Abstract
Swine enteric viral infections are responsible for substantial economic losses in the pork industry worldwide. Porcine epidemic diarrhea (PEDV) is one of the main causative agents of diarrhea in lactating pigs, and reports of PEDV coinfection with other enteric viruses highlight the importance of viral interactions for disease presentation and outcomes. Using next-generation sequencing (NGS) and sequence analyses from samples taken from piglets with acute diarrhea, we explored the possible interactions between PEDV and other less reported pathogens. PEDV coinfection with porcine kobuvirus (PKV) was detected in 36.4% (27/74) of samples. Full genomes from porcine coronavirus and kobuvirus were obtained, as was a partial porcine sapovirus genome (PSaV). The phylogenetic results show the clustering of these strains corresponding to the geographical relationship. To our knowledge, this is the first full genome and isolation report for porcine kobuvirus in México, as well as the first phylogenetic analysis for porcine sapovirus in the country. The NGS approach provides a better perspective of circulating viruses and other pathogens in affected production units.
1. Introduction
Diarrheic diseases in swine have strong economic impacts on production units around the world. In recent years, porcine coronaviruses have represented some of the principal causes of these diseases; however, reports of naturally occurring coinfection of coronaviruses with other viral agents highlight the importance of less characterized viruses on disease severity and outcome [1,2].
Porcine epidemic diarrhea virus (PEDV) induces an enteric disease with high mortality rates in suckling piglets characterized by acute diarrhea and severe dehydration. It was first identified in Europe in 1978 and in Asia in 1982. Since then, PEDV has had a great impact on the Asian swine industry. In May 2013, the presence of PEDV was reported in México along with the United States and Canada, resulting in heavy economic losses for the swine production units [3,4].
PEDV is a member of the Coronaviridae family, subfamily Orthocoronavirinae, and genus Alphacoronavirus and is an enveloped virus approximately 120 nm in diameter with a single-stranded, positive-sense RNA genome of ≈28 kb starting with a 292 nt untranslated region (UTR) divided into six open reading frames (ORFs). The PEDV genome encodes for 16 non-structural proteins (nsp); four structural proteins: spike (S), envelope (E), membrane (M), and nucleocapsid (N); and one accessory protein, ORF3 [5,6].
By contrast, porcine kobuvirus 1 (PKV) is a member of the Kobuvirus genus responsible for gastrointestinal disease, although it has been detected in fecal samples of animals without clinical signs [7]. PKV was discovered in Hungary in 2007 and has been reported in Thailand, China, Japan, Korea, Brazil, the Netherlands, and the United States [7,8,9,10,11,12,13]. This virus is a member of the Picornaviridae family, Kobuvirus genus, and Aichivirus C species [14]. Kobuviruses are non-enveloped icosahedral viruses of approximately 30–32 nm. The viral genome consists of 8.2 kb of nucleotides in positive-sense, single-stranded RNA with a poly (A) tail. The genome encodes a viral polyprotein that is divided into the non-structural protein L, three structural capsid proteins (VP0, VP3, and VP1), and seven non-structural proteins [15].
PKV coinfections have been reported for astrovirus, circovirus, rotavirus A, transmissible gastroenteritis virus (TGV), and PEDV [13,16,17,18]. At the same time, PEDV coinfection has also been reported for the same viral agents [2,19,20], including mixed infections of multiple viruses [21]. PEDV and PKV coinfection has been reported in the USA and China, ranging from 15% [8,22] to 81.55% among intestinal samples [23].
Another less reported virus in swine is the porcine sapovirus (PSaV), a member of the Caliciviridae family. PSaV is a 35-nm non-enveloped virus with a 7.3 kb single-stranded, positive-sense RNA genome. The genome consists of ORF1, encoding the non-structural proteins and the capsid protein (VP1) that is used for genogroup classification, and ORF2, encoding the minor structural protein (VP2) [24]. PSaV has a wide geographical distribution and has been detected in 62% of pigs in the USA with a rate of coinfection with PKV of 10.8% and with PEDV of 13.5% [25,26].
The coinfection of viral diarrheic agents represents a potential complication for disease identification, control, and prevention, particularly in locations with no previous report of these viruses. In this study, we explored next-generation sequencing (NGS) as a tool for the identification of viral pathogens during an outbreak of diarrhea in a commercial farm, making the detection and characterization of natural viral coinfection in diarrheic piglets possible.
2. Materials and Methods
2.1. Sample Collection
Samples were collected from 10 different farms distributed in four Mexican states, and all tested animals presented clinical manifestations compatible with gastrointestinal disease at the time of sampling. The commercial farrow-to-finisher units tested were located in the states of Nuevo León, Guanajuato, and Estado de México, and samples from the state of Chiapas belonged to backyard swine herds. The production located in Estado de México had a history of previous diarrheic outbreaks caused by PEDV. For the rest of the tested farms, no previous outbreaks caused by PEDV were reported.
A total of 68 rectal swabs from adult swine and six intestinal tissue samples from piglets in litters presenting clinical signs were taken following euthanasia according to the American Veterinary Medical Association (AVMA) Guidelines for the Euthanasia of Animals: 2013 Edition on the farm. The samples were aliquoted and stored at −80 °C in sterile plastic tubes with Dulbecco’s modified Eagle’s medium (DMEM) until processing.
2.2. RNA Extraction
RNA extraction of the tissue samples was performed using the RNeasy Mini Kit (Cat no. 74,904 Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. Approximately 30 mg of tissue samples were disrupted in frozen mortars and Buffer RLT inside a laminar flow cabin. The quantification of extracted RNA was performed with a Qubit RNA HS Assay Kit (Cat no: Q32852 Invitrogen). After extraction, quantification, and quality assessment, the extractions were divided for use in molecular testing and sequencing.
2.3. Molecular Testing
cDNA was synthesized using Superscript VILO (Invitrogen, Hercules, CA, USA) and PCR for PEDV detection was performed using Platinum™ Taq DNA Polymerase High Fidelity (Cat no: 11,304,011 Invitrogen™, Hercules, CA, USA) following the protocol described by Li et al. [27] The reaction was conducted under the following conditions: 94 °C for 2 min, (94 °C for 1 min, 55 °C for 1 min, and 72 °C for 90 min) for 40 cycles, with a final period of 10 min at 72 °C using the following primers targeting the PEDV M gene: 5′-AGTCTTACATGCGAATTGACC-3′ and 5′-AGCTGACAGAAGCCATAAAGT-3′.
For the detection of PKV, the protocol described by Liu et al. [28] was employed using the primers PK-F10 5′-GGTGGACTCATTGAGTAC-3′ and PK-R10 5′-CCTCCCTGGGTGCAGCTTC-3. The samples were also tested for the presence of Deltacoronavirus using the protocol used by Hu et al. with the primers forward 5′-CGCGTAATCGTGTGATCTATGT-3′ and reverse 5′-CCGGCCTTTGAAGTGGTTAT-3′ [29]. Two PEDV- and PKV-positive intestinal tissue samples of piglets with acute clinical signs were selected for whole genome sequencing and sent to the next-generation sequencing core facility at the Biotechnology Institute, UNAM in cold chain transport.
2.4. Sequencing and Bioinformatics Analyses
RNA sequencing of positive samples was performed at the next-generation sequencing core facility at the Biotechnology Institute, UNAM. RNA concentrations were measured (Genova Nano micro-volume spectrophotometer 737,501, Jenway, Staffordshire, UK) and sequencing libraries were constructed using the TruSeq Stranded mRNA Sample Preparation Kit (Illumina, San Diego, CA, USA) using 5 uL of RNA. The samples were then deep sequenced on the Illumina NextSeq500 system, generating paired-end reads of 75 bases. After quality assessments by FASTQC (Babraham Bioinformatics, UK) [30] and adapter elimination using Cutadapt v1.11, the sequence reads were first filtered using Bowtie2 Alignment software against the Sus scrofa reference genome (GCF_000003025.6).
The remaining data were assembled with the SPAdes genome assembler v3.12.0 (Center for Algorithmic Biotechnology, St. Petersburg, Russia) and SSPACE v3.0 tool (BaseClear B.V., The Netherlands) [31,32,33]. The assembled contigs were subject to taxonomic classification using the Kraken metagenomic classification pipeline. To identify contigs corresponding to viral genomes, Basic Local Alignment Search Tool (BLASTn) was used against a viral genome database obtained from the NCBI website (available from: https://ftp.ncbi.nlm.nih.gov) (accessed on 20 February 2020) [34,35]. Finally, the original reads were mapped to the viral contigs using the BBMap Alignment software to verify the coverage [25]. Prediction of homologous recombination events was performed using RDP4 (Recombination Detection Program) [26]. The analyses were executed at the Center for Genomic Sciences, UNAM facilities.
2.5. Phylogenetic Analysis
Phylogenetic trees were constructed using the maximum likelihood algorithm with SH-like branch support using the PhyML software v3.3.21090321 (Université de Montpellier, France) [36] and iTOL v6.1.1 (University of Würzburg, Germany) [37], with the general time reversible model selected by the sJModelTest v2.1.10 software (University of Vigo, Spain) [38] for all trees.
For the phylogenetic analyses, complete genome sequences of PKV, as well as sequences corresponding to PEDV and the capsid coding region of PSaV, were downloaded from the NCBI database. Then, representative sequences of clusters were selected by CD-HIT-EST [22], and sequences from México were included if available. The representative sequences and obtained genomes were aligned using MAFFT v7.45 (Osaka University, Japan) [39].
For PKV, only the coding sequence (CDS) was used for the phylogenetic reconstruction due to the variability in the lengths of the available sequences. For the PSaV phylogeny, the capsid protein coding region was employed.
2.6. Viral Culture
The two original intestinal samples that tested positive for PKV and PEDV by RT-PCR and confirmed by sequencing were propagated separately for virus isolation. Intestinal samples were prepared as 10% homogenates in DMEM and passed through 0.45 µm filters (GE Healthcare, Chicago, IL, USA). The resultant supernatants were inoculated with Vero cells (African green monkey kidney) cultured at 37 °C with 5% CO2 in Dulbecco’s Modified Eagle’s Media (DMEM; GIBCO, Gaithersburg, MD, USA). Cytopathic effects (CPEs) were evaluated on a daily basis and analyzed for viral replication by RT-PCR after every passage.
3. Results
3.1. Prevalence of Porcine Kobuvirus and Porcine Coronavirus in Diarrheic Pigs
Coinfection of PKV and PEDV was detected in 80% (8/10) of the analyzed farms located in Nuevo León, Guanajuato, and Estado de México states. All the farms presented clinical manifestations at the moment of sample collection. Of the samples used in this study, 51.4% (38/74) were positive for PKV and 67.6% (50/74) were positive for PEDV. Overall, PKV and PEDV coinfection was detected in 36.4% (27/74) of samples. The Deltacoronavirus genome presence was only detected in one farm located in Guanajuato with no PEDV or PKV coinfection.
3.2. Viral Genome Detection
Positive PEDV and PKV intestinal samples belonging to two piglets with acute clinical signs were selected for next-generation sequencing. After the elimination of low-quality reads, an average of 7,029,014 reads per sample were obtained. These reads were assembled, and contigs were taxonomically annotated using Kraken and BLASTn. The Kraken results show the identification of known swine pathogens classified in families Coronaviridae, Picornaviridae, and Caliciviridae; other viral agents of families Tospoviridae, Bromoviridae, and Marseilleviridae were also detected. The assembled contigs of swine viruses and their length are shown in Table 1.

Table 1.
The number of mapped reads and contigs assembled for tissue samples of diarrheic pigs. The swine viral families identified in the analyzed samples.
At the species level, sample 205H showed coinfection by PEDV, PKV, and PSaV, while sample 103H showed evidence of PEDV and PKV coinfection, but no presence of PSaV. Two complete PEDV genomes (28,029 and 28,034 nt) were assembled in addition to one full-length sequence corresponding to porcine kobuvirus Aichivirus C (8116 nt), a partial porcine kobuvirus Aichivirus C sequence (580 nt), and a partial porcine sapovirus sequence (2977 nt) (Table 2). BLASTn analyses using an NCBI viral genome database showed high coverage and identity between both PEDV genomes and a USA strain (KF272920.1).
Table 2.
Percentage identities of the assembled contigs for swine viral families.
3.3. Genome Analysis of Porcine Kobuvirus
PEDV and Aichivirus C coinfections were identified in the two analyzed samples. A complete sequence of the porcine kobuvirus genome (GenBank Accession no. MT211964) was obtained in sample 205H, and a contig of 10% of the length of the genome was also generated for sample 103H (Table 2). Phylogenetic analysis based on the complete polyprotein gene revealed that the strain detected in this study formed a cluster with isolate OH/RV11/2011 from the USA and Chgz/2015 (Figure 1).

Figure 1.
Phylogenetic tree using the full CDS of PKV. The tree was inferred using the maximum likelihood method and GTR model with gamma distribution and SH-like branch support using PhyML software v3.3.21 (Université de Montpellier, France). The sequence obtained in this study is marked in color.
Sequence analyses of the polyprotein gene revealed a nucleotide identity range of 89.70–90.66% and an amino acid identity between 96.91 and 97.84% to reference strains from the USA and China (GenBank: MF506730.1, KC424640.1), respectively. The previously reported 90 nt motif duplex present in strains from Hungary, Spain, and China and the insertion present at position 135–137 nt of US/OH/RV50/2011 were not present in the assembled sequences [14,21]. No evidence of recombination was found.
3.4. Genome Analysis of the Porcine Epidemic Diarrhea Virus
For both samples, full-length coronavirus genomes were assembled. These sequences presented a ≈99.6% identity to local Mexican sequences. Full genome phylogenetic analysis showed that the assembled genomes were located in a cluster with other previously reported no-indel Mexican strains (Figure 2). ORF3 of the two genomes consisted of 224 aa, with a high nucleotide identity (≈99.8%) to local strains, and, as expected from the phylogeny results, did not have the deletion present in attenuated strains [40,41]. However, the two ORF3 sequences showed mutations at the amino acid level: V → A at amino acid position (aa) 21, V → I at aa 54, F → V at aa 80, and A → T at aa 100.
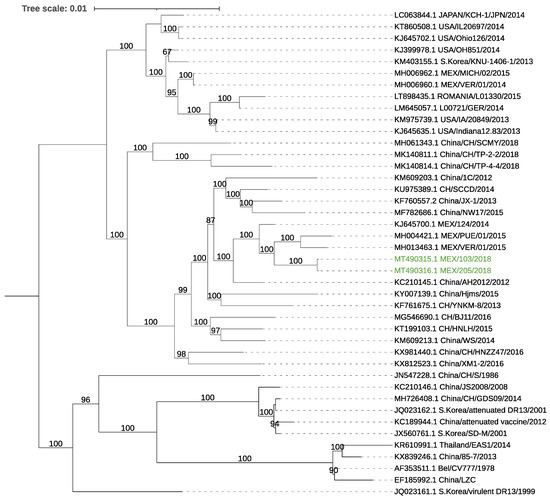
Figure 2.
Phylogenetic tree using the full genome sequences of PEDV. The tree was inferred using the maximum likelihood method and GTR model with gamma distribution and SH-like branch support using PhyML software v3.3.21 (Université de Montpellier, France). The sequences obtained in this study are marked in color.
The analysis of the S gene of both samples showed a nucleotide similarity with local strains of 99.47–99.66% and 99.42–99.78% at the amino acid level (MH004420.1, KJ645708.1) and a 99.66% nt/96.64% aa identity to the reference strain CV777 (NC_003436.1), with most amino acid changes located in the S1 domain. In addition, multiple mutations were located among regions containing neutralizing epitopes at residues 592–607: G → S at aa 594, A → E at aa 605, and in the epitope region 636–789: Y → S at aa 766, N → D 707, N → S at aa 719, N → S at aa 724, and Y → S at aa 766 of the reference genome CV777 S protein. The obtained sequences were uploaded to the GenBank database: MT490315 (103H), MT490316 (205H).
3.5. Sequence Analysis of PSaV
A sequence of 2977 nt corresponding to PSaV partial ORF 1 and ORF 2 was assembled from one of the samples, displaying high nucleotide homology (91.13%) to an available Sapovirus GIII sequence (KX688107.1). For VP2, the similarity was 93.36% at the nucleotide level and 97.08% at the amino acid level to the USA 2015 sequence (MK965901.1). The capsid gene presented a homology of 97% at the amino acid level with the GIII 2019 USA strain (MK965900.1). Phylogenetic analysis based on the capsid gene region of PSaV showed a clustering with samples from the USA and China (Figure 3).

Figure 3.
Phylogenetic tree for the capsid coding region of porcine Sapovirus. The tree was inferred using the maximum likelihood method and GTR model with gamma distribution and SH-like branch support using PhyML software v3.3.21 (Université de Montpellier, France). The sequence obtained in this study is marked in color.
3.6. Viral Culture
Following inoculation and passage with PKV-positive samples,
a cytopathic effect was visible 3 to 4 days
post-infection (p.i.). After harvesting, PKV RNA was consistently detected in two isolates after several passages. Porcine coronavirus RNA was not detected by RT-PCR in the cells or supernatants in passages in cell culture, and no cell fusion or syncytia characteristic of porcine coronavirus replication was observed (Figure 4).

Figure 4.
Representative cytopathic effect (CPE) after 5 days post-infection. (a) Mock-infected Vero cells. (b) Cytopathic effect of Vero cells after passage 9 from infection with gastric content obtained from a diarrheic piglet 10X.
4. Discussion
Since the introduction of PEDV in México in 2013, it has become one of the primary causative agents for diarrheic manifestations in swine productions. Additionally, PKV was detected in pigs from the majority of analyzed herds. Although PEDV was identified as being responsible for the majority of the outbreaks in the analyzed production units, we were able to identify the presence of PKV in most of these farms. PKV was unknown to be circulating in México and had not been considered in the control measures at the affected farms due to the lack of previous reports in the country.
The presence of both viruses in the same herds with similar clinical signs could indicate a high association of kobuvirus infection with diarrhea but not necessarily as the causative agent since no affected farm was solely positive for PKV. In addition to coronavirus infection by PEDV, diarrheic outbreaks in Mexican porcine farms have been attributed to Deltacoronavirus infection; however, the only farm positive for Deltacoronavirus in this study was negative for coinfection with PEDV and PKV [42].
PKV has been detected in pigs with and without clinical symptoms [43], with the highest prevalence occurring in suckling pigs and 40% positivity in non-diarrheic piglets, as reported by Park, 2010 [13]. It is possible that PKV is an opportunistic pathogen of gastroenteritis in pigs when primary infection by PEDV or other pathogens occurs; however, the possible effects in disease outcome, recovery times, and daily weight gain in surviving animals are unknown. Although the PEDV and PKV coinfection pathogenesis remains unclear, Zhao et al. reported a higher prevalence of this coinfection over the prevalence of infections by PEDV alone in China, which could indicate a possible greater impact and dissemination for coinfection cases [18].
Based on the whole genome phylogenetic analyses, both PEDV and PKV showed a close relation to strains reported in México, China, and the USA. No local sequences for PKV were available at the time of this study. Another agent detected in the NGS analyses was PSaV. The PSaV capsid analysis reflected the same geographical association as PKV and PEDV. The clustering of these viruses reflects the close commercial relation of the swine industry between the USA and México. To our knowledge, this is the first full genome report of porcine kobuvirus in México, as well as the first phylogenetic analysis for porcine PSaV in the country.
The taxonomic classification of de novo assembled contigs by BLASTn reported the presence of several viral agents in the samples, mainly members of Coronaviridae, Picornaviridae, and Caliciviridae families in sample 205H known to cause similar clinical presentations. However, the assembly of full-length genomes for PKV in sample 103H was not possible, and PSaV was not detected. The difference in the detected pathogens may derive from the disparity in the sequencing efficiency, as both animals were housed in close proximity in the same area. Additional analyses based on contigs and filtered reads detected the presence of known commensal and environmental viruses coherent with the type of sample used—likely originating from the digested intestinal content.
During an active outbreak in a production unit, diagnosis is restricted to a small array of agents known to be present in the geographical region; thus, the most prevalent causes such as PEDV are likely to be investigated and controlled, leaving possible secondary infections undiagnosed, which could lead to complications, particularly for feedback-based control. While the implementation of regular viral metagenomics as a regular diagnostic tool could be prohibitive for most farms, an NGS analysis of representative samples from farms could provide a better perspective and understanding of the circulating viruses and other pathogens in the production unit and could be available from a commercial provider. Further studies must be conducted to estimate the prevalence of PKV and PSaV in swine production units and their clinical implications.
Author Contributions
Formal analysis, M.-E.G.-H.; funding acquisition, R.-E.S.-S.; investigation, M.-E.G.-H. and R.-E.S.-S.; methodology, C.S.-J.; project administration, M.-E.T.-O. and R.-E.S.-S.; supervision, L.L.-A.-B., S.-L.A.-E. and B.I.T.-R.; validation, L.L.-A.-B.; writing—original draft, M.-E.G.-H.; writing—review and editing, M.-E.T.-O., L.L.-A.-B., C.S.-J. and R.-E.S.-S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica PAPIIT-IN221218 and CONACyT.
Institutional Review Board Statement
The study was conducted according to the guidelines of and requirements indicated by the Institutional Committee for the Care and Use of Experimental Animals (CICUAE- FMVZ-UNAM).
Data Availability Statement
The data presented in this study are openly available in in the GenBank at https://www.ncbi.nlm.nih.gov/genbank/ (accessed on 20 February 2020), Reference numbers: MT490316.1, MT726205.1, MT211964.1, MT490315.1.
Acknowledgments
The authors would like to thank MC. Elvia Lazo, MC. Susana Juarez and MVZ Selene Ferrnández Hernández for their support in the laboratory work, and Stefan A. for English revisions. Thanks go to the Post Graduate program in Ciencias de la Producción y de la Salud Animal and CONACyt for the scholarship received.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sun, Y.G.; Li, R.; Xie, S.; Qiao, S.; Li, Q.; Chen, X.X.; Deng, R.; Zhang, G. Identification of a novel linear B-cell epitope within the collagenase equivalent domain of porcine epidemic diarrhea virus spike glycoprotein. Virus Res. 2019, 266, 34–42. [Google Scholar] [CrossRef]
- Zhang, F.; Luo, S.; Gu, J.; Li, Z.; Li, K.; Yuan, W.; Ye, Y.; Li, H.; Ding, Z.; Song, D.; et al. Prevalence and phylogenetic analysis of porcine diarrhea associated viruses in southern China from 2012 to 2018. BMC Vet. Res. 2019, 15, 470. [Google Scholar] [CrossRef]
- Stevenson, G.W.; Hoang, H.; Schwartz, K.J.; Burrough, E.R.; Sun, D.; Madson, D.; Cooper, V.L.; Pillatzki, A.; Gauger, P.; Schmitt, B.J.; et al. Emergence of Porcine epidemic diarrhea virus in the United States: Clinical signs, lesions, and viral genomic sequences. J. Vet. Diagn. Investig. 2013, 25, 649–654. [Google Scholar] [CrossRef]
- Trujillo-Ortega, M.E.; Beltrán-Figueroa, R.; García-Hernández, M.E.; Juárez-Ramírez, M.; Sotomayor-González, A.; Hernández-Villegas, E.N.; Becerra-Hernández, J.F.; Sarmiento-Silva, R.E. Isolation and characterization of porcine epidemic diarrhea virus associated with the 2014 disease outbreak in Mexico: Case report. BMC Vet. Res. 2016, 12, 132. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.; Wyler, R. Quantitation, biological and physicochemical properties of cell culture-adapted porcine epidemic diarrhea coronavirus (PEDV). Vet. Microbiol. 1989, 20, 131–142. [Google Scholar] [CrossRef]
- Marthaler, D.; Jiang, Y.; Otterson, T.; Goyal, S.; Rossow, K.; Collins, J. Complete genome sequence of porcine epidemic diarrhea virus strain USA/Colorado/2013 from the United States. Genome Announc. 2013, 1, e00555-13. [Google Scholar] [CrossRef]
- Reuter, G.; Boldizsár, Á.; Kiss, I.; Pankovics, P. Candidate new species of Kobuvirus in porcine hosts. Emerg. Infect. Dis. 2008, 14, 1968. [Google Scholar] [CrossRef]
- Sisay, Z.; Wang, Q.; Oka, T.; Saif, L. Prevalence and molecular characterization of porcine enteric caliciviruses and first detection of porcine kobuviruses in US swine. Arch. Virol. 2013, 158, 1583–1588. [Google Scholar] [CrossRef][Green Version]
- Yu, J.M.; Jin, M.; Zhang, Q.; Li, H.Y.; Li, D.D.; Xu, Z.Q.; Li, J.S.; Cui, S.X.; Yang, S.H.; Liu, N.; et al. Candidate porcine Kobuvirus, China. Emerg. Infect. Dis. 2009, 15, 823. [Google Scholar] [CrossRef] [PubMed]
- Barry, A.F.; Ribeiro, J.; Alfieri, A.F.; van der Poel, W.H.M.; Alfieri, A.A. First detection of kobuvirus in farm animals in Brazil and the Netherlands. Infect. Genet. Evol. 2011, 11, 1811–1814. [Google Scholar] [CrossRef] [PubMed]
- Khamrin, P.; Maneekarn, N.; Kongkaew, A.; Kongkaew, S.; Okitsu, S.; Ushijima, H. Porcine kobuvirus in piglets, Thailand. Emerg. Infect. Dis. 2009, 15, 2075. [Google Scholar] [CrossRef]
- Khamrin, P.; Maneekarn, N.; Hidaka, S.; Kishikawa, S.; Ushijima, K.; Okitsu, S.; Ushijima, H. Molecular detection of kobuvirus sequences in stool samples collected from healthy pigs in Japan. Infect. Genet. Evol. 2010, 10, 950–954. [Google Scholar] [CrossRef]
- Park, S.J.; Kim, H.K.; Moon, H.J.; Song, D.S.; Rho, S.M.; Han, J.Y.; Nguyen, V.G.; Park, B.K. Molecular detection of porcine kobuviruses in pigs in Korea and their association with diarrhea. Arch. Virol. 2010, 155, 1803–1811. [Google Scholar] [CrossRef]
- Reuter, G.; Boldizsár, Á.; Pankovics, P. Complete nucleotide and amino acid sequences and genetic organization of porcine kobuvirus, a member of a new species in the genus Kobuvirus, family Picornaviridae. Arch. Virol. 2009, 154, 101. [Google Scholar] [CrossRef]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef]
- Jackova, A.; Sliz, I.; Mandelik, R.; Salamunova, S.; Novotny, J.; Kolesarova, M.; Vlasakova, M.; Vilcek, S. Porcine kobuvirus 1 in healthy and diarrheic pigs: Genetic detection and characterization of virus and co-infection with rotavirus A. Infect. Genet. Evol. 2017, 49, 73–77. [Google Scholar] [CrossRef]
- Zhou, W.; Ullman, K.; Chowdry, V.; Reining, M.; Benyeda, Z.; Baule, C.; Juremalm, M.; Wallgren, P.; Schwarz, L.; Zhou, E.; et al. Molecular investigations on the prevalence and viral load of enteric viruses in pigs from five European countries. Vet. Microbiol. 2016, 182, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.P.; Yang, Z.; Lin, W.D.; Wang, W.Y.; Yang, J.; Jin, W.J.; Qin, A.J. The rate of co-infection for piglet diarrhea viruses in China and the genetic characterization of porcine epidemic diarrhea virus and porcine kobuvirus. Acta Virol. 2016, 60, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Ruan, H.; Qiao, S.; Deng, R.; Zhang, G. Co-infection status of porcine circoviruses (PCV2 and PCV3) and porcine epidemic diarrhea virus (PEDV) in pigs with watery diarrhea in Henan province, central China: Co-infection of PCV2, PCV3 and PEDV. Microb. Pathog. 2020, 142, 104047. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Kang, B.K.; Lee, C.S.; Song, D.S. Impact of porcine group A rotavirus co-infection on porcine epidemic diarrhea virus pathogenicity in piglets. Res. Vet. Sci. 2008, 84, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, L.; Zheng, Y.; Zhang, J.; Guo, B.; Yoon, K.J.; Gauger, P.C.; Harmon, K.M.; Main, R.G.; Li, G. Metagenomic analysis of the RNA fraction of the fecal virome indicates high diversity in pigs infected by porcine endemic diarrhea virus in the United States. Virol. J. 2018, 15, 95. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Li, C.; Qi, S.; Yang, D.; Jiang, N.; Yin, B.; Guo, D.; Kong, F.; Yuan, D.; Feng, L.; et al. A molecular epidemiological investigation of PEDV in China: Characterization of co-infection and genetic diversity of S1-based genes. Transbound. Emerg. Dis. 2020, 67, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Di Bartolo, I.; Tofani, S.; Angeloni, G.; Ponterio, E.; Ostanello, F.; Ruggeri, F.M. Detection and characterization of porcine caliciviruses in Italy. Arch. Virol. 2014, 159, 2479–2484. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; Lawrence Berkeley National Lab.(LBNL): Berkeley, CA, USA, 2014. [Google Scholar]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Li, Z.-L.; Zhu, L.; Ma, J.-Y.; Zhou, Q.-F.; Song, Y.-H.; Sun, B.-L.; Chen, R.-A.; Xie, Q.-M.; Bee, Y.-Z. Molecular characterization and phylogenetic analysis of porcine epidemic diarrhea virus (PEDV) field strains in south China. Virus Genes 2012, 45, 181–185. [Google Scholar] [CrossRef]
- Liu, P.; Du, R.J.; Wang, L.; Han, J.; Liu, L.; Zhang, Y.L.; Xia, J.K.; Lu, F.M.; Zhuang, H. Management of Hepatitis E Virus (HEV) zoonotic transmission: Protection of rabbits against HEV challenge following immunization with HEV 239 vaccine. PLoS ONE 2014, 9, e87600. [Google Scholar] [CrossRef]
- Hu, H.; Jung, K.; Vlasova, A.N.; Chepngeno, J.; Lu, Z.; Wang, Q.; Saif, L.J. Isolation and characterization of porcine deltacoronavirus from pigs with diarrhea in the United States. J. Clin. Microbiol. 2015, 53, 1537–1548. [Google Scholar] [CrossRef]
- Andrews, S. FASTQC A Quality Control tool for High Throughput Sequence Data. 2015. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 13 April 2021).
- Wang, Q.H.; Souza, M.; Funk, J.A.; Zhang, W.; Saif, L.J. Prevalence of noroviruses and sapoviruses in swine of various ages determined by reverse transcription-PCR and microwell hybridization assays. J. Clin. Microbiol. 2006, 44, 2057–2062. [Google Scholar] [CrossRef]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.A.; Korobeynikov, A.; Lapidus, A.; Prjibelski, A.D.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. 2013, 20, 714–737. [Google Scholar] [CrossRef] [PubMed]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.; Korobeynikov, A.; Lapidus, A.; Prjibelsky, A.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling genomes and mini-metagenomes from highly chimeric reads. In Research in Computational Molecular Biology; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Lethiec, F.; Duroux, P.; Gascuel, O. PHYML Online—A web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 2005, 33 (Suppl. 2), W557–W559. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Lara-Romero, R.; Gómez-Núñez, L.; Cerriteño-Sánchez, J.L.; Márquez-Valdelamar, L.; Mendoza-Elvira, S.; Ramírez-Mendoza, H.; Rivera-Benítez, J.F. Molecular characterization of the spike gene of the porcine epidemic diarrhea virus in Mexico, 2013–2016. Virus Genes 2018, 54, 215–224. [Google Scholar] [CrossRef]
- Chen, Q.; Gauger, P.C.; Stafne, M.R.; Thomas, J.T.; Madson, D.M.; Huang, H.; Zheng, Y.; Li, G.; Zhang, J. Pathogenesis comparison between the United States porcine epidemic diarrhoea virus prototype and S-INDEL-variant strains in conventional neonatal piglets. J. Gen. Virol. 2016, 97, 1107–1121. [Google Scholar] [CrossRef]
- Pérez-Rivera, C.; Ramírez-Mendoza, H.; Mendoza-Elvira, S.; Segura-Velázquez, R.; Sánchez-Betancourt, J.I. First report and phylogenetic analysis of porcine deltacoronavirus in Mexico. Transbound. Emerg. Dis. 2019, 66, 1436–1441. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lan, X.; Yang, B. Molecular Epidemiological Investigation of Porcine kobuvirus and Its Coinfection Rate with PEDV and SaV in Northwest China. BioMed Res. Int. 2016, 2016, 1–6. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).