Prediction of the Spatial Origin of Puumala Virus Infections Using L Segment Sequences Derived from a Generic Screening PCR †


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
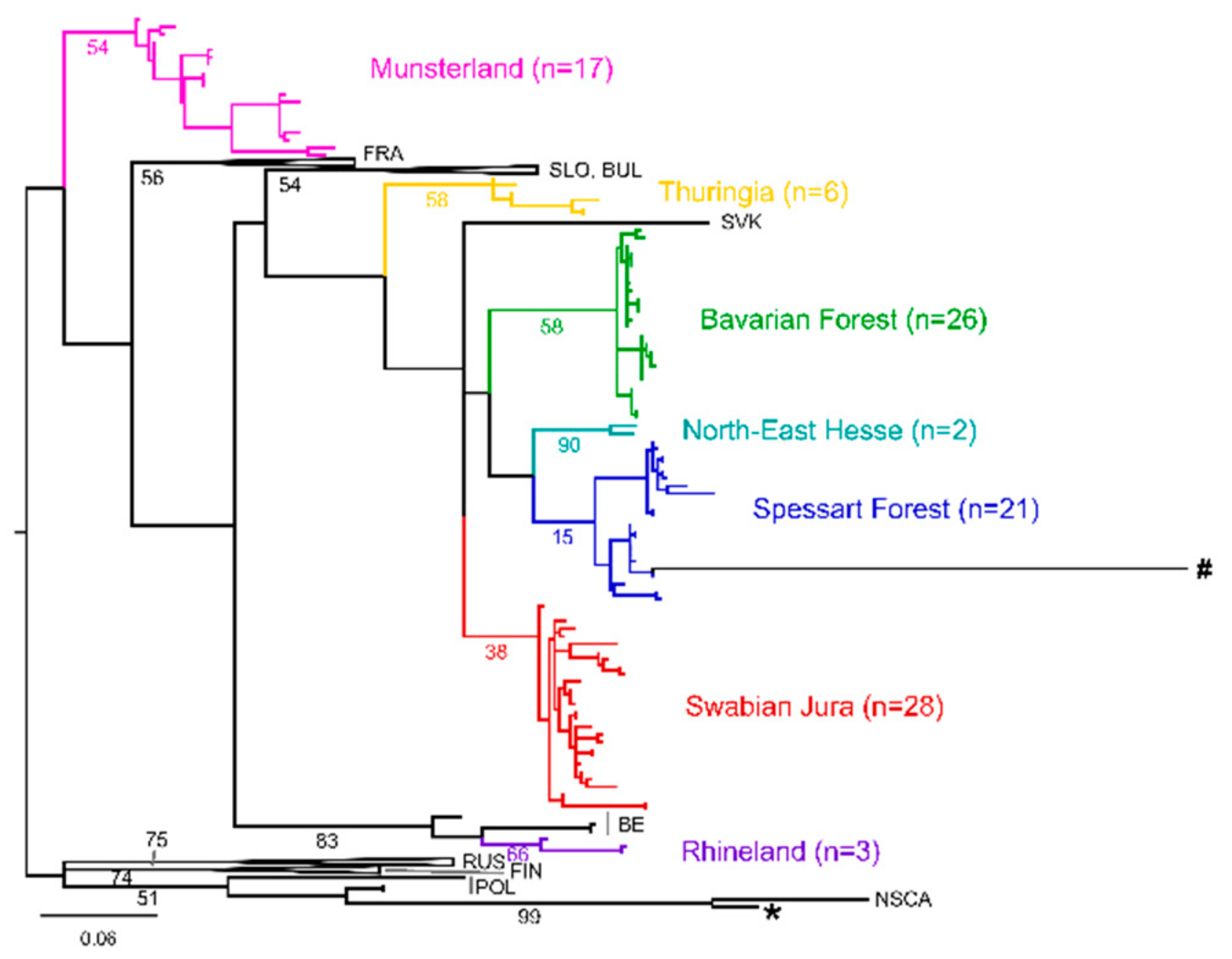
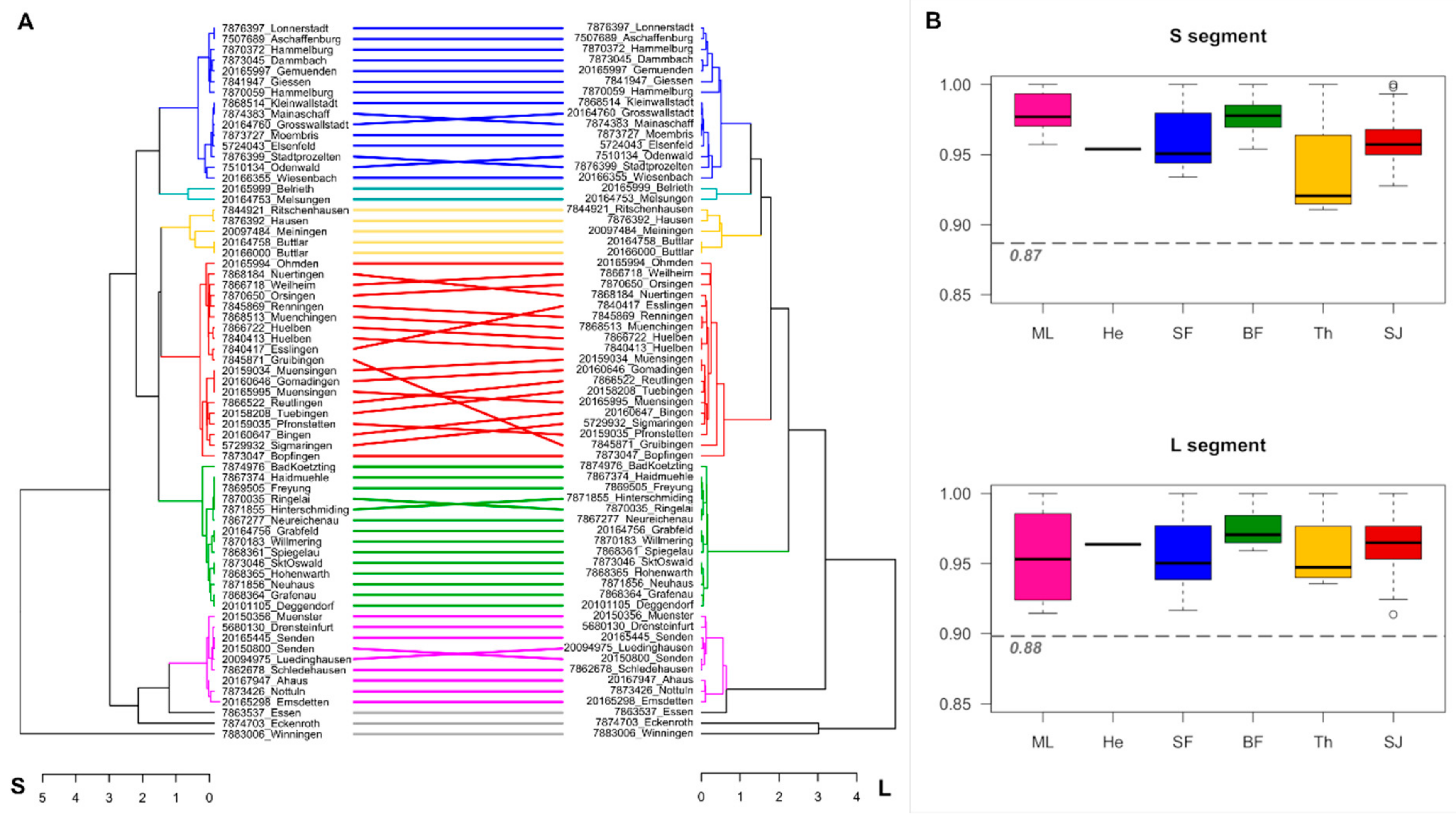
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Latus, J.; Schwab, M.; Tacconelli, E.; Pieper, F.-M.; Wegener, D.; Dippon, J.; Müller, S.; Zakim, D.; Segerer, S.; Kitterer, D.; et al. Clinical Course and Long-Term Outcome of Hantavirus-Associated Nephropathia Epidemica, Germany. Emerg. Infect. Dis. 2015, 21. [Google Scholar] [CrossRef] [PubMed]
- Krüger, D.H.; Ulrich, R.G.; Hofmann, J. Hantaviruses as Zoonotic Pathogens in Germany. Dtsch. Arztebl. Int. 2013, 110, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Vaheri, A.; Henttonen, H.; Voutilainen, L.; Mustonen, J.; Sironen, T.; Vapalahti, O. Hantavirus Infections in Europe and Their Impact on Public Health. Rev. Med. Virol. 2013, 23, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Reil, D.; Binder, F.; Freise, J.; Imholt, C.; Beyrer, K.; Jacob, J.; Krüger, D.H.; Hofmann, J.; Dreesman, J.; Ulrich, R.G. Hantaviren in Deutschland: Aktuelle Erkenntnisse Zu Erreger, Reservoir, Verbreitung Und Prognosemodellen Hantaviruses in Germany: Current Knowledge on Pathogens, Reservoirs, Distribution and Forecast Models. Berliner und Münchener Tierärtzliche Wochenschrift 2018, 131, 453–464. [Google Scholar] [CrossRef]
- Faber, M.; Krüger, D.H.; Auste, B.; Stark, K.; Hofmann, J.; Weiss, S. Molecular and Epidemiological Surveillance of Human Puumala and Dobrava-Belgrade Hantavirus Infections, Germany, 2001–2017. Eurosurveillance 2019, (in press).
- Schilling, S.; Emmerich, P.; Klempa, B.; Auste, B.; Schnaith, E.; Schmitz, H.; Krüger, D.H.; Günther, S.; Meisel, H. Hantavirus Disease Outbreak in Germany: Limitations of Routine Serological Diagnostics and Clustering of Virus Sequences of Human and Rodent Origin. J. Clin. Microbiol. 2007, 45, 3008–3014. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, J.; Hofmann, J.; Enders, M.; Tewald, F.; Oehme, R.M.; Rosenfeld, U.M.; Sheikh Ali, H.; Schlegel, M.; Essbauer, S.; Osterberg, A.; et al. Multiple Synchronous Outbreaks of Puumala Virus, Germany, 2010. Emerg. Infect. Dis. 2012, 18, 1461–1464. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, J.; Meisel, H.; Klempa, B.; Vesenbeckh, S.M.; Beck, R.; Michel, D.; Schmidt-Chanasit, J.; Ulrich, R.G.; Grund, S.; Enders, G.; et al. Hantavirus Outbreak, Germany, 2007. Emerg. Infect. Dis. 2008, 14, 850–852. [Google Scholar] [CrossRef]
- Klempa, B.; Fichet-Calvet, E.; Lecompte, E.; Auste, B.; Aniskin, V.; Meisel, H.; Denys, C.; Koivogui, L.; Ter Meulen, J.; Krüger, D.H. Hantavirus in African Wood Mouse, Guinea. Emerg. Infect. Dis. 2006, 12, 838–840. [Google Scholar] [CrossRef]
- Weber de Melo, V.; Sheikh Ali, H.; Freise, J.; Kühnert, D.; Essbauer, S.; Mertens, M.; Wanka, K.M.; Drewes, S.; Ulrich, R.G.; Heckel, G. Spatiotemporal Dynamics of Puumala Hantavirus Associated with Its Rodent Host, Myodes Glareolus. Evol. Appl. 2015, 8, 545–559. [Google Scholar] [CrossRef]
- Castel, G.; Couteaudier, M.; Sauvage, F.; Pons, J.-B.; Murri, S.; Plyusnina, A.; Pontier, D.; Cosson, J.-F.; Plyusnin, A.; Marianneau, P.; et al. Complete Genome and Phylogeny of Puumala Hantavirus Isolates Circulating in France. Viruses 2015, 7, 5476–5488. [Google Scholar] [CrossRef] [PubMed]
- Bowen, M.D.; Gelbmann, W.; Ksiazek, T.G.; Nichol, S.T.; Nowotny, N. Puumala Virus and Two Genetic Variants of Tula Virus Are Present in Austrian Rodents. J. Med. Virol. 1997, 53, 174–181. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView Version 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Lefort, V.; Longueville, J.-E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [PubMed]
- RStudio Team. RStudio: Integrated Development Environment for R; RStudio, Inc.: Boston, MA, USA, 2016; Available online: http://www.rstudio.com (accessed on 2 July 2018).
- Paradis, E.; Schliep, K. Ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in {R}. Bioinformatics 2018, 35, 526–528. [Google Scholar] [CrossRef]
- Galili, T. Dendextend: An R Package for Visualizing, Adjusting, and Comparing Trees of Hierarchical Clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef]
- Becker, R.A.; Wilks, A.R.; Brownrigg, R. Maps: Draw Geographical Map. 2018. Available online: https://cran.r-project.org/package=maps (accessed on 10 May 2019).
- Korva, M.; Knap, N.; Rus, K.R.; Fajs, L.; Grubelnik, G.; Bremec, M.; Knapič, T.; Trilar, T.; Županc, T.A. Phylogeographic Diversity of Pathogenic and Non- Pathogenic Hantaviruses in Slovenia. Viruses 2013, 5, 3071–3087. [Google Scholar] [CrossRef]
- Razzauti, M.; Plyusnina, A.; Henttonen, H.; Plyusnin, A. Microevolution of Puumala Hantavirus during a Complete Population Cycle of Its Host, the Bank Vole (Myodes Glareolus). PLoS ONE 2013, 8, e64447. [Google Scholar] [CrossRef] [PubMed]
- Szabó, R.; Radosa, L.; Ličková, M.; Sláviková, M.; Heroldová, M.; Stanko, M.; Pejčoch, M.; Osterberg, A.; Laenen, L.; Schex, S.; et al. Phylogenetic Analysis of Puumala Virus Strains from Central Europe Highlights the Need for a Full-Genome Perspective on Hantavirus Evolution. Virus Genes 2017, 53, 913–917. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiss, S.; Klempa, B.; Tenner, B.; Kruger, D.H.; Hofmann, J. Prediction of the Spatial Origin of Puumala Virus Infections Using L Segment Sequences Derived from a Generic Screening PCR. Viruses 2019, 11, 694. https://doi.org/10.3390/v11080694
Weiss S, Klempa B, Tenner B, Kruger DH, Hofmann J. Prediction of the Spatial Origin of Puumala Virus Infections Using L Segment Sequences Derived from a Generic Screening PCR. Viruses. 2019; 11(8):694. https://doi.org/10.3390/v11080694
Chicago/Turabian StyleWeiss, Sabrina, Boris Klempa, Beate Tenner, Detlev H. Kruger, and Jörg Hofmann. 2019. "Prediction of the Spatial Origin of Puumala Virus Infections Using L Segment Sequences Derived from a Generic Screening PCR" Viruses 11, no. 8: 694. https://doi.org/10.3390/v11080694
APA StyleWeiss, S., Klempa, B., Tenner, B., Kruger, D. H., & Hofmann, J. (2019). Prediction of the Spatial Origin of Puumala Virus Infections Using L Segment Sequences Derived from a Generic Screening PCR. Viruses, 11(8), 694. https://doi.org/10.3390/v11080694