Leyssen, P.; Neyts, J.; Jochmans, D. Remdesivir, Molnupiravir and Nirmatrelvir remain active against SARS-CoV-2 Omicron and other variants of concern. Antivir. Res. 2022, 198, 105252. [Google Scholar] [CrossRef]
- Ali, N. Recent Evidence and Possible Therapy against COVID-19-Mediated Hepatic Dysfunction. J. Environ. Pathol. Toxicol. Oncol. 2021, 40, 33–41. [Google Scholar] [CrossRef]
- Magro, G. COVID-19: Review on latest available drugs and therapies against SARS-CoV-2. Coagulation and inflammation cross-talking. Virus Res. 2020, 286, 198070. [Google Scholar] [CrossRef]
- Te Velthuis, A.J.W.; Zubkova, T.G.; Shaw, M.; Mehle, A.; Boltz, D.; Gmeinwieser, N.; Stammer, H.; Milde, J.; Muller, L.; Margitich, V. Enisamium Reduces Influenza Virus Shedding and Improves Patient Recovery by Inhibiting Viral RNA Polymerase Activity. Antimicrob. Agents Chemother. 2021, 65, e02605–e02620. [Google Scholar] [CrossRef]
- Boltz, D.; Peng, X.; Muzzio, M.; Dash, P.; Thomas, P.G.; Margitich, V. Activity of enisamium, an isonicotinic acid derivative, against influenza viruses in differentiated normal human bronchial epithelial cells. Antivir. Chem. Chemother. 2018, 26, 2040206618811416. [Google Scholar] [CrossRef]
- Holubovska, O.; Bojkova, D.; Elli, S.; Bechtel, M.; Boltz, D.; Muzzio, M.; Peng, X.; Sala, F.; Cosentino, C.; Mironenko, A.; et al. Enisamium is an inhibitor of the SARS-CoV-2 RNA polymerase and shows improvement of recovery in COVID-19 patients in an interim analysis of a clinical trial. medRxiv 2021. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe COVID-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef]
- Horby, P.W.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Emberson, J.; Palfreeman, A.; Raw, J.; Elmahi, E.; Prudon, B.; et al. Lopinavir–ritonavir in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2020, 396, 1345–1352. [Google Scholar] [CrossRef]
- Consortium, W.H.O.S.T.; Pan, H.; Peto, R.; Henao-Restrepo, A.M.; Preziosi, M.P.; Sathiyamoorthy, V.; Abdool Karim, Q.; Alejandria, M.M.; Hernandez Garcia, C.; Kieny, M.P.; et al. Repurposed Antiviral Drugs for COVID-19-Interim WHO Solidarity Trial Results. N. Engl. J. Med. 2021, 384, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Worlock, K.B.; Huang, N.; Lindeboom, R.G.H.; Butler, C.R.; Kumasaka, N.; Conde, C.D.; Mamanova, L.; Bolt, L.; Richardson, L.; et al. Local and systemic responses to SARS-CoV-2 infection in children and adults. Nature 2022, 602, 321–327. [Google Scholar] [CrossRef]
- Wark, P.A.B.; Pathinayake, P.S.; Kaiko, G.; Nichol, K.; Ali, A.; Chen, L.; Sutanto, E.N.; Garratt, L.W.; Sohal, S.S.; Lu, W.; et al. ACE2 expression is elevated in airway epithelial cells from older and male healthy individuals but reduced in asthma. Respirology 2021, 26, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Shan, B.; Shao, M.; Zhang, Q.; Hepler, C.; Paschoal, V.A.; Barnes, S.D.; Vishvanath, L.; An, Y.A.; Jia, L.; Malladi, V.S.; et al. Perivascular mesenchymal cells control adipose-tissue macrophage accrual in obesity. Nat. Metab. 2020, 2, 1332–1349. [Google Scholar] [CrossRef]
- Singh, M.V.; Chapleau, M.W.; Harwani, S.C.; Abboud, F.M. The immune system and hypertension. Immunol. Res. 2014, 59, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107. [Google Scholar] [CrossRef]
- Chen, Y.; Li, T.; Ye, Y.; Chen, Y.; Pan, J. Impact of Fundamental Diseases on Patients With COVID-19. Disaster Med. Public Health Prep. 2020, 14, 776–781. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| SARS-CoV-2 Proteins | Target | Mechanism | References |
|---|---|---|---|
| Nsp1 | STAT1; STAT2; IRF3; Tyk2; 18S rRNA; 40S ribosomal and primosomal subunit | Translation inhibition by interfering with nuclear export of host mRNA; blocks IRF3 nuclear translocation; inhibition of STAT1 phosphorylation; reduced expression of STAT2 and Tyk2 | [76,77,78,79,80,81,82,83,84,85,86] |
| Nsp3 | NF-kB; ISG15; IRF3; PARP9/DTX3L | Cleaves ISG15 from IRF3; inhibits IFN-I promoters, IRF3 and NF-kB binding sites; reverses PARP9/DTX3L-dependent ADP-ribosylation | [87,88,89] |
| Nsp5 | IRF3; STAT1; STAT2; RIG-I; MAVS | Cleaves off the 10 most-N-terminal amino acids from RIG-I; promotes the ubiquitination and proteosome-mediated degradation of MAVS; inhibits blocking the nucleus translocation of phosphorylated IRF3; induces phospho-STAT1/2 accumulation impairing type I IFN signaling | [88,90,91,92] |
| Nsp6 | STAT1; STAT2; IRF3; TBK1 | Suppresses phosphorylation of IRF3 by binding to TBK1; inhibits STAT1 and STAT2 phosphorylation | [83] |
| Nsp8 | 7SL RNA Component of SRP54 | disrupts protein trafficking for secretion or membrane integration | [76] |
| Nsp9 | 7SL RNA Component of SRP19 | disrupts protein trafficking for secretion or membrane integration | [76] |
| Nsp10 | IRF3; NF-kB | Impairs the activity of IFNA4 and IFNB1, IRF3 binding and NF-kB binding, and suppresses cytokines production | [88] |
| Nsp13 | STAT1; STAT2; TBK1; IRF3 | Inhibits TBK-1, STAT1 and STAT2 phosphorylation; blocks IRF3 nuclear translocation | [83,88,93] |
| Nsp14 | IRF3; IFNAR1 | Inhibits IRF3 nuclear translocation; induces lysosomal degradation of IFNAR1; inhibits host cellular translation via ExoN and N7-MTase activities | [88,93,94] |
| Nsp15 | IRF3; early autophagosome | Inhibits IRF3 nuclear translocation; inhibits de novo autophagy induction | [88] |
| Nsp16 | U1 and U2 splicing RNAs | Suppresses host mRNA splicing through binding to the pre-mRNA recognition domains of the U1 and U2 splicing RNAs | [76] |
| S | ACE2 | Evades host cells and induces syncytia formation through binding to ACE2 receptor | [95,96,97] |
| E | Autophagosome | Blocks autophagic turnover | [88] |
| M | RIG-I; MDA-5; MAVS; autophagosome | Impairs MAVS aggregation and recruitment of downstream components; induces LC3B accumulation in the perinuclear space; suppresses type I and III IFN expression by targeting RIG-I/MDA-5 signaling | [88,98,99] |
| N | TBK1; IRF3; RIG-I | Binds to with the RIG-I protein at its DExD/H domain and suppresses IFN-β production; impairs TBK1/IRF3 association and IRF3 nuclear translocation | [100,101] |
| Orf3a | STAT1; lysosomes; autophagosomes | Inhibits STAT1 phosphorylation, blocking the fusion of lysosomes with autophagosomes | [83,88] |
| Orf3b | IRF3 | Inhibits IRF3 nuclear translocation | [102] |
| Orf6 | IRF3; STAT1; KPNA2; ISGF3 | Interacts with KPNA2; blocks STAT1, IRF3 and ISGF3 nuclear translocation | [83,93,103,104] |
| Orf7a | STAT2; lysosomes | Inhibits STAT2 phosphorylation; decreases lysosomes acidification | [83,88] |
| Orf7b | STAT1; STAT2 | Inhibits STAT1 and STAT2 phosphorylation | [83] |
| Orf8 | NF-κB; MHC-Ι molecules | Mediates their lysosomal degradation of MHC-I molecules; inhibits NF-κB-responsive promoter | [93,105,106] |
| Orf9b | RIG-I; MDA-5; MAVS; NEMO; TRIF; TBK1; IRF3; STING | Interrupts K63-linked polyubiquitination of NEMO and inhibits IFN signaling; interacts with RIG-I, MDA-5, MAVS, TRIF, STING, and TBK1 and impedes the phosphorylation and nuclear translocation of IRF3 | [107,108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Chi, S.; Dmytruk, K.; Dmytruk, O.; Tan, S. Coronaviral Infection and Interferon Response: The Virus-Host Arms Race and COVID-19. Viruses 2022, 14, 1349. https://doi.org/10.3390/v14071349
Liu Q, Chi S, Dmytruk K, Dmytruk O, Tan S. Coronaviral Infection and Interferon Response: The Virus-Host Arms Race and COVID-19. Viruses. 2022; 14(7):1349. https://doi.org/10.3390/v14071349
Chicago/Turabian StyleLiu, Qi, Sensen Chi, Kostyantyn Dmytruk, Olena Dmytruk, and Shuai Tan. 2022. "Coronaviral Infection and Interferon Response: The Virus-Host Arms Race and COVID-19" Viruses 14, no. 7: 1349. https://doi.org/10.3390/v14071349
APA StyleLiu, Q., Chi, S., Dmytruk, K., Dmytruk, O., & Tan, S. (2022). Coronaviral Infection and Interferon Response: The Virus-Host Arms Race and COVID-19. Viruses, 14(7), 1349. https://doi.org/10.3390/v14071349