
Borneol Ester Derivatives as Entry Inhibitors of a Wide Spectrum of SARS-CoV-2 Viruses

 , , , , ,
, , , , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Biological Experiments
2.1.1. Cell Cultures
2.1.2. Plasmids
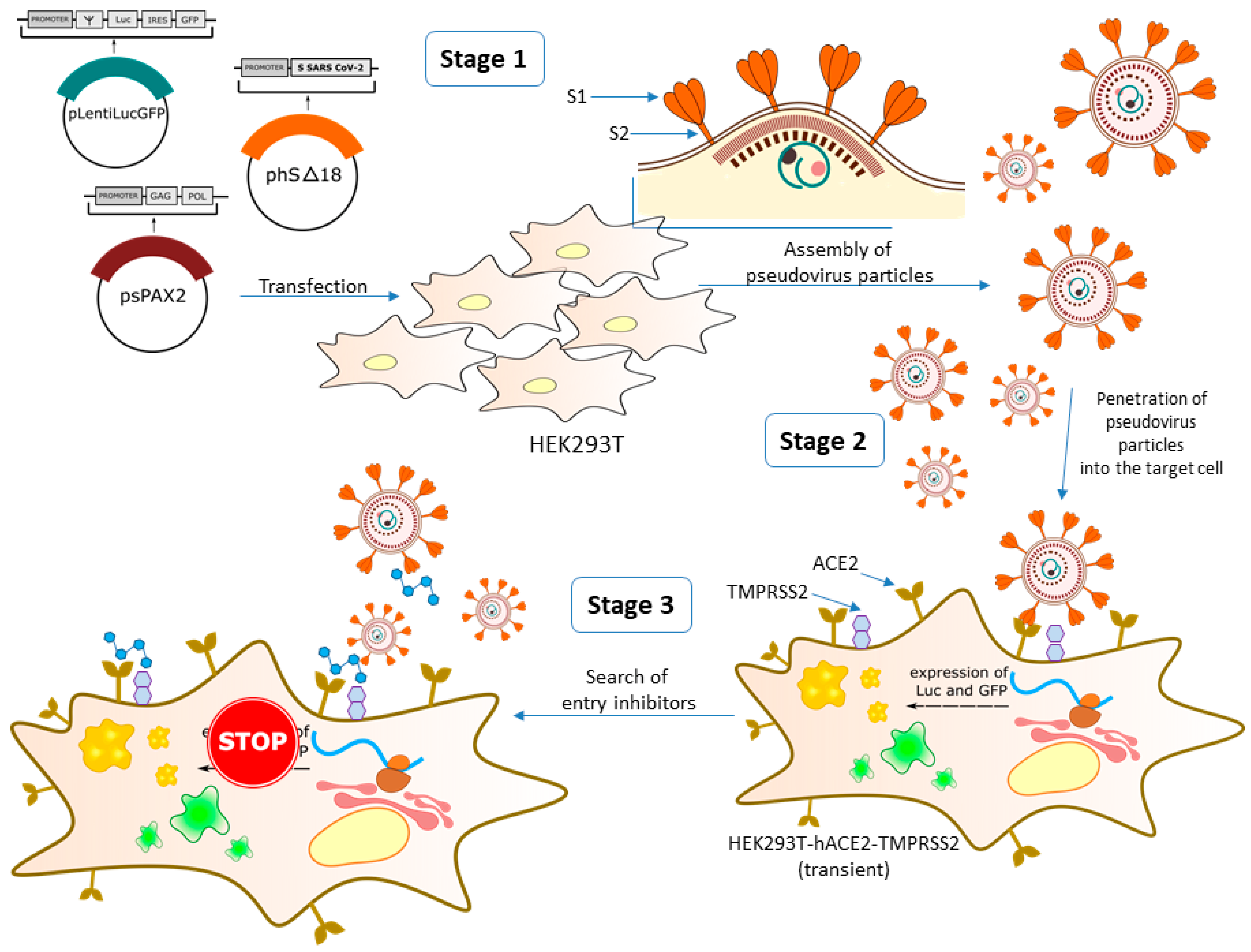
2.1.3. Preparation of SARS-CoV-2 Pseudotyped Lentiviral Particles
2.1.4. Pseudovirus Entry Assay
2.1.5. Determination of Cytotoxicity of Compounds on HEK293T Cells
2.1.6. Determination of Semi-Inhibitory Concentrations of Compounds against Lenti-S SARS-CoV-2 Pseudoviruses and Calculation of Selectivity Index (SI) Values
2.1.7. ELISA-Based Competitive Inhibition of the RBD/ACE2 Interaction
2.1.8. Evaluation of the Antiviral Activities againts SARS-CoV-2 Viruses
2.1.9. Statistical Analysis
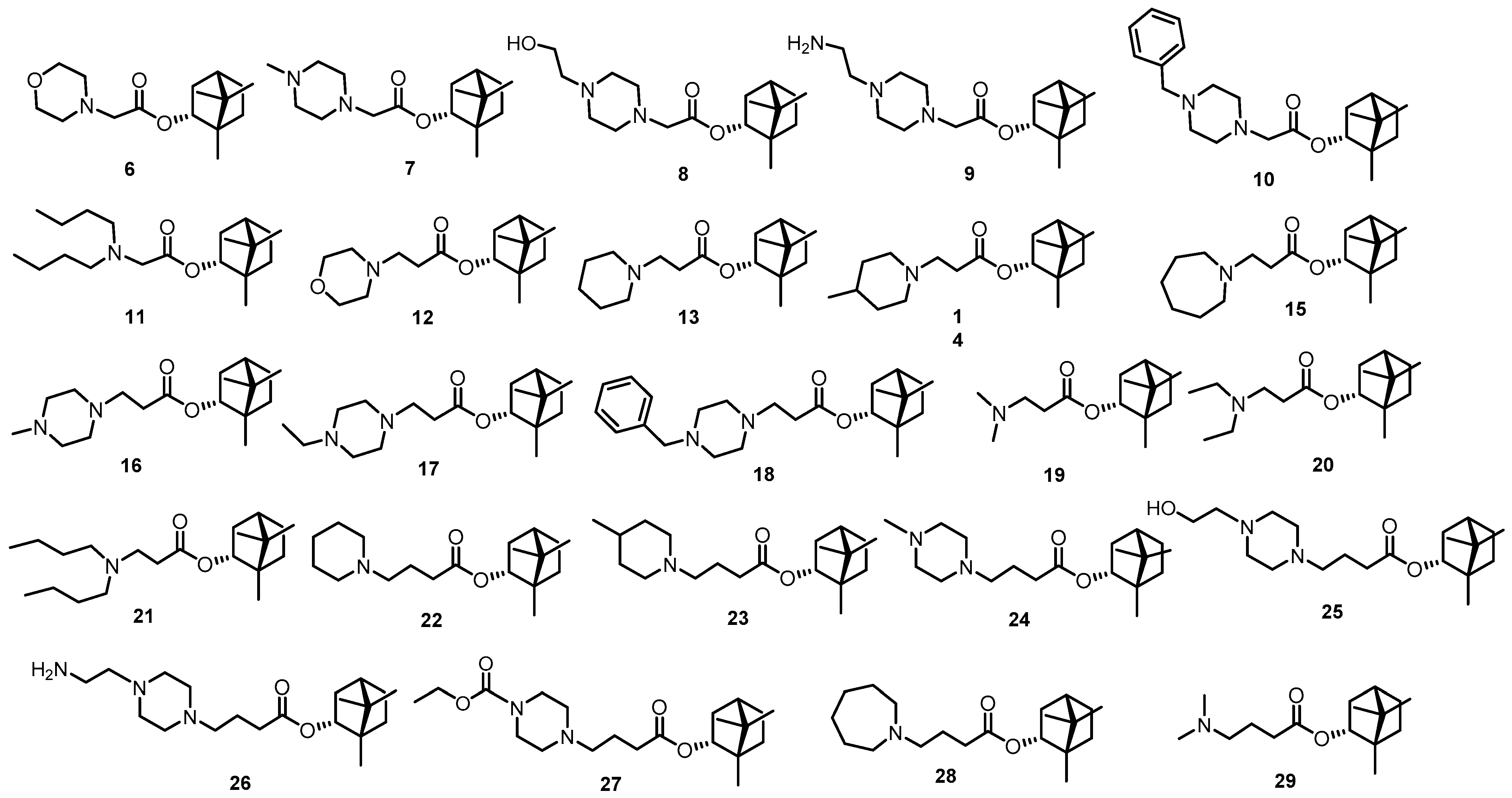
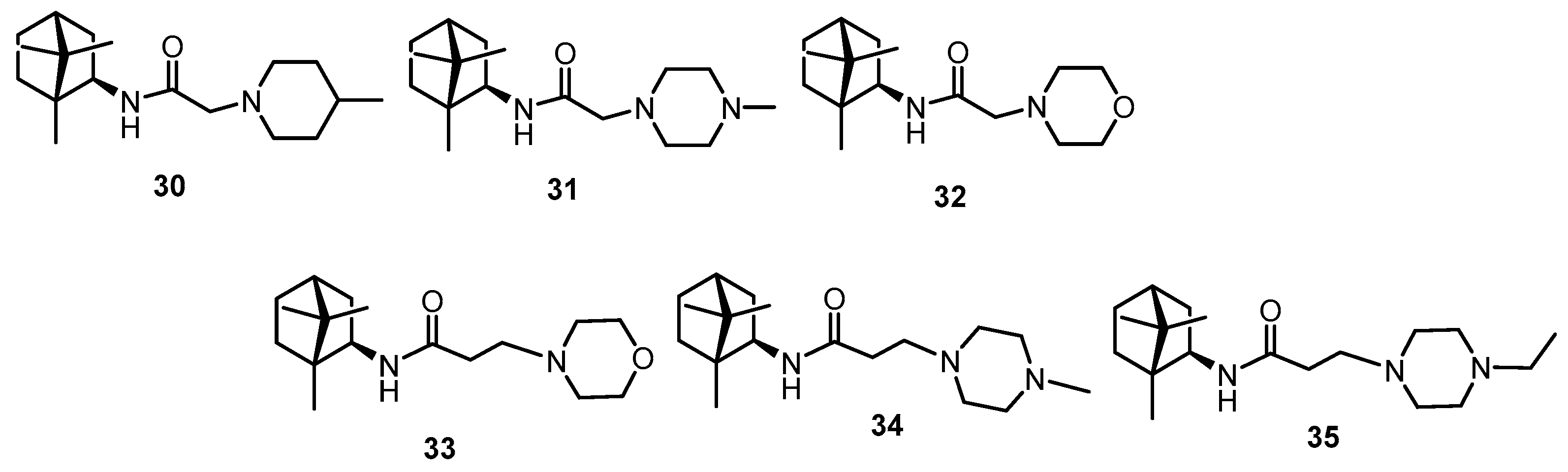
2.2. Synthesis of Compounds
2.3. Molecular Dynamics and Docking
2.3.1. Ligand and Protein Preparation
2.3.2. Possible Ligand Binding Site Search
2.3.3. Molecular Docking and Dynamics Simulations
3. Results and Discussion
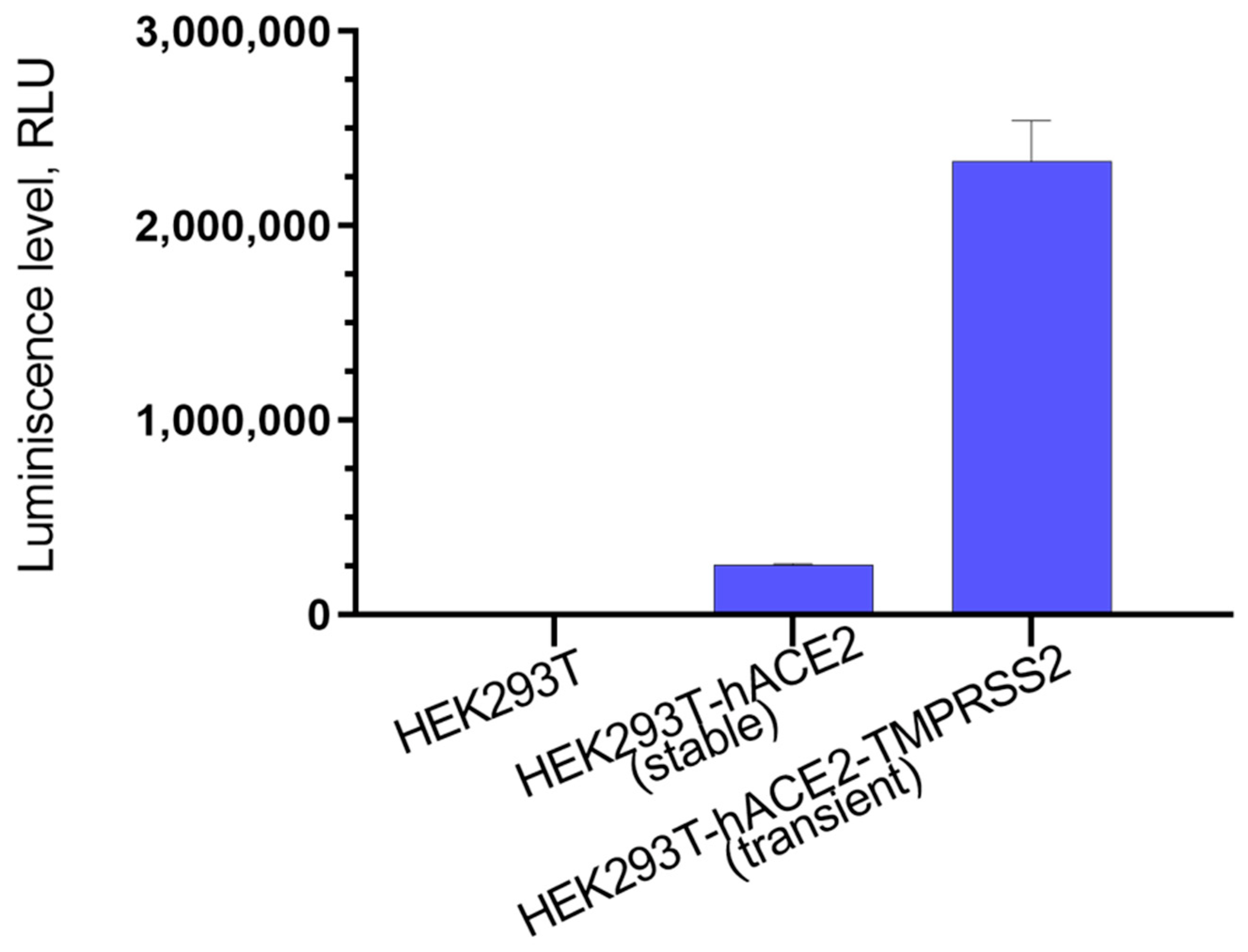
3.1. Development of a Pseudovirus System
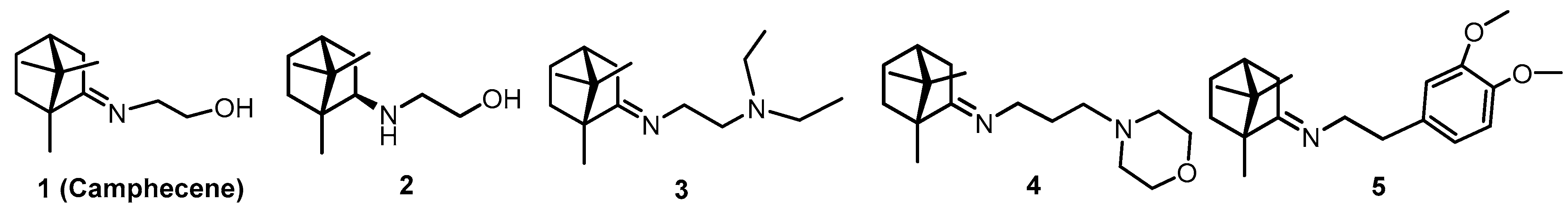
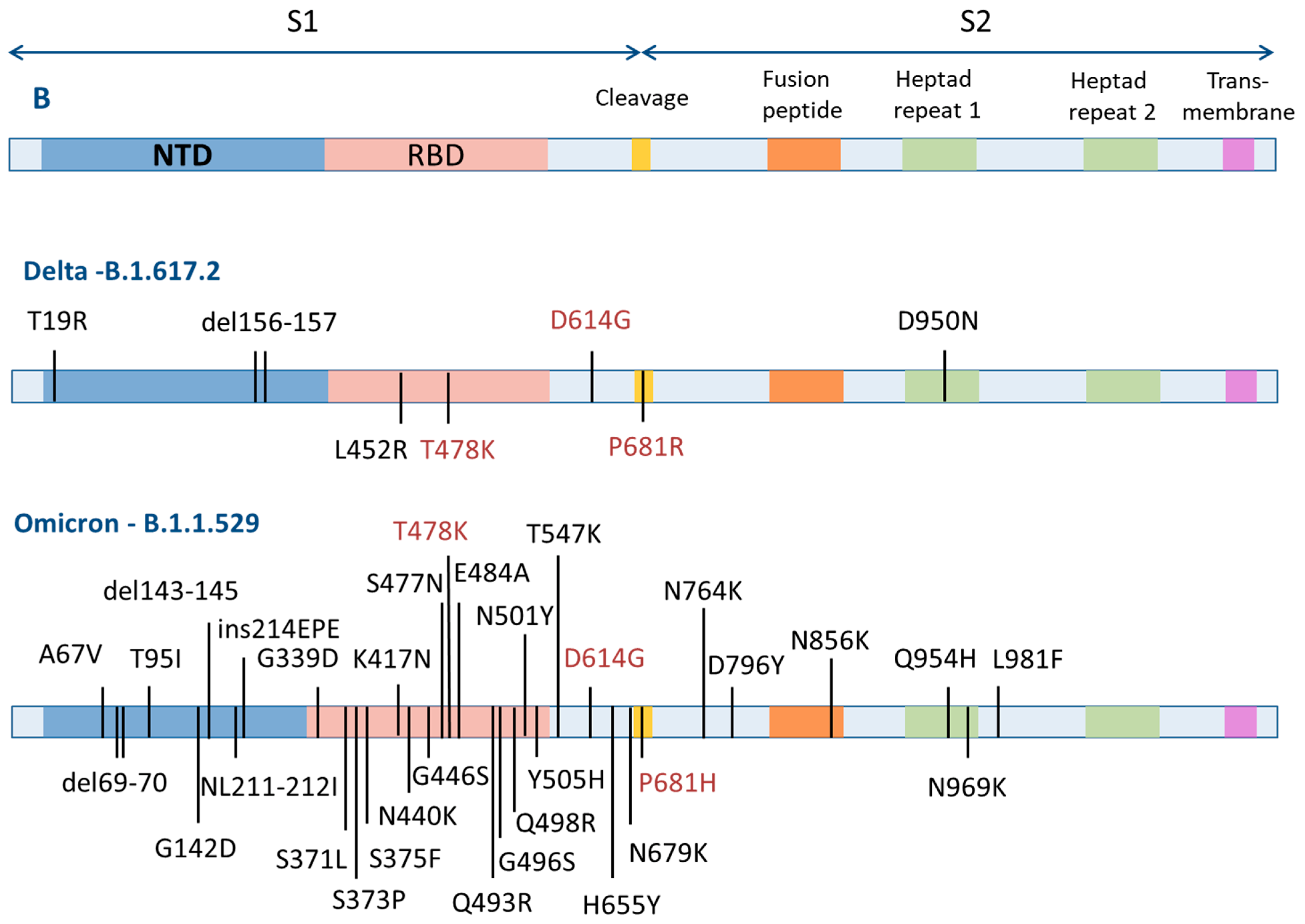
3.2. Choice of Research Objects
3.3. Testing with the Use of Pseudo-Viral Systems
3.4. Antiviral Activity against SARS-CoV-2 Viruses
3.5. ELISA-Based Competitive Inhibition of the RBD/ACE2 Interaction
3.6. Molecular Modeling Study
3.6.1. Finding and Reasoning about the Binding Site of Potential Entry Inhibitors

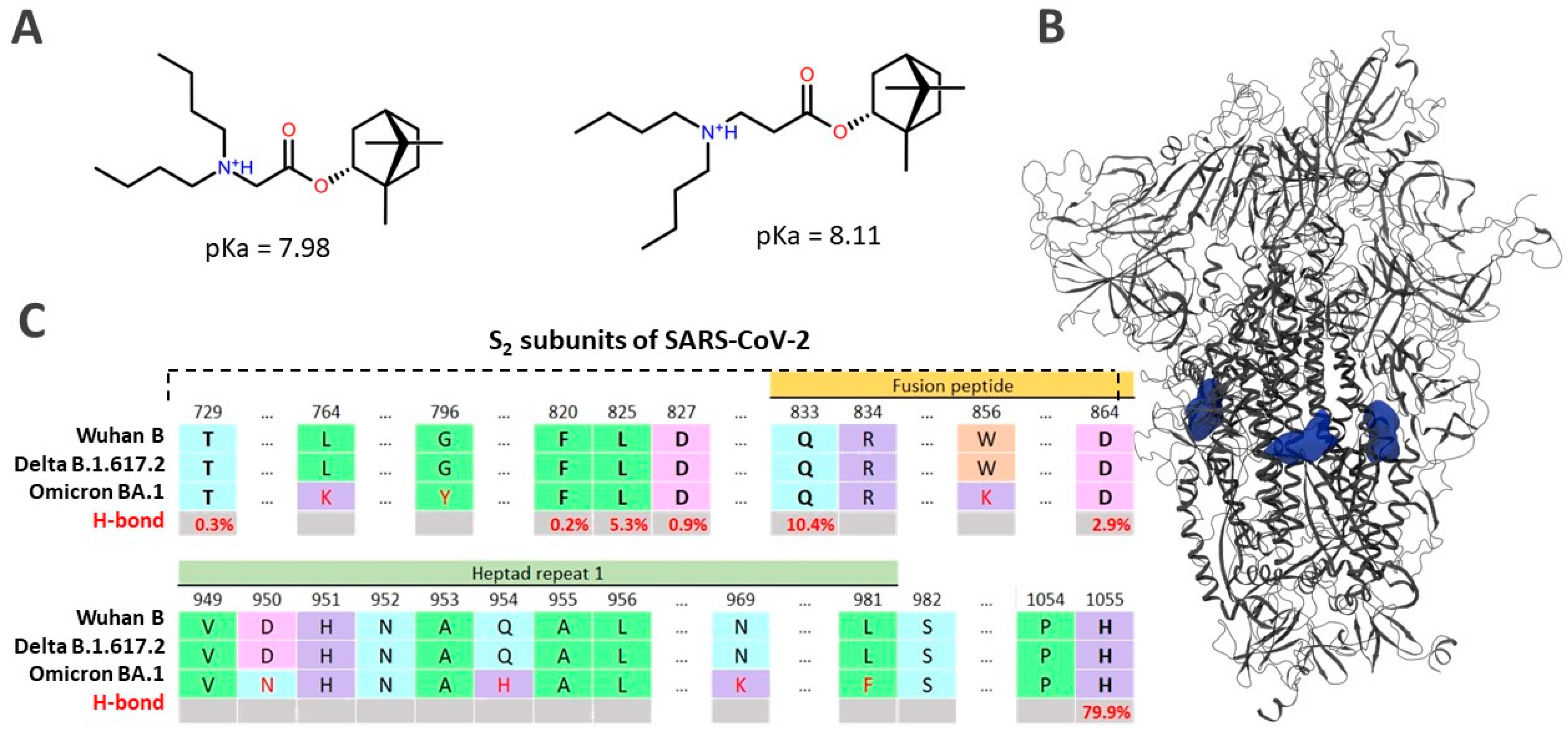
3.6.2. Protonation of Ligands
3.6.3. Molecular Dynamics Simulation
3.6.4. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, H.; Stratton, C.W.; Tang, Y. Outbreak of pneumonia of unknown etiology in Wuhan, China: The mystery and the miracle. J. Med. Virol. 2020, 92, 401–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. COVID-19 Weekly Epidemiological Update; World Health Organisation: Geneva, Switzerland, 2022; pp. 1–23. [Google Scholar]
- Forni, G.; Mantovani, A. COVID-19 vaccines: Where we stand and challenges ahead. Cell Death Differ. 2021, 28, 626–639. [Google Scholar] [CrossRef] [PubMed]
- Novikov, F.N.; Stroylov, V.S.; Svitanko, I.V.; Nebolsin, V.E. Molecular basis of COVID-19 pathogenesis. Russ. Chem. Rev. 2020, 89, 858–878. [Google Scholar] [CrossRef]
- Mann, R.; Perisetti, A.; Gajendran, M.; Gandhi, Z.; Umapathy, C.; Goyal, H. Clinical Characteristics, Diagnosis, and Treatment of Major Coronavirus Outbreaks. Front. Med. 2020, 7, 766. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Adil, S.F.; Alkhathlan, H.Z.; Tahir, M.N.; Saif, S.; Khan, M.; Khan, S.T. COVID-19: A Global Challenge with Old History, Epidemiology and Progress So Far. Molecules 2020, 26, 39. [Google Scholar] [CrossRef]
- Golob, J.L.; Lugogo, N.; Lauring, A.S.; Lok, A.S. SARS-CoV-2 vaccines: A triumph of science and collaboration. JCI Insight 2021, 6, e149187. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Qin, C.; Liu, M.; Liu, J. Effectiveness and safety of SARS-CoV-2 vaccine in real-world studies: A systematic review and meta-analysis. Infect. Dis. Poverty 2021, 10, 132. [Google Scholar] [CrossRef]
- Rolland, M.; Gilbert, P.B. Sieve analysis to understand how SARS-CoV-2 diversity can impact vaccine protection. PLoS Pathog. 2021, 17, e1009406. [Google Scholar] [CrossRef]
- Han, X.; Ye, Q. The variants of SARS-CoV-2 and the challenges of vaccines. J. Med. Virol. 2022, 94, 1366–1372. [Google Scholar] [CrossRef]
- Finsterer, J. Neurological side effects of SARS-CoV-2 vaccinations. Acta Neurol. Scand. 2022, 145, 5–9. [Google Scholar] [CrossRef]
- Yan, W.; Zheng, Y.; Zeng, X.; He, B.; Cheng, W. Structural biology of SARS-CoV-2: Open the door for novel therapies. Signal Transduct. Target. Ther. 2022, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Cho, C.-C.D.; Geng, Z.Z.; Shaabani, N.; Ma, X.R.; Vatansever, E.C.; Alugubelli, Y.R.; Ma, Y.; Chaki, S.P.; Ellenburg, W.H.; et al. Evaluation of SARS-CoV-2 Main Protease Inhibitors Using a Novel Cell-Based Assay. ACS Cent. Sci. 2022, 8, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, Z.; He, X.; Gebre, M.S.; Bondzie, E.A.; Wan, H.; Jacob-Dolan, C.; Martinez, D.R.; Nkolola, J.P.; Baric, R.S.; et al. Deletion of the SARS-CoV-2 Spike Cytoplasmic Tail Increases Infectivity in Pseudovirus Neutralization Assays. J. Virol. 2021, 95, e00044-21. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, X.-E. Construction and applications of SARS-CoV-2 pseudoviruses: A mini review. Int. J. Biol. Sci. 2021, 17, 1574–1580. [Google Scholar] [CrossRef]
- Rawson, J.M.O.; Duchon, A.; Nikolaitchik, O.A.; Pathak, V.K.; Hu, W.S. Development of a cell-based luciferase complementation assay for identification of sars-cov-2 3clpro inhibitors. Viruses 2021, 13, 173. [Google Scholar] [CrossRef]
- Fu, W.; Chen, Y.; Wang, K.; Hettinghouse, A.; Hu, W.; Wang, J.-Q.; Lei, Z.-N.; Chen, Z.-S.; Stapleford, K.A.; Liu, C. Repurposing FDA-approved drugs for SARS-CoV-2 through an ELISA-based screening for the inhibition of RBD/ACE2 interaction. Protein Cell 2021, 12, 586–591. [Google Scholar] [CrossRef]
- Reiners, N.; Schnurra, C.; Trawinski, H.; Kannenberg, J.; Hermsdorf, T.; Aebischer, A.; Schöneberg, T.; Reiche, S.; Jassoy, C. Performance of a SARS CoV-2 antibody ELISA based on simultaneous measurement of antibodies against the viral nucleoprotein and receptor-binding domain. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 2645–2649. [Google Scholar] [CrossRef]
- Wang, Y.; Long, X.; Ding, X.; Fan, S.; Cai, J.; Yang, B.-J.; Zhang, X.; Luo, R.; Yang, L.; Ruan, T.; et al. Novel nucleocapsid protein-targeting phenanthridine inhibitors of SARS-CoV. Eur. J. Med. Chem. 2022, 227, 113966. [Google Scholar] [CrossRef]
- Schmidt, F.; Weisblum, Y.; Muecksch, F.; Hoffmann, H.-H.; Michailidis, E.; Lorenzi, J.C.C.; Mendoza, P.; Rutkowska, M.; Bednarski, E.; Gaebler, C.; et al. Measuring SARS-CoV-2 neutralizing antibody activity using pseudotyped and chimeric viruses. J. Exp. Med. 2020, 217, e20201181. [Google Scholar] [CrossRef]
- Sokolova, A.S.; Yarovaya, O.I.; Shernyukov, A.V.; Gatilov, Y.V.; Razumova, Y.V.; Zarubaev, V.V.; Tretiak, T.S.; Pokrovsky, A.G.; Kiselev, O.I.; Salakhutdinov, N.F. Discovery of a new class of antiviral compounds: Camphor imine derivatives. Eur. J. Med. Chem. 2015, 105, 263–273. [Google Scholar] [CrossRef]
- Sokolova, A.S.; Yarovaya, O.I.; Shernyukov, A.V.; Pokrovsky, M.A.; Pokrovsky, A.G.; Lavrinenko, V.A.; Zarubaev, V.V.; Tretiak, T.S.; Anfimov, P.M.; Kiselev, O.I.; et al. New quaternary ammonium camphor derivatives and their antiviral activity, genotoxic effects and cytotoxicity. Bioorganic Med. Chem. 2013, 21, 6690–6698. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.S.; Yarovaya, O.I.; Semenova, M.D.; Shtro, A.A.; Orshanskaya, I.R.; Zarubaev, V.V.; Salakhutdinov, N.F. Synthesis and in vitro study of novel borneol derivatives as potent inhibitors of the influenza A virus. Med. Chem. Commun. 2017, 8, 960–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolova, A.S.; Yarovaya, O.I.; Zybkina, A.V.; Mordvinova, E.D.; Shcherbakova, N.S.; Zaykovskaya, A.V.; Baev, D.S.; Tolstikova, T.G.; Shcherbakov, D.N.; Pyankov, O.V.; et al. Monoterpenoid-based inhibitors of filoviruses targeting the glycoprotein-mediated entry process. Eur. J. Med. Chem. 2020, 207, 112726. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.S.; Yarovaya, O.I.; Bormotov, N.I.; Shishkina, L.N.; Salakhutdinov, N.F. Discovery of a New Class of Inhibitors of Vaccinia Virus Based on (−)-Borneol from Abies sibirica and (+)-Camphor. Chem. Biodivers. 2018, 15, e1800153. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Benton, D.J.; Wrobel, A.G.; Roustan, C.; Borg, A.; Xu, P.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. The effect of the D614G substitution on the structure of the spike glycoprotein of SARS-CoV. Proc. Natl. Acad. Sci. USA 2021, 118, e2022586118. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, C.; Zhang, C.; Wang, Y.; Hong, Q.; Xu, S.; Li, Z.; Yang, Y.; Huang, Z.; Cong, Y. Structural basis for SARS-CoV-2 Delta variant recognition of ACE2 receptor and broadly neutralizing antibodies. Nat. Commun. 2022, 13, 871. [Google Scholar] [CrossRef]
- Ni, D.; Lau, K.; Turelli, P.; Raclot, C.; Beckert, B.; Nazarov, S.; Pojer, F.; Myasnikov, A.; Stahlberg, H.; Trono, D. Structural analysis of the Spike of the Omicron SARS-COV-2 variant by cryo-EM and implications for immune evasion. bioRxiv 2021, arXiv:2021.12.27.474250. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Watson, M.A.; Greenwood, J.R.; Philipp, D.M. Multiconformation, Density Functional Theory-Based p K a Prediction in Application to Large, Flexible Organic Molecules with Diverse Functional Groups. J. Chem. Theory Comput. 2016, 12, 6001–6019. [Google Scholar] [CrossRef] [PubMed]
- Vankadari, N. Arbidol: A potential antiviral drug for the treatment of SARS-CoV-2 by blocking trimerization of the spike glycoprotein. Int. J. Antimicrob. Agents 2020, 56, 105998. [Google Scholar] [CrossRef]
- Borisevich, S.S.; Khamitov, E.M.; Gureev, M.A.; Yarovaya, O.I.; Rudometova, N.B.; Zybkina, A.V.; Mordvinova, E.D.; Shcherbakov, D.N.; Maksyutov, R.A.; Salakhutdinov, N.F. Simulation of Molecular Dynamics of SARS-CoV-2 S-Protein in the Presence of Multiple Arbidol Molecules: Interactions and Binding Mode Insights. Viruses 2022, 14, 119. [Google Scholar] [CrossRef]
- Bowers, K.J.; Sacerdoti, F.D.; Salmon, J.K.; Shan, Y.; Shaw, D.E.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; et al. Molecular dynamics—Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing-SC ’06, Tampa, FL, USA, 11–17 November 2006; ACM Press: New York, NY, USA, 2006; p. 84. [Google Scholar]
- Suenaga, A.; Okimoto, N.; Hirano, Y.; Fukui, K. An Efficient Computational Method for Calculating Ligand Binding Affinities. PLoS ONE 2012, 7, e42846. [Google Scholar] [CrossRef] [Green Version]
- Repasky, M.P.; Shelley, M.; Friesner, R.A. Flexible Ligand Docking with Glide. In Current Protocols in Bioinformatics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; pp. 1–36. [Google Scholar]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Zarubaev, V.V.; Garshinina, A.V.; Tretiak, T.S.; Fedorova, V.A.; Shtro, A.A.; Sokolova, A.S.; Yarovaya, O.I.; Salakhutdinov, N.F. Broad range of inhibiting action of novel camphor-based compound with anti-hemagglutinin activity against influenza viruses in vitro and in vivo. Antivir. Res. 2015, 120, 126–133. [Google Scholar] [CrossRef]
- Zarubaev, V.V.; Pushkina, E.A.; Borisevich, S.S.; Galochkina, A.V.; Garshinina, A.V.; Shtro, A.A.; Egorova, A.A.; Sokolova, A.S.; Khursan, S.L.; Yarovaya, O.I.; et al. Selection of influenza virus resistant to the novel camphor-based antiviral camphecene results in loss of pathogenicity. Virology 2018, 524, 69–77. [Google Scholar] [CrossRef]
- Borisevich, S.S.; Gureev, M.A.; Yarovaya, O.I.; Zarubaev, V.V.; Kostin, G.A.; Porozov, Y.B.; Salakhutdinov, N.F. Can molecular dynamics explain decreased pathogenicity in mutant camphecene-resistant influenza virus? J. Biomol. Struct. Dyn. 2021, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zarubaev, V.V.; Garshinina, A.V.; Volobueva, A.S.; Slita, A.V.; Yarovaya, O.I.; Bykov, V.V.; Leonov, K.A.; Motov, V.S.; Khazanov, V.A.; Salakhutdinov, N.F. Optimization of application schedule of camphecene, a novel anti-influenza compound, based on its pharmacokinetic characteristics. Fundam. Clin. Pharmacol. 2022, 36, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.S.; Yarovaya, O.I.; Baev, D.S.; Shernyukov, A.V.; Shtro, A.A.; Zarubaev, V.V.; Salakhutdinov, N.F. Aliphatic and alicyclic camphor imines as effective inhibitors of influenza virus H1N. Eur. J. Med. Chem. 2017, 127, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.S.; Kovaleva, K.S.; Yarovaya, O.I.; Bormotov, N.I.; Shishkina, L.N.; Serova, O.A.; Sergeev, A.A.; Agafonov, A.P.; Maksuytov, R.A.; Salakhutdinov, N.F. (+)-Camphor and (−)-borneol derivatives as potential anti-orthopoxvirus agents. Arch. Pharm. 2021, 354, 2100038. [Google Scholar] [CrossRef] [PubMed]
- Kononova, A.A.; Sokolova, A.S.; Cheresiz, S.V.; Yarovaya, O.I.; Nikitina, R.A.; Chepurnov, A.A.; Pokrovsky, A.G.; Salakhutdinov, N.F. N-Heterocyclic borneol derivatives as inhibitors of Marburg virus glycoprotein-mediated VSIV pseudotype entry. Med. Chem. Comm. 2017, 8, 2233–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingo, P.; de Benito, N. Alpha variant SARS-CoV-2 infection: How it all starts. EBioMedicine 2021, 74, 103703. [Google Scholar] [CrossRef]
- Twohig, K.A.; Nyberg, T.; Zaidi, A.; Thelwall, S.; Sinnathamby, M.A.; Aliabadi, S.; Seaman, S.R.; Harris, R.J.; Hope, R.; Lopez-Bernal, J.; et al. Hospital admission and emergency care attendance risk for SARS-CoV-2 delta (B.1.617.2) compared with alpha (B.1.1.7) variants of concern: A cohort study. Lancet Infect. Dis. 2021, 22, 35–42. [Google Scholar] [CrossRef]
- Sharma, V.; Rai, H.; Gautam, D.N.S.; Prajapati, P.K.; Sharma, R. Emerging evidence on Omicron (B.1.1.529) SARS-CoV-2 variant. J. Med. Virol. 2022, 94, 1876–1885. [Google Scholar] [CrossRef]
- Vanmechelen, B.; Logist, A.; Wawina-Bokalanga, T.; Verlinden, J.; Martí-Carreras, J.; Geenen, C.; Slechten, B.; Cuypers, L.; André, E.; Baele, G.; et al. Identification of the First SARS-CoV-2 Lineage B.1.1.529 Virus Detected in Europe. Microbiol. Resour. Announc. 2022, 11, 9–12. [Google Scholar] [CrossRef]
- Sokolova, A.S.; Yarovaya, O.I.; Baranova, D.V.; Galochkina, A.V.; Shtro, A.A.; Kireeva, M.V.; Borisevich, S.S.; Gatilov, Y.V.; Zarubaev, V.V.; Salakhutdinov, N.F. Quaternary ammonium salts based on (-)-borneol as effective inhibitors of influenza virus. Arch. Virol. 2021, 166, 1965–1976. [Google Scholar] [CrossRef]
- Russell, R.J.; Kerry, P.S.; Stevens, D.J.; Steinhauer, D.A.; Martin, S.R.; Gamblin, S.J.; Skehel, J.J. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc. Natl. Acad. Sci. USA 2008, 105, 17736–17741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479–480, 498–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020, 6, 1722–1734. [Google Scholar] [CrossRef]
- Chen, Z.; Cui, Q.; Caffrey, M.; Rong, L.; Du, R. Small molecule inhibitors of influenza virus entry. Pharmaceuticals 2021, 14, 587. [Google Scholar] [CrossRef] [PubMed]
- Kadam, R.U.; Wilson, I.A. Structural basis of influenza virus fusion inhibition by the antiviral drug Arbidol. Proc. Natl. Acad. Sci. USA 2017, 114, 206–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Cao, R.; Zhang, H.; Liu, J.; Xu, M.; Hu, H.; Li, Y.; Zhao, L.; Li, W.; Sun, X.; et al. The anti-influenza virus drug, arbidol is an efficient inhibitor of SARS-CoV-2 in vitro. Cell Discov. 2020, 6, 28. [Google Scholar] [CrossRef]
- Cai, L.; Guo, X.; Cao, Y.; Ying, P.; Hong, L.; Zhang, Y.; Yi, G.; Fu, M. Determining available strategies for prevention and therapy: Exploring COVID-19 from the perspective of ACE2 (Review). Int. J. Mol. Med. 2021, 47, 1–15. [Google Scholar] [CrossRef]
- Yang, H.; Chang, J.C.; Guo, Z.; Carney, P.J.; Shore, D.A.; Donis, R.O.; Cox, N.J.; Villanueva, J.M.; Klimov, A.I.; Stevens, J. Structural Stability of Influenza A(H1N1)pdm09 Virus Hemagglutinins. J. Virol. 2014, 88, 4828–4838. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | CC50 a | Wuhan Lineages B | Delta Lineage B.1.617.2 | ||
|---|---|---|---|---|---|
| µM | IC50 b µM | SI c | IC50 b µM | SI c | |
| 1 | 2950 | >500 | - | NT d | − |
| 2 | 1260 | >500 | - | NT | − |
| 3 | 1260 | >500 | - | NT | − |
| 4 | 1080 | >440 | - | NT | − |
| 5 | 660 | >200 | - | NT | − |
| 6 | 982 | 188.6 ± 21.1 | 5 | NT | − |
| 7 | 1455 | >500 | - | NT | − |
| 8 | 1045 | >500 | - | NT | − |
| 9 | 346 | >200 | - | NT | − |
| 10 | 362 | 15.6 ± 2.1 | 23 | >100 | − |
| 11 | 186 | 17.7 ± 3.2 | 10 | 16.0 ± 4.5 | 11 |
| 12 | 396 | 21.3 ± 4.3 | 18 | >100 | − |
| 13 | 960 | >200 | - | NT | − |
| 14 | 540 | >200 | - | NT | − |
| 15 | 538 | >200 | - | NT | − |
| 16 | 350 | >200 | - | NT | − |
| 17 | 855 | >200 | - | NT | − |
| 18 | 292 | 17.7 ± 3.7 | 17 | >100 | − |
| 19 | 1015 | >200 | NT | − | |
| 20 | 935 | >200 | NT | − | |
| 21 | 743 | 25.8 ± 4.2 | 29 | 14.2 ± 2.9 | 53 |
| 22 | 407 | >200 | - | NT | − |
| 23 | 350 | >200 | - | NT | − |
| 24 | 348 | 155 | 2 | NT | − |
| 25 | 438 | 196 | 2 | NT | − |
| 26 | 179 | 59.8 ± 7.1 | 3 | NT | − |
| 27 | 166 | 52.6 ± 8.3 | 3 | NT | − |
| 28 | 1080 | 190 | 5 | NT | − |
| 29 | 490 | >200 | - | NT | − |
| 30 | 417 | 41.2 ± 5.2 | 10 | >100 | − |
| 31 | 1525 | 156 | 9 | NT | − |
| 32 | 820 | >200 | - | NT | − |
| 33 | 2160 | >200 | - | NT | − |
| 34 | 1140 | >200 | - | NT | − |
| 35 | 1260 | 190 | 6 | NT | − |
| Arbidol | 14.8 | 7.8 ± 3.1 | 2 | NT | − |
| Agent | Wuhan Lineages B a | Delta Lineage B.1.617.2 b | Omicron Lineage B.1.1.529 c | ||||
|---|---|---|---|---|---|---|---|
| CC50 d, µM | IC50 e, µM | SI f | IC50, µM | SI | IC50, µM | SI | |
| 10 | 110.5 ± 11.2 | NA | - | NA | - | NA | - |
| 11 | 313.5 ± 14.1 | 13.0 ± 2.1 * | 24 * | 28.2 ± 3.1 * | 11 * | 14.5 ± 2.1 * | 22 * |
| 9.6 ± 1.4 ** | 32 ** | 17.6 ± 2.1 ** | 17 ** | 7.7 ± 1.8 ** | 40 ** | ||
| 12 | 261.3 ± 16.3 | NA | - | NA | - | 160.5 ± 18.2 * | 1.6 * |
| 18 | 103.8 ± 13.1 | NA | - | NA | - | NT | - |
| 21 | 336.6 ± 16.6 | 51.1 ± 6.2 * | 6 * | 10.9 ± 1.8 * | 30 * | 10.9 ± 1.5 * | 30 * |
| 4.7 ± 6.2 ** | 71 ** | 3.5 ± 6.2 ** | 96 ** | 3.3 ± 6.2 ** | 102 ** | ||
| Remdesivir | 710.9 ± 21.2 | 13.7 ± 2.2 * | 51 * | 7.3 ± 1.5 * | 97 * | 6.6 ± 1.1 * | 107 * |
| 3.8 ± 0.42 ** | 186 ** | 2.1 ± 0.16 ** | 338 ** | 2.0 ± 0.13 ** | 356 ** | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yarovaya, O.I.; Shcherbakov, D.N.; Borisevich, S.S.; Sokolova, A.S.; Gureev, M.A.; Khamitov, E.M.; Rudometova, N.B.; Zybkina, A.V.; Mordvinova, E.D.; Zaykovskaya, A.V.; et al. Borneol Ester Derivatives as Entry Inhibitors of a Wide Spectrum of SARS-CoV-2 Viruses. Viruses 2022, 14, 1295. https://doi.org/10.3390/v14061295
Yarovaya OI, Shcherbakov DN, Borisevich SS, Sokolova AS, Gureev MA, Khamitov EM, Rudometova NB, Zybkina AV, Mordvinova ED, Zaykovskaya AV, et al. Borneol Ester Derivatives as Entry Inhibitors of a Wide Spectrum of SARS-CoV-2 Viruses. Viruses. 2022; 14(6):1295. https://doi.org/10.3390/v14061295
Chicago/Turabian StyleYarovaya, Olga I., Dmitriy N. Shcherbakov, Sophia S. Borisevich, Anastasiya S. Sokolova, Maxim A. Gureev, Edward M. Khamitov, Nadezda B. Rudometova, Anastasiya V. Zybkina, Ekaterina D. Mordvinova, Anna V. Zaykovskaya, and et al. 2022. "Borneol Ester Derivatives as Entry Inhibitors of a Wide Spectrum of SARS-CoV-2 Viruses" Viruses 14, no. 6: 1295. https://doi.org/10.3390/v14061295