Manipulation of Epithelial Differentiation by HPV Oncoproteins


1
Department of Otorhinolaryngology: Head and Neck Surgery, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA 19104, USA
2
Department of Microbiology, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA 19104, USA
Viruses 2019, 11(4), 369; https://doi.org/10.3390/v11040369
Submission received: 18 March 2019
/
Revised: 18 April 2019
/
Accepted: 20 April 2019
/
Published: 22 April 2019
(This article belongs to the Special Issue Viruses Ten-Year Anniversary)
{kind=link}
{kind=link}
Abstract
:Papillomaviruses replicate and cause disease in stratified squamous epithelia. Epithelial differentiation is essential for the progression of papillomavirus replication, but differentiation is also impaired by papillomavirus-encoded proteins. The papillomavirus E6 and E7 oncoproteins partially inhibit and/or delay epithelial differentiation and some of the mechanisms by which they do so are beginning to be defined. This review will outline the key features of the relationship between HPV infection and differentiation and will summarize the data indicating that papillomaviruses alter epithelial differentiation. It will describe what is known so far and will highlight open questions about the differentiation-inhibitory mechanisms employed by the papillomaviruses.
1. Introduction
Papillomaviruses and their host species have evolved together for tens of millions of years, and it is thought that the viruses have both adapted to specific host niches and co-speciated with their hosts [1]. Papillomavirus infections are highly specific for an individual vertebrate species and restricted to a specific tissue: the stratified squamous epithelium [2]. A highly regulated differentiation program enables stratified epithelia to constantly self-renew and papillomavirus replication is enabled by epithelial differentiation [3,4]. Differentiation is required for replication of the papillomavirus DNA genome and for late viral gene expression, and the progression of epithelial cells from basal layers to eventual desquamation enables papillomavirus assembly and egress. Papillomavirus replication requires epithelial differentiation, but also delays it. To enable viral genome replication in infected epithelial cells that would have otherwise committed to a terminal differentiation program, the virus must promote proliferation in the differentiating cell. This is said to ‘uncouple’ proliferation from differentiation. Papillomaviruses inhibit differentiation via a variety of mechanisms, some of which overlap with the pro-proliferative targets and others that are distinct.
The human papillomavirus (HPV) E6 and E7 proteins have been the most frequently characterized with respect to their effects on cellular proliferation and, to some degree, on differentiation. Some of the pro-proliferative mechanisms employed by HPV E6 and E7 are shared by other DNA tumor viruses that do not specifically infect stratified epithelia. For example, several DNA tumor viruses inactivate the retinoblastoma and p53 tumor suppressors to promote proliferation and evade apoptosis [5]. Other viral activities are unique to the papillomavirus-encoded proteins, and these unique activities have allowed the adaptation of papillomaviruses to their specific species and to specific biological niches [1]. The ability to control epithelial differentiation is one such specific activity. HPVs have been recognized to alter differentiation for many years, but recent advances have begun to clarify the mechanisms by which they do so.
This review will summarize the studies that have established the effects of HPV-encoded proteins, particularly HPV E6 and E7, on epithelial differentiation. It will highlight some of the mechanisms by which HPV E6 and E7 delay and impair differentiation. Impaired differentiation is a hallmark of some cancers and could contribute to HPV-mediated carcinogenesis. Open questions related to HPV control of epithelial differentiation and the possibility of differentiation therapy for HPV-positive cancers will be discussed.
2. Differentiation-Dependent HPV Biology
HPV have circular double-stranded DNA genomes of ~8 kbp that encode seven to nine open reading frames (ORF) depending on the specific virus. Genetic diversity among the papillomaviruses (PV) is high. Well over 200 HPV genotypes and hundreds more animal PV genotypes have been identified and sequenced and many more are pending characterization [6,7]. Individual PV genotypes differ from one another by at least 10% at the DNA sequence level and they are commonly arranged into a phylogeny based upon the sequence of the L1 gene [2]. The known HPV group into five genera (alpha, beta, gamma, mu, and nu) in an L1-based phylogenetic tree and the majority of the HPV are in genus alpha, beta, or gamma. The genera are further subdivided into species on the basis of sequence similarity. PV that differ by less than 10% are referred to as subtypes and variants. Genus alpha HPV are predominately, but not exclusively, mucosal-tropic, whereas the genus beta and genus gamma HPV infect cutaneous epithelia.
Several features of the productive HPV life cycle depend on the biology of the stratified squamous epithelium. Upon initial infection, HPV particles bind to heparan sulfate proteoglycans present on the cell membrane or in the extracellular matrix [8,9]. A subsequent conformational change in L1 and L2 capsid proteins enables their cleavage by furin or other host enzymes. This enables virus entry into basal epithelial cells via a slow process involving endocytosis and uptake into endosomes. The L2 protein is required to deliver the viral genome to the nucleus in a process recently shown to require a cell-penetrating peptide (CPP) sequence conserved in L2 [10]. The ability of L2 to penetrate the endosomal membrane and to bind to the retromer complex allows for trafficking of the viral particle within the endosome.
Following deposition of the HPV DNA in the nucleus, a burst of viral genome amplification establishes HPV genomes in basal epithelial cells. Viral DNA replication requires the HPV E1 and E2 proteins as well as components of the host cell DNA replication machinery [2,11]. In a productive infection, viral genomes are maintained over the lifetime of the infected cell, replicating once per cell cycle. Maintenance replication requires tethering of HPV genomes to host mitotic chromatin via the viral E2 protein, enabling partitioning of replicated genomes into newly divided daughter cells. When basal cells begin the highly regulated process of epithelial differentiation, PV genomes are maintained in the differentiating cell. Finally, the viral genomes are amplified to a high copy number during the vegetative, or productive, stage of virus replication. Regulation of the switch from maintenance to vegetative PV DNA replication is incompletely understood but requires epithelial differentiation. The HPV E6 and E7 proteins are not themselves directly involved in DNA replication, but they promote cell cycle progression in the otherwise differentiating epithelial cells. This enables expression of the cellular machinery required for viral DNA replication.
HPV transcription occurs from two major promoters: an early promoter present in the ~1 kbp viral non-coding region (variously referred to as the long coding region (LCR) or the upstream regulatory region (URR)) and a downstream late promoter. These have been extensively characterized in HPV31, where they are present at nucleotide 97 (p97) and nucleotide 742 (p742), respectively. The corresponding sites in HPV16 are p97 and p670 and in HPV18 are p55 and p811 [3,12,13,14,15]. Transcription from the HPV genome is unidirectional and is controlled by several mechanisms that are each related to differentiation. HPV early and late promoter activity is regulated by differentiation, with relatively little early promoter activity in basal cells and a differentiation-dependent increase in late viral promoter activity following viral DNA replication [13]. In addition, viral transcripts are subject to numerous splicing events, some of which are regulated by differentiation. For high-risk HPV, E6 and E7 are transcribed as a bicistronic early transcript and splicing of E6 is required for E7 translation [16,17]. In contrast, low-risk genus alpha HPV and genus beta HPV transcribe E6 and E7 from two distinct promoters and do not exhibit the corresponding splice in E6 [18]. Another major splicing event is a differentiation-dependent splice that is required to produce the E1^E4 transcript encoding the E4 protein. Although it is encoded in the viral early region, E4 is expressed from the late RNA transcript following the onset of vegetative DNA replication and is important for virion production and infectivity [19,20]. In summary, the late stages of HPV replication are dependent on keratinocyte differentiation and the ways in which differentiation enables replication have been incompletely defined. Cellular differentiation is required for HPV replication, and intricate links between virus and host differentiation-dependent biology control many aspects of HPV replication. Although beyond the scope of this review, many aspects of differentiation-dependent HPV gene regulation have been studied and summarized comprehensively elsewhere [3].
3. Keratinocyte Differentiation and Models of HPV Replication
3.1. Stages of Keratinocyte Differentiation
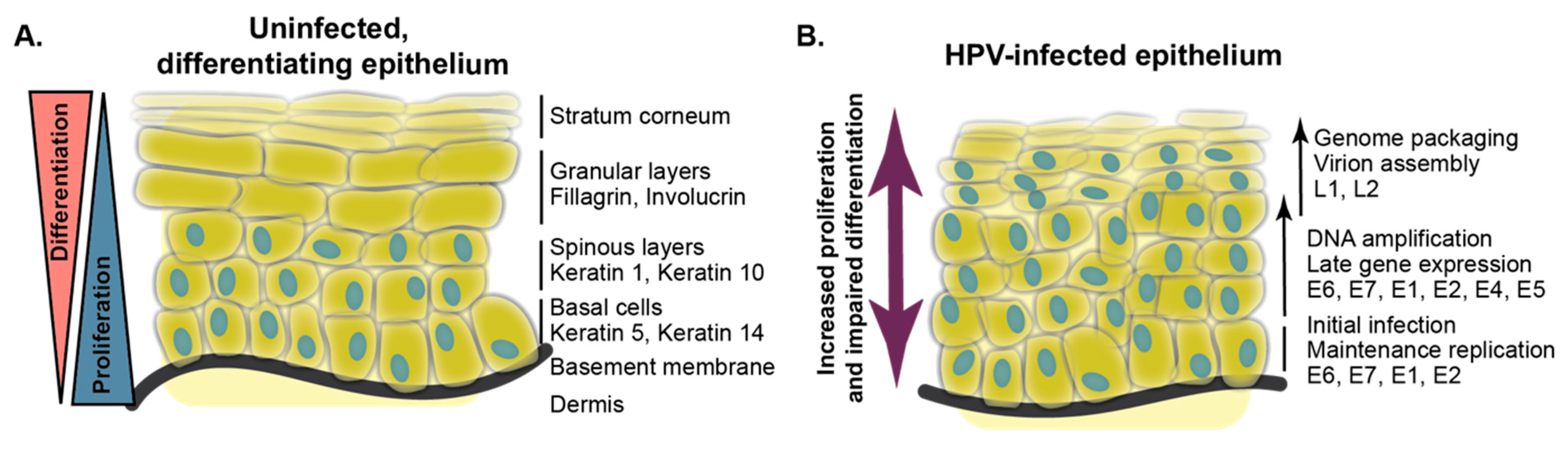
Epithelial differentiation is a constantly occurring regenerative process that forms the protective exterior barrier between an organism and its surrounding environment. Best studied in mammalian skin, the process of epidermal differentiation originates in proliferative basal keratinocytes that are separated from the dermis by a specialized form of extracellular matrix (ECM), the basement membrane [21]. Laminins on the basement membrane attach to integrins, such as α6β4 and α3β1, on the basal cell surface to tether basal cells to the ECM. Adherens junctions connect basal cells to one another. Proliferative basal epithelial cells express keratin 5 and keratin 14.
The first transition in the differentiation program is the delamination of a basal cell and its progression to a suprabasal (spinous) state [22,23]. Suprabasal keratinocytes express keratin 1 and keratin 10, which encode intermediate filament proteins that enable the formation of a cell–cell network connected by desmosomes. The next most distal layers to the ECM are the granular layer and the stratum corneum, respectively, and later markers of epithelial differentiation expressed in these layers include involucrin, loricrin, and fillagrin (Figure 1). By the time the keratinocyte differentiation program is complete, the once-proliferative cell exists as a squame, a dead cellular sac lacking a nucleus or other organelles. It is highly crosslinked and encased in a waterproof lipid network providing the exterior barrier function. Although many of the same cellular markers are expressed in the same order in mucosal versus cutaneous epithelia, it is important to note that differences in gene expression in these distinct tissues exist and enable their biological differences [24,25].
3.2. Differentiation Models
Several aspects of the differentiation-dependent HPV life cycle can be modeled in keratinocyte culture systems [26,27,28]. Human keratinocytes that harbor episomal (circular) and/or integrated HPV genomes have been both isolated from patient samples and generated experimentally. Keratinocytes can be induced to differentiate in monolayer culture using treatment with an increased concentration of calcium (typically 1.2–1.5 mM CaCl2), with or without the withdrawal of growth factors and/or the addition of serum. For many of the HPV-episome-containing cell lines, calcium treatment induces viral DNA amplification, transcription from the late promoter, and differentiation-dependent viral transcript splicing events. Culture in methylcellulose-based semisolid medium can promote many of the same changes. Three-dimensional cell culture systems (organotypic “raft” cultures) have been employed by HPV researchers for several decades. In such cultures, keratinocytes are grown to confluence on a matrix (often a collagen matrix containing mouse fibroblasts), then lifted to the air–liquid interface and allowed to stratify and differentiate for a period of 1–2 weeks. For cells that contain viral episomes, this allows for somewhat heterogenous differentiation and mimics some aspects of the viral life cycle. HPV virions are typically produced from organotypic cultures only rather inefficiently, especially in the case of immortalized cell lines. New PV model systems are sure to enable additional investigation. For example, a recently discovered murine papillomavirus has prompted additional studies on the relationship between cutaneous PV biology and epithelial differentiation [29,30,31,32,33,34].
Experiments in these systems have demonstrated that differentiation is required for HPV replication. Some specific cellular pathways that regulate epithelial differentiation have been found to regulate HPV genome amplification and late transcription. Protein kinase C (PKC) inhibitors limit HPV late activities, and efficient HPV virion production from organotypic raft cultures requires PKC activation [35,36]. Other experiments in keratinocyte differentiation models have begun to illustrate the effects of the HPV gene products on epithelial differentiation. These studies are reviewed in detail herein.
4. HPV Carcinogenic Activity and the E6 and E7 Oncoproteins
Just over one dozen mucosal HPV, primarily found in genus alpha species 7 and 9, are the “high-risk” HPV genotypes that cause nearly all cervical cancer and many other anogenital cancers [37,38]. High-risk HPV infection is responsible for approximately 5% of the worldwide cancer burden. In regions with limited access to screening and prevention programs, cervical cancer remains a major cause of mortality. Cancers of the oropharynx can be caused by high-risk HPV infection, predominantly by HPV16. In contrast to many other cancer types, the incidence of HPV-positive head and neck squamous cell carcinoma (HNSCC) is increasing in many populations, and in the United States alone there are now more cases of HPV-positive HNSCC than of cervical cancer [39]. Screening and prevention programs for HPV-positive oropharyngeal cancer and for HPV-associated genital cancers other than cervical cancer do not exist, nor are pharmacological approaches available to treat existing HPV infections. ‘Low-risk’ HPV, such as HPV6 and HPV11, cause lesions that rarely progress to cancer, but infection with low-risk HPV can cause genital warts, laryngeal papillomas, and other mucosal lesions [2]. Comparing high- to low-risk HPV has provided a wealth of information about the carcinogenic activity of the high-risk HPV proteins [40]. Many of the cellular reprogramming events required to enable HPV replication in the differentiating keratinocyte are likely to be conserved among diverse HPV types. Consequently, biological activities that are specific to high-risk HPV may account for their carcinogenic potential.
The contributions of the HPV E6 and E7 oncogenes to carcinogenesis have been studied extensively. A few years after zur Hausen and colleagues published the initial reports that cervical cancer samples contain human papillomavirus DNA, zur Hausen’s and other groups found that the HPV E6 and E7 ORFs are specifically preserved in cervical cancer cells and that they have transforming activity in rodent cells [41,42,43,44,45,46,47,48,49,50]. Further molecular analysis of the E6 and E7 gene products led to a model in which E7 proteins bind to retinoblastoma family proteins, including RB1, releasing E2F transcription factors and allowing passage through the G1/S checkpoint [51,52,53]. HPV E7 also promote proliferation by inhibiting the CDK inhibitors p21WAF1/CIP1 and p27KIP1 [54,55,56]. A consequence of high-risk HPV E7 expression is p53 stabilization [57]. To counter the apoptotic signaling that would otherwise be triggered by p53 stabilization, high-risk E6 proteins recruit the cellular ubiquitin ligase E6AP/UBE3A to enable p53 degradation [58,59]. In addition, E6 and E7 trigger genomic instability [60,61,62,63]. Oncogenic transformation by HPV results from persistent, long-term dysregulated expression of high-risk HPV E6 and E7 proteins [64,65].
This existing model does not account for all of the available information concerning the activities of HPV E6 and E7 [66]. Most HPV E7 proteins bind to RB1 and promote E2F-dependent gene expression, but HPV E7 proteins have varying ability to transform cells. Low-risk E7 proteins have very little transforming activity [67] and HPV1 E7 binds RB1 with high affinity but does not transform [68,69]. For several decades, HPV researchers have speculated that high-risk HPV E7 proteins have an RB1-independent transforming activity. This is supported by at least two lines of evidence. First, mutational analysis shows that alterations in the HPV16 E7 N- and C-termini result in a protein that is competent to bind RB1 but impaired for transformation [70,71,72,73,74,75]. Second, mouse models of cervical cancer and head and neck cancer provide evidence for an RB1-independent transforming activity of HPV E7 in vivo. In the oral model, <40% of mice develop tumors when RB1 is deleted, but the addition of HPV16 E7 to the RB1 knockout results in >90% tumor incidence [76]. In the cervical model, HPV16 E7 can induce cancer even when the transgenic mouse expresses an RB1 allele that cannot be bound by E7 [77,78]. Overall, there is not a consensus on which cellular targets might account for the RB1-independent transforming activity. Some recent findings address the initiation of carcinogenesis, accounting for the difference between high-risk and low-risk HPV E7 by demonstrating that high-risk HPV E7 upregulate the cell cycle inhibitors (CKI) p16INK4A/p14ARF epigenetically [79,80,81]. RB1 degradation by high-risk HPV E7 is proposed to enable evasion of the oncogene-induced senescence (OIS) that would result from p16INK4A induction.
A recent sequencing study highlights the critical contribution of HPV16 E7 to cervical carcinogenesis [82]. In this report, targeted sequencing of over 5000 patient samples from benign and precancerous HPV-positive cervical lesions revealed remarkable variability of HPV16 DNA sequences among patients. Depending on the cohort, in ~75–85% of patient samples the major HPV16 species detected had an HPV16 sequence unique among the thousands of patients. Furthermore, the variation in E6 was not the same as the variation in E7. In asymptomatic individuals, E6 and E7 sequences were both highly variable. In contrast, in precancerous lesions E6 sequences varied significantly, whereas E7 sequences were almost completely conserved. This suggests that if a lesion is to undergo malignant progression, there is a strong preference for the maintenance of the prototypical E7 sequence and that E7 is the high-risk HPV oncogene that is the major driver of oncogenic progression [82,83]. This result in human samples is consistent with the earlier finding that in a mouse model comparing HPV16 E6 and E7, it is E7 that is the dominant driver of estrogen-induced cervical carcinogenesis [84].
Updated models of oncogenic transformation by high-risk HPV have the potential to incorporate the new information provided by high-throughput studies and to account for activities of the viral oncoproteins that are not included in existing models. It is often stated that HPV oncoproteins act to uncouple proliferation from differentiation [4]. However, it is not clear whether HPV oncoproteins must impair differentiation to cause cancer and if so, what cellular targets are involved. Future studies should address whether the carcinogenic activity of specific HPV genotypes is related to their differentiation-inhibitory activities. An understanding of HPV-mediated carcinogenesis that includes regulation of differentiation might address some of the shortcomings of the existing model. If cellular targets required for HPV oncoproteins to modulate differentiation could be identified, these might provide tractable therapeutic targets.
5. HPV-Associated Cancer and Differentiation
The recognition that a subset of HNSCC are caused by HPV has enabled the comparison of HPV-positive to HPV-negative tumors at a similar anatomical site. A finding from pathology reports is that HPV-positive HNSCC tend to be poorly differentiated compared to HPV-negative HNSCC [85,86]. A recent analysis from my laboratory has demonstrated that this difference is also reflected by gene expression in HPV-positive relative to HPV-negative HNSCC [87]. Using data from The Cancer Genome Atlas (TCGA), we found that with respect to repressed genes, HPV-positive and HPV-negative cancers differ primarily in gene sets related to epithelial differentiation. Furthermore, we analyzed data from a comparison of primary human foreskin keratinocytes (HFK) to HFK expressing HPV16 E6 and E7 and found that the same downregulation of differentiation gene expression was dependent on the viral oncogenes [87,88]. Overall, these data indicate that HPV-positive cancers are poorly differentiated relative to HPV-negative cancers and that this is a consequence of E6/E7 expression.
Some previous studies in the pathology literature are consistent with this hypothesis even though they did not directly assess HPV status. A 1996 study observed a large range of involucrin expression in HNSCC and reported that poorly differentiated HNSCC are more likely to distantly metastasize [89]. Propensity for metastasis is a feature of HPV-positive HNSCC. A more recent analysis proposes that the relationship between HPV status and tumor differentiation is more pronounced in certain subtypes of HNSCC. Keck and colleagues described three subtypes of HNSCC: (i) a basal subtype that did not include any HPV-positive tumors and in which all the samples were well-keratinized and differentiated, (ii) an inflamed/mesenchymal subtype that includes some HPV-positive and some HPV-negative tumors and in which the HPV-positive cancers are nonkeratinizing and poorly differentiated, and (iii) a classical subtype in which HPV-positive and HPV-negative samples are not distinguished by differentiation status [90]. Keratin 19 appears to be expressed more predominantly in epithelial carcinomas compared to normal cells and it is highly expressed in HPV-positive compared to HPV-negative oropharyngeal cancers [91,92,93].
6. High-Risk HPV Oncoproteins Alter Epithelial Differentiation
6.1. Delay of Differentiation in Organotypic Culture
Soon after observing that high-risk HPV oncogene DNA was preserved in cervical cancer cells, researchers began to conduct studies on the oncogenes and on the entire HPV genome in differentiation models in keratinocytes. They observed that HPV DNA-positive HFK or SCC-13 cells could stratify in raft cultures and that the cells expressed keratins but did not differentiate normally. Hematoxylin and eosin staining on HPV-positive raft cultures indicates that the abnormal morphology resulting from HPV DNA is comparable to the abnormal morphology in a cervical or penile intraepithelial neoplasia [94]. Initial studies on HPV16 were soon extended to include HPV18. In normal epithelium, proliferative cells are restricted to the basal epithelial layers, but in raft cultures expressing both HPV18 E6 and E7 or the complete HPV18 genome, parabasal cells extend throughout the stratified epithelium [94,95,96,97,98]. More recent studies in organotypic raft cultures have examined additional HPV types. A comparison of nine established high-risk HPV types and two probable high-risk HPV types by Schutze and colleagues determined that all of these viruses alter differentiation in an organotypic raft culture as assessed by keratin 10 staining [99].
Researchers have examined high-risk HPV E6 or E7 alone and/or used mutant genomes that do not express E6 or E7 in order to assess the individual contributions of the oncoproteins. The strongest effects on differentiation occur when E6 and E7 are expressed together. Organotypic cultures that express high-risk HPV E7 consistently exhibit an increase in proliferation in suprabasal cells. Several of the initial reports concluded that high-risk HPV E7 has more differentiation-inhibiting activity than HPV E6 [62,96,97,100,101]. This was confirmed by recent work using organotypic cultures of HaCat cells in which intact HPV16 genomes promote hyperproliferation and partially inhibit differentiation [102]. Cells harboring mutant genomes that did not express HPV16 E7 formed rafts in which keratin 14 expression was not restricted to basal cells and in which involucrin expression was increased. Compared to HPV16 E6, HPV16 E7 had the stronger effect on promoting hyperproliferation and disturbing normal epithelial differentiation. However, high-risk HPV E6 also exhibit differentiation inhibitory activity. This appears to predominate in basal cells, but in some cases HPV16 E6 can impair differentiation in organotypic cultures [103,104]. Relatively few studies have assessed low-risk HPV oncogenes in these assays, but those that have suggest that low-risk HPV E7 have less ability to impair differentiation than high-risk HPV E7 [62,96]. Some mutational analysis has assessed which activity of high-risk HPV E7 might mediate its effects on differentiation (see Section 6.4).
6.2. Downregulation of Differentiation-Related Cellular Gene Expression
Transcriptional studies on normal and HPV-DNA-containing cells and on HPV-positive and negative cancer cells illustrate that HPV gene expression impairs epithelial differentiation. Consistent with the analysis of raft cultures, human cells harboring high-risk HPV genomes express lower levels of differentiation marker genes and both high-risk HPV E6 and E7 likely contribute to this repression. Most work has focused on the HPV16 oncogenes, with some analysis of HPV18 and minimal examination of low-risk HPV. Early cDNA array experiments examined a few thousand cellular genes and compared normal cells to cells expressing HPV16 E6 and E7 [105]. The strongest effects of the oncoproteins on gene expression were in cells induced to differentiate via growth factor withdrawal and calcium treatment. When the oncogenes were analyzed separately, introducing HPV16 E7 downregulated some markers of keratinocyte differentiation and further introduction of HPV16 E6 augmented the downregulation some of these markers and moderated the repression of others [106].
Additional transcriptional studies on HPV16 immortalized cell lines and on cervical cancer cell lines demonstrate that the repression of differentiation is maintained over time in HPV-positive cells. Wan and colleagues compared normal cells to early passage HPV16-immortalized and to late passage HPV16-immortalized cells and noted that downregulation of epithelial structural genes is reflected both in the early and late cell models [107]. These authors also found that some HPV-mediated changes in keratin expression were reflected in an immunohistochemistry database containing HPV16-positive samples. In a subsequent study, comparing HPV16- and HPV18-positive cervical cancer cells to normal cells indicated that several hundred genes were up- or downregulated in the cancer cells. The downregulated genes included differentiation-related transcripts such as serine protease inhibitors, TGFβ1, and keratin 16 [108]. Advances in microarray technology enabled the analysis of more transcripts and patient samples. A 2006 study analyzed 29 cervical cancer samples, most of them HPV16-positive, and 18 normal controls, again finding that genes downregulated in cervical cancer included many markers of epithelial differentiation plus other genes related to immune signaling [109]. The downregulated genes related to keratinocyte differentiation included keratins, involucrin, serine proteases and protease inhibitors, and desmoglein 1. A general limitation of these early microarray studies is that the ability to detect transcripts was not matched by computational pathway analysis tools.
More recent analyses have combined transcriptional data with pathway analysis. Overall, pathways related to cell cycle, cellular assembly and organization, DNA replication, and growth and proliferation are dysregulated by HPV16 E6 and E7. Basal keratins, such as keratin 5, are relatively unaffected by HPV16 E6 and E7, but HPV16 E7 tends to downregulate individual genes related to epithelial differentiation and a similar effect is observed with HPV16 E6 alone or with HPV16 E6 and E7 expressed together [110]. Recently, Klymenko and colleagues used RNA sequencing and pathway analysis to compare the response to calcium-mediated differentiation in normal immortalized keratinocytes (NIKS) versus NIKS harboring episomal HPV16 genomes (NIKS-16) [111]. Many downregulated genes in this analysis are related to epithelial barrier function, cytoskeleton, and cell adhesion, indicating that HPV16 is able to repress the epithelial differentiation program even in the presence of a strong differentiation-promoting signal. Differentiated cells harboring HPV16 DNA also exhibit a gene expression signature reflecting downregulation of immune-function-related pathways. In this and in other studies, HPV gene expression appears to have the strongest effects on relatively early markers of epithelial differentiation, such as keratin 10, compared to a more modest downregulation of expression of late differentiation proteins such as involucrin. At least one study has examined the effects of HPV16 E6 and E7 on an involucrin promoter–luciferase reporter construct with similar results [112].
6.3. HPV E6 and Differentiation
6.3.1. MAML1, Notch, and Genus Beta HPV E6
Genus beta HPV infect the cutaneous epithelium. A subset of beta HPV cause hyperproliferative lesions in the skin of patients with the genetic disorder Epidermodysplasia Verruciformis, and links between beta HPV and skin cancer have also been proposed [113]. Several recent studies have defined the cellular binding partners of the genus beta versus genus alpha HPV oncoproteins. Somewhat unexpectedly, these efforts revealed that the cellular proteins bound by genus beta HPV E6 proteins are distinct from those targeted by genus alpha HPV E6 [114,115]. Two large-scale proteomics efforts identified MAML1 as a binding partner of genus beta HPV E6. Rozenblatt-Rosen and colleagues identified HPV5 and HPV8 E6 in complex with MAML1 [115]. Our own analysis of eight beta HPV E6 and eight genus alpha HPV E6 confirmed that the beta HPV E6–MAML1 interaction is conserved across HPV genus beta and not shared by genus alpha HPV E6 [114]. Subsequent analyses found that the bovine papillomavirus type 1 (BPV1) E6 protein also binds to MAML1 [116,117]. BPV1 E6 has activity in transformation assays but does not engage p53, and so the molecular basis of its transforming activity has been the subject of some investigation [118,119,120,121]. Although some genus beta HPV E6 bind and/or stabilize p53, this is not conserved among genus beta HPV E6 and the effect on p53 transcriptional activity varies among the beta HPV E6 [122]. Consequently, the identification of MAML1 as a conserved interactor of genus beta HPV E6 sparked interest for its potential to explain biological properties of the cutaneous HPV.
Elegant structural studies on papillomavirus E6 proteins demonstrate how HPV E6 achieve specific binding to their cellular targets [123,124,125]. PV E6 engage several of their cellular targets by binding to an acidic leucine-containing motif (‘LxxLL’) in the cellular protein. Cellular proteins that contain the LxxLL motif and that are targeted by one or more PV E6 proteins include E6AP, MAML1, and paxillin [120,121,126,127,128,129]. The LxxLL peptide is bound by a hydrophobic pocket in E6 formed by two zinc-binding domains separated by an alpha-helical linker. Differences in the residues that form the pocket in E6 confer specificity for a particular LxxLL motif [125]. This model has been validated by an extraordinarily comprehensive analysis of diverse human and animal papillomavirus E6 proteins [130]. Most PV E6 tested in that study bind MAML1 and inhibit a GAL-MAML1 transcriptional reporter, whereas some genus alpha HPV E6 bind E6AP.
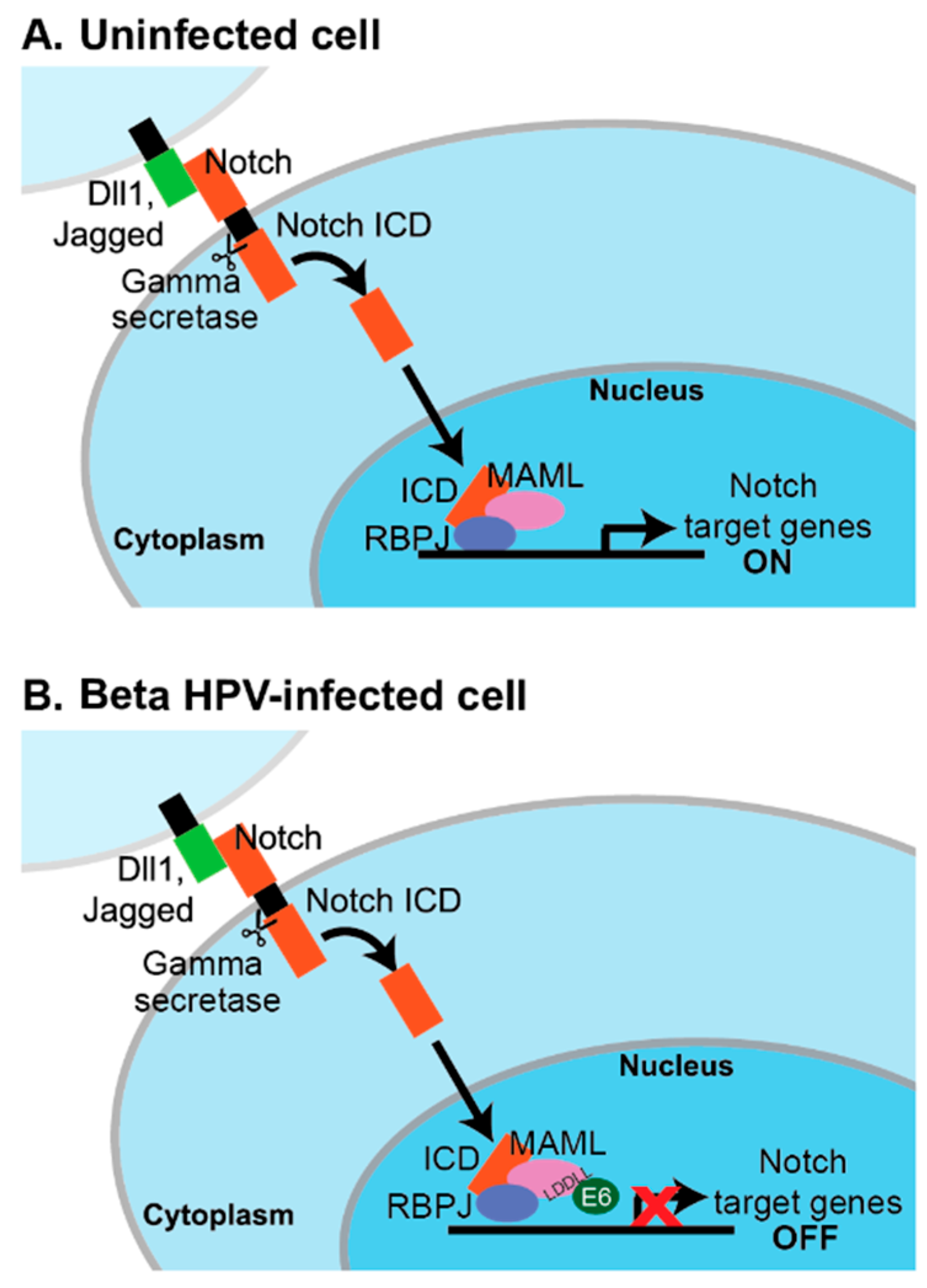
Several signaling pathways are affected by MAML1, including those involving Notch, p53, MEF2C, and beta-catenin [131,132]. However, the predominant effect of beta HPV E6 binding to MAML1 appears to be on NOTCH signaling. NOTCH was first characterized as an oncogene in T cells, but it is a tumor suppressor in the skin where it serves to promote differentiation [133,134]. NOTCH1-4 comprise a family of single-pass transmembrane proteins that are activated upon interaction with a Delta-like or Jagged ligand. Such interaction and subsequent cleavage of NOTCH by metalloprotease and gamma-secretase allows for the release of the NOTCH intracellular domain (ICD) and its translocation to the nucleus. In the nucleus, the NOTCH ICD forms a complex with CSL DNA-binding proteins (CSL: CBF1/RBP-J (mammalian), Su(H) (Drosophila), and Lag-1 (Caenorhabditis elegans)), displacing the repressors of CSL activity and allowing for the recruitment of transcriptional co-activators. RBP-J/CBF1 is the mammalian repressor and MAML1 is one such co-activator [131,135,136]. The HPV proteomics experiments and further validation studies indicate that the beta HPV E6–MAML1 complex can also include RBP-J and the NOTCH ICD [32,114,116,137].
Several groups used NOTCH-MAML1 reporter assays and analyzed endogenous NOTCH-responsive transcripts to find that beta HPV E6 inhibit signaling in response to a NOTCH stimulus [116,117,137]. The LxxLL motif bound by beta HPV E6 proteins is at the C-terminus of MAML1, which is the region important for its transcriptional co-activator activity [138]. The working model is that beta HPV E6 do not impair the formation of the MAML1–RBP-J–ICN complex at NOTCH-responsive promoters, but block the recruitment of additional components required for transcription [137] (Figure 2). Some details of this model remain to be tested. For example, CBP and EP300 can be co-activating components of a MAML1–NOTCH signaling complex, but only a small subset of beta HPV E6 bind to CBP and EP300, whereas a large number of E6 bind to MAML1 and inhibit a MAML1-dependent transcriptional reporter [114,130]. Proteins encoded by other viruses, including Epstein–Barr virus (EBV), Kaposi’s Sarcoma Herpesvirus (KSHV), and adenovirus, are known to interact with the Notch/RBP-J complex and to regulate transcription from both viral and host cellular promoters [139,140,141,142,143,144,145].
These results have been interpreted to mean that beta HPV E6 proteins bind MAML1 to repress epithelial differentiation. Presumably, this would enable maintenance of a more proliferative state in the beta HPV infected epithelial cell and a cellular environment that promotes persistence and viral DNA replication. Meyers and colleagues demonstrated that HPV8 E6 significantly inhibits the calcium-responsive transcription of HES1 and the calcium-dependent inhibition of involucrin in keratinocytes [137]. This inhibition occurs to a comparable degree as inhibiting NOTCH cleavage with a gamma-secretase inhibitor, but the use of a Notch pathway targeted transcriptional array suggests that the effects of HPV8 E6 are not identical to the effects of chemical gamma-secretase inhibition.
The lack of robust life cycle models for the genus beta HPV means that these predictions regarding differentiation have not been tested in organotypic cultures. However, a recently discovered mouse papillomavirus, MmuPV1, shows promise as a model of cutaneous PV biology [29]. MmuPV1 E6 binds to MAML1 and inhibits Notch signaling. Single amino acid changes in MmuPV1 E6 impair binding to MAML1 and limit the ability of MmuPV1 to cause papillomas in nude mice [32]. The interaction of cutaneous PV E6 proteins with MAML1 contributes to the ability of these E6 to impair epithelial differentiation. The ability to test this hypothesis in an animal model will further elucidate the relationship between differentiation inhibition and papillomavirus-associated disease.
6.3.2. Genus Alpha HPV E6
The effects of genus alpha HPV E6 on differentiation and the mechanisms by which they might act are less well established than for genus beta HPV E6. A differentiation-inhibitory effect of HPV16 E6 was identified in early studies that measured the ability of oncoproteins to induce resistance to calcium-mediated differentiation [146]. Later versions of this assay demonstrated some activity for both E6 and E7, but indicated that they acted more efficiently together [147]. The transcriptional studies already introduced suggest that HPV16 E6 downregulates the transcription of genes related to epithelial differentiation, particularly in monolayer culture. Variants of HPV16 E6 are mostly impaired with respect to their ability to restrict differentiation [104,148], although now with the advent of high-quality structures for HPV16 E6 it may be helpful to reassess whether such variants are stably expressed and able to engage their cellular targets.
Like genus beta HPV E6, HPV16 E6 has been suggested to alter Notch signaling, although the underlying mechanism(s) are less clear than for the cutaneous HPV E6. MAML1 was initially identified in a yeast-two-hybrid screen for binding partners of HPV16 E6 [138], but the HPV16 E6–MAML1 interaction was not subsequently observed in any of the recent proteomic analyses of HPV E6 [114,115]. It has been proposed that HPV16 E6 inhibits NOTCH dependent on degradation of p53 or dependent on TAp63β [149,150,151,152]. Kranjec and colleagues recently reported that HPV16 E6 promotes proliferation of high-density keratinocyte cultures and that it acts primarily in basal cells through the combined inactivation of p53 and NOTCH [153]. They propose that HPV16 E6 inhibits NOTCH cleavage and decreases NOTCH transcription dependent on p53 degradation, although the details of such a mechanism remain to be elucidated.
6.3.3. Notch and SCC
Inactivation of the NOTCH signaling pathway by cutaneous HPV is consistent with the tumor-suppressive role of NOTCH in the skin [154,155]. NOTCH signaling is activated in epithelial cells that have divided and left the basal layer, and this occurs concomitant with or dependent on a decrease in p63 levels [144]. The specific NOTCH target genes responsible for induction of the differentiation program remain to be determined.
Studies in cervical cancer cell lines indicate that NOTCH signaling is dysregulated in cervical cancer, although there is not yet a consensus on whether manipulating NOTCH signaling in these cells promotes or inhibits cancer cell proliferation [156,157,158,159,160,161]. The finding that genes in the NOTCH pathway are frequently mutated in HNSCC led to the conclusion that NOTCH is tumor suppressive in HNSCC [162,163]. As it is in cervical cancer, the actual relationship may be more complex. A variety of NOTCH mutations have now been observed in various populations of HNSCC patients, some activating and some inactivating [164]. A recent study examined the differences in NOTCH1 expression in HPV-positive versus HPV-negative HNSCC and concluded that there is an association between NOTCH1 mutation and HPV status. Specifically, tumors wild-type for NOTCH1 are more likely to be HPV-positive than NOTCH1-mutated tumors [165]. More investigation will be required to clarify whether high-risk HPV oncoproteins inactivate NOTCH signaling.
6.3.4. TGFβ
In addition to NOTCH signaling, other pathways impact epithelial differentiation and are affected by HPV E6. TGFβ signaling begins with the TGFβ ligand binding to serine/threonine kinase receptor complexes. The intracellular binding partners of the TGFβ receptor are receptor-associated SMAD (R-SMAD) and co-activator SMAD (co-SMAD) proteins. Phosphorylation events allow for the assembly of an R-SMAD/co-SMAD complex on target gene promoters. The complex can transactivate the promoters of target genes, such as p21CIP1 (CDKN1A), p27KIP1 (CDKN1B), and p15INK4B (CDKN2B). The net effect of activation of these cell cycle inhibitors is cell cycle arrest in G1. TGFβ signaling controls epithelial differentiation and can have context-dependent tumor suppressive or oncogenic activities. The pathway is frequently mutated in epithelial tumors such that its activity is lost [166,167,168,169], but its activation can also promote the epithelial–mesenchymal transition and thus drive invasion [170].
One of the early transcriptional studies on gene expression changes induced by HPV16 E6 and E7 noted that the viral oncoproteins alter the cellular response to TGFβ [105]. The authors of this study found overlap between genes induced by the viral oncoproteins and genes induced by TGFβ and proposed that HPV16 E6 and E7 altered expression of TGFβ and TGFβ-inducible genes as a result of inhibiting differentiation. Consistent with this, co-expression of HPV16 E6 and E7 decreased secretion of TGFβ2 after treatment with the differentiation-inducing agent all-trans retinoic acid (ATRA).
The mechanistic basis for effects on TGFβ signaling has been more clearly established for the genus beta HPV E6 proteins. Mendoza and colleagues found in a yeast-two-hybrid experiment that HPV5 E6, but not HPV9 E6, bound to SMAD3 [171]. HPV5 E6 was consequently able to repress TGFβ signaling by binding SMAD3 and perhaps to destabilize both SMAD3 and SMAD4. HPV5 E6 but not HPV5 E7 was able to repress a SMAD-dependent luciferase reporter, indicating that this effect is specific for certain HPV oncoproteins.
More recent experiments have further elucidated the interaction between genus beta HPV E6 and TGFβ signaling. The HPV E6 proteomics experiments identified several genus beta HPV E6 in complex with SMAD2 and SMAD3 [114,115]. Meyers and colleagues went on to determine that beta HPV E6 oncoproteins do not alter the TGFβ-dependent changes in SMAD2/3 phosphorylation nor do they affect SMAD2/3 nuclear localization [32]. The beta HPV E6 proteins do appear to interfere with the formation of the SMAD-containing transcriptional complex at the p15INK4B (CDKN2B) promoter after TGFβ treatment.
6.4. HPV E7 and Differentiation
6.4.1. Inhibition of Differentiation by HPV E7
The pro-proliferative effect of HPV E7 has been documented extensively [172]. This section will highlight the evidence that HPV E7 can delay or impair cellular differentiation. Although increased proliferation could indirectly restrict differentiation, there is evidence for direct effects of HPV E7 on the epithelial differentiation program independent of effects on proliferation. This is supported by work in organotypic culture, in monolayer cells, and in mouse models.
Epithelial differentiation in organotypic culture is impaired by episomal HPV16 DNA dependent on the HPV16 E7 ORF [95,100]. A recent study directly compared E5, E6, or E7-null HPV16 genomes and found that although loss of any one of the three oncoproteins alters epithelial morphology, the HPV-positive rafts lacking E7 were particularly impaired [102]. HPV16 E7-null rafts exhibit a striking loss of keratin 14 staining in suprabasal layers. Further analysis of HPV16 E7 mutants indicates that the induction of suprabasal DNA synthesis by HPV16 E7 is separate from its ability to perturb differentiation [173]. HPV16 E7 ∆PTLHE is altered in conserved region 1 (CR1) and can bind to but not degrade the pocket proteins RB1, RBL1/p107, and RBL2/p130. It is particularly impaired in limiting differentiation in raft cultures, leading to the conclusion that pocket protein degradation is required for the inhibition of differentiation but not for HPV16 E7 to promote suprabasal DNA synthesis [173]. Several cutaneous HPV E7 also exhibit evidence of differentiation inhibition in organotypic culture [174].
In monolayer culture, HPV16-E7-expressing cells resist differentiation induced by calcium treatment [56]. p21CIP1 links differentiation and proliferation in keratinocytes and the differentiation-inhibitory effect of HPV16 E7 has been ascribed to its ability to bind to p21CIP1 and block the inactivation of cyclin/cdk complexes [54,56]. In a monolayer culture assay that measures the ability of HPV16 E7 to inhibit myoblast differentiation, HPV16 E7 decreases expression of a differentiation marker and both HPV16 E7 ∆PTLHE and RB1-binding domain mutants are impaired in this activity [72]. Low-risk mucosal HPV E7 proteins have been examined less frequently for their ability to alter differentiation, but at least one study reported that HPV6 E7 can reduce involucrin protein expression after calcium treatment [175].
Several experiments in mice indicate that HPV16 E7 inhibits differentiation and suggest that this is independent of RB1 binding [78,176]. Balsitis and colleagues developed a mouse model to separate the RB1-dependent and RB1-independent activities of HPV16 E7. They used transgenic mice that express HPV16 E7 under the control of the keratin 14 promoter, then crossed them to mice harboring a mutant RB1 that cannot bind to HPV E7. Strikingly, in this model, even when HPV16 E7 cannot bind to RB1, it can still delay terminal differentiation and induce hyperplasia. This indicates that the ability of HPV16 E7 to delay or restrict differentiation is at least partially independent of its ability to bind RB1. A similar study in the mouse cervix emphasizes the importance of RB1-independent effects of HPV16 E7 in the mucosal epithelium [77].
6.4.2. Differentiation Pathways Altered by HPV E7
Some cellular pathways related to differentiation are known to be dysregulated by HPV E7. HPV16 E7 can abrogate some effects of TGFβ treatment, such as TGFβ-mediated inhibition of the myc promoter [177]. Studies on TGFβ also highlight the differences between mucosal and cutaneous HPV oncoproteins. The SMAD-TGFβ reporter that was repressed by genus beta HPV E6 but not beta HPV E7 is affected differently by high-risk HPV16 oncoproteins [171]. Specifically, it was repressed by high-risk HPV E7 but not high-risk HPV E6. There is at least one report that HPV16 E7 can block binding of SMAD3 to its target sequence [178], but it should be noted that HPV E7 did not bind to SMAD proteins in any of the recent proteomic studies.
High-risk HPV E7 modulate levels of p63, a master regulator of epidermal development in epithelial cells [179]. An upstream regulator of p63 is miR-203, which increases in abundance as keratinocytes differentiate. p63 protein levels decrease upon epithelial differentiation, but in the presence of HPV31 genomes or HPV31 E7, miR-203 expression decreases and p63 levels are higher than in control cells [180]. p63 is required for HPV31 late functions, such as genome amplification and late viral gene expression. In HPV-positive keratinocytes depleted of p63, the levels of cell cycle regulatory proteins, such as cyclin A, cyclin B1, cdk1, and cdc25c, were reduced and late viral activities were impaired [181]. p63 protein levels are elevated in cervical cancer cell lines, dependent on HPV E7, and there is a good correlation between high p63 staining and HPV positivity in cervical lesions [182,183].
6.4.3. HPV E7 and PTPN14
The mutational analyses of HPV16 E7 suggest that its differentiation-inhibitory activity could be independent of RB1 binding, but the RB1-independent activities of high-risk HPV have remained poorly defined. The proteomic analyses published by several groups have suggested a new RB1-independent activity of high-risk HPV E7 [114,115]. Diverse HPV E7 bind to the putative tumor suppressor PTPN14 and high-risk HPV E7 target PTPN14 for proteasome-mediated degradation using the ubiquitin ligase UBR4 [184,185]. The E7/PTPN14/UBR4 complex is distinct from the E7/RB1 complex and PTPN14 degradation correlates with the oncogenic activity of papillomavirus E7. The HPV16 E7 N-terminus is required for binding to the ubiquitin ligase UBR4 and targeting of the putative tumor suppressor PTPN14 for degradation [185].
Our recent work on PTPN14 and high-risk HPV E7 led us to discover that the loss of PTPN14 from human keratinocytes results in a striking downregulation of the epithelial differentiation program [87]. An N-terminal variant of HPV16 E7 that cannot degrade PTPN14 is impaired in keratinocyte immortalization and in restricting epithelial differentiation. We hypothesize that this finding could be related to the earlier work that described a defect in differentiation inhibition for an N-terminal HPV16 E7 deletion mutant. We propose that degradation of PTPN14 represents a critical RB1-independent function of high-risk HPV E7, that the effect of PTPN14 degradation is to limit differentiation, and that activity is essential for HPV-mediated oncogenic transformation.
Mechanistically, we do not yet understand how PTPN14 degradation could limit epithelial differentiation. PTPN14 is a non-receptor protein tyrosine phosphatase that negatively regulates the transcriptional co-activator YAP1 [186,187,188,189,190]. HPV E7 does not repress the canonical YAP1 targets CTGF and CYR61 as a result of PTPN14 degradation in human keratinocytes [87]. However, YAP1 has been reported to repress epithelial differentiation [191] and future experiments could assess whether this process is impacted by the high-risk HPV E7-mediated degradation of PTPN14.
6.5. HPV E5 and Differentiation
Most effort to understand how HPV manipulates epithelial differentiation has focused on the E6 and E7 oncogenes and on signaling pathways, including Notch and TGFβ. Additional HPV-encoded proteins likely impact differentiation and additional differentiation-related signaling pathways are altered during HPV infection. For example, a recent study comparing HPV18 genomes to HPV18 genomes that do not express the E5 protein found that E5 contributes to a repression of involucrin expression upon calcium-mediated differentiation [192].
7. Differentiation Therapy
Differentiation therapy, the induction of differentiation by a chemical agent, has been extremely successful in specific instances in the clinic. For example, acute promyelocytic leukemia can often be cured by treatment with ATRA and arsenic [193,194]. Although the mechanism underlying this cure is now recognized to be more complicated than initial reports suggested, the promise of differentiation induction as a relatively non-toxic therapeutic approach remains high [195]. ATRA has also shown promise in mouse models of non-melanoma skin cancer and in reducing proliferation of human squamous cell carcinoma cells [196,197,198]. HPV-positive cervical lesions, particularly lower-grade lesions, have shown some response to treatment with ATRA and other studies support the idea that ATRA could inhibit the growth of HPV-positive cell lines and/or tumors [199,200,201,202,203]. Further investigation of the role of differentiation therapy in treating HPV infections and HPV-associated cancers is important. No currently available therapeutic agents can treat the millions of new HPV infections that result in hundreds of thousands of new premalignant lesions and HPV-positive cancers each year. Disrupting any essential step in the carcinogenic progression of an HPV-positive lesion would have therapeutic potential, and an inhibition of differentiation by HPV oncoproteins may be such an essential step.
8. Conclusions and Open Questions
HPV-encoded proteins reprogram host cells to promote virus replication and enable persistent infection. In particular, HPV E6 and E7 drive the establishment of a pro-proliferative environment in the infected cell. They promote cell cycle re-entry in differentiating keratinocytes and restrict the epithelial differentiation program. Years of research on HPV E6 and E7 have enabled groundbreaking discoveries on the mechanisms that normally control the cell cycle and on how cell growth control pathways are dysregulated in cancer.
HPV E6 and E7 lack enzymatic activity, meaning that their reprogramming activities are enabled by interactions with host cellular proteins. Unbiased proteomic analyses have begun to reveal the host cellular targets that are shared among and/or unique to a subset of the HPV oncoproteins. Some of these targets appear to be used by the HPV oncoproteins to manipulate the differentiation program. For example, the genus beta HPV E6 proteins engage MAML1 and SMAD to modulate signaling in the NOTCH and TGFβ pathways [32,116,117,166]. We now understand that there are several mechanisms used by cutaneous HPV E6 to inhibit keratinocyte differentiation that are not shared by the genus alpha mucosal HPV.
Studies on the mucosal HPV oncoproteins indicate that both high-risk HPV E6 and E7 have differentiation inhibitory activity. The accumulated evidence suggests that high-risk HPV E7 have a stronger inhibitory effect on differentiation relative to high-risk HPV E6, particularly in three-dimensional culture models. Furthermore, some mutational analyses are consistent with the idea that high-risk HPV E7 has a differentiation-inhibitory effect that is not dependent on its ability to bind RB1. PTPN14 is one host cell target of high-risk HPV E7 that is targeted independent of RB1 and shows early promise as a regulator of keratinocyte differentiation, but more work to understand how high-risk HPV repress differentiation is warranted. In addition, most of the work on how genus alpha HPV oncoproteins affect differentiation has been on the high-risk HPV. It remains unclear whether high-risk HPV oncoproteins, particularly high-risk HPV E7, have a greater capacity to decrease differentiation than their low-risk counterparts. An additional challenge to directly comparing high-risk HPV E6 and E7 comes from the fact that in cells that contain the HPV genome, E7-encoding transcripts are more abundant than E6-encoding transcripts [16,17]. A direct comparison of the differentiation-inhibitory activities of the two proteins would require that they are expressed at equal levels. HPV E6 and E7 also regulate miRNAs, which could themselves impact keratinocyte differentiation [88,204].
Taken together, the results summarized in this review support the idea that cutaneous HPV E6 could have a greater differentiation-inhibitory capacity than cutaneous HPV E7, whereas mucosal HPV E7 is the more potent inhibitor of differentiation compared to mucosal E6. This hypothesis should be tested rigorously. Others have noted that when comparing HPV E6 and E7, E6 is the more potent oncoprotein in the skin whereas E7 is more potent in the mucosal epithelium [40]. It will be interesting to determine whether this difference is related to the ability of the oncoproteins to limit epithelial differentiation and to consider whether the distinct differentiation-related targets of the cutaneous versus mucosal HPV oncoproteins represent their having evolved to target biological differences in the tissues.
A final open question is whether inhibition of differentiation is required for HPV-mediated oncogenic transformation. It would seem logical that all HPV, not only high-risk HPV, must limit differentiation to enable viral genome replication in the stratified epithelium. However, differentiation is not only a normal process occurring in stratified epithelia, it is also a keratinocyte stress response. Oncogene-induced differentiation (OID) is the process by which keratinocytes differentiate in response to tumor suppressor (including p53) inactivation [205,206] or oncogene overexpression [207,208,209,210,211]. The importance of OID is emphasized by the observation that epithelial cancers frequently have mutated p53 but that p53 mutation is rarely sufficient to drive skin cancer [212,213]. Future studies should investigate whether high-risk HPV oncoproteins trigger differentiation as a result of tumor suppressor inactivation and if so, whether the differentiation-inhibitory activities of the oncoproteins are required to evade OID.
Funding
Work in the White laboratory is supported by American Cancer Society Research Scholar Grant 131661-RSG-18-048-01-MPC.
Acknowledgments
This review was made possible by helpful discussions with members of my laboratory. I apologize for any instances in which work was not cited due to space constraints.
Conflicts of Interest
The author declares no conflict of interest.
References
- Van Doorslaer, K. Evolution of the papillomaviridae. Virology 2013, 445, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Howley, P.M.; Schiller, J.T.; Lowy, D.R. Papillomaviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2013; pp. 1662–1703. [Google Scholar]
- Graham, S.V. Keratinocyte Differentiation-Dependent Human Papillomavirus Gene Regulation. Viruses 2017, 9, 245. [Google Scholar] [CrossRef]
- Moody, C. Mechanisms by which HPV Induces a Replication Competent Environment in Differentiating Keratinocytes. Viruses 2017, 9, 261. [Google Scholar] [CrossRef] [PubMed]
- Howley, P.M.; Livingston, D.M. Small DNA tumor viruses: Large contributors to biomedical sciences. Virology 2009, 384, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K.; Tan, Q.; Xirasagar, S.; Bandaru, S.; Gopalan, V.; Mohamoud, Y.; Huyen, Y.; McBride, A.A. The Papillomavirus Episteme: A central resource for papillomavirus sequence data and analysis. Nucleic Acids Res 2013, 41, D571–D578. [Google Scholar] [CrossRef]
- Van Doorslaer, K.; Gopalan, V.; Bandaru, S.; Mahmoud, Y.; Tan, Q.; Xirasaga, S.; McBride, A.A. Papillomavirus Episteme (PaVE). Available online: http://pave.niaid.nih.gov/ (accessed on 25 February 2019).
- Campos, S.K. Subcellular Trafficking of the Papillomavirus Genome during Initial Infection: The Remarkable Abilities of Minor Capsid Protein L2. Viruses 2017, 9, 370. [Google Scholar] [CrossRef]
- Aksoy, P.; Gottschalk, E.Y.; Meneses, P.I. HPV entry into cells. Mutat. Res. Rev. Mutat. Res. 2017, 772, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Monteiro da Silva, G.; Deatherage, C.; Burd, C.; DiMaio, D. Cell-Penetrating Peptide Mediates Intracellular Membrane Passage of Human Papillomavirus L2 Protein to Trigger Retrograde Trafficking. Cell 2018, 174, 1465–1476.e13. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. Mechanisms and strategies of papillomavirus replication. Biol. Chem. 2017, 398, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Grassmann, K.; Rapp, B.; Maschek, H.; Petry, K.U.; Iftner, T. Identification of a differentiation-inducible promoter in the E7 open reading frame of human papillomavirus type 16 (HPV-16) in raft cultures of a new cell line containing high copy numbers of episomal HPV-16 DNA. J. Virol. 1996, 70, 2339–2349. [Google Scholar]
- Wang, X.; Liu, H.; Ge, H.; Ajiro, M.; Sharma, N.R.; Meyers, C.; Morozov, P.; Tuschl, T.; Klar, A.; Court, D.; et al. Viral DNA Replication Orientation and hnRNPs Regulate Transcription of the Human Papillomavirus 18 Late Promoter. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Bodily, J.M.; Meyers, C. Genetic analysis of the human papillomavirus type 31 differentiation-dependent late promoter. J. Virol. 2005, 79, 3309–3321. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Meyers, C.; Wang, H.K.; Chow, L.T.; Zheng, Z.M. Construction of a full transcription map of human papillomavirus type 18 during productive viral infection. J. Virol. 2011, 85, 8080–8092. [Google Scholar] [CrossRef]
- Brant, A.C.; Majerciak, V.; Moreira, M.A.M.; Zheng, Z.M. HPV18 Utilizes Two Alternative Branch Sites for E6*I Splicing to Produce E7 Protein. Virol. Sin. 2019. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Tao, M.; McCoy, J.P., Jr.; Zheng, Z.M. The E7 oncoprotein is translated from spliced E6*I transcripts in high-risk human papillomavirus type 16- or type 18-positive cervical cancer cell lines via translation reinitiation. J. Virol. 2006, 80, 4249–4263. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.L.; Caodaglio, A.S.; Sichero, L. Regulation of HPV transcription. Clinics 2018, 73, e486s. [Google Scholar] [CrossRef]
- Biryukov, J.; Myers, J.C.; McLaughlin-Drubin, M.E.; Griffin, H.M.; Milici, J.; Doorbar, J.; Meyers, C. Mutations in HPV18 E1^E4 Impact Virus Capsid Assembly, Infectivity Competence, and Maturation. Viruses 2017, 9, 385. [Google Scholar] [CrossRef]
- Doorbar, J. The E4 protein; structure, function and patterns of expression. Virology 2013, 445, 80–98. [Google Scholar] [CrossRef] [Green Version]
- Gonzales, K.A.U.; Fuchs, E. Skin and Its Regenerative Powers: An Alliance between Stem Cells and Their Niche. Dev. Cell 2017, 43, 387–401. [Google Scholar] [CrossRef] [Green Version]
- Blanpain, C.; Fuchs, E. Epidermal homeostasis: A balancing act of stem cells in the skin. Nat. Rev. Mol. Cell Biol. 2009, 10, 207–217. [Google Scholar] [CrossRef]
- Fuchs, E.; Green, H. Changes in keratin gene expression during terminal differentiation of the keratinocyte. Cell 1980, 19, 1033–1042. [Google Scholar] [CrossRef]
- Gibbs, S.; Ponec, M. Intrinsic regulation of differentiation markers in human epidermis, hard palate and buccal mucosa. Arch. Oral Biol. 2000, 45, 149–158. [Google Scholar] [CrossRef]
- Baek, J.H.; Lee, G.; Kim, S.N.; Kim, J.M.; Kim, M.; Chung, S.C.; Min, B.M. Common genes responsible for differentiation and senescence of human mucosal and epidermal keratinocytes. Int. J. Mol. Med. 2003, 12, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.T. Model systems to study the life cycle of human papillomaviruses and HPV-associated cancers. Virol. Sin. 2015, 30, 92–100. [Google Scholar] [CrossRef]
- Fehrmann, F.; Laimins, L.A. Human papillomavirus type 31 life cycle: Methods for study using tissue culture models. Methods Mol. Biol. 2005, 292, 317–330. [Google Scholar]
- McLaughlin-Drubin, M.E.; Meyers, C. Propagation of infectious, high-risk HPV in organotypic “raft” culture. Methods Mol. Med. 2005, 119, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Ingle, A.; Ghim, S.; Joh, J.; Chepkoech, I.; Bennett Jenson, A.; Sundberg, J.P. Novel laboratory mouse papillomavirus (MusPV) infection. Vet. Pathol. 2011, 48, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Viarisio, D.; Mueller-Decker, K.; Kloz, U.; Aengeneyndt, B.; Kopp-Schneider, A.; Grone, H.J.; Gheit, T.; Flechtenmacher, C.; Gissmann, L.; Tommasino, M. E6 and E7 from beta HPV38 cooperate with ultraviolet light in the development of actinic keratosis-like lesions and squamous cell carcinoma in mice. PLoS Pathog. 2011, 7, e1002125. [Google Scholar] [CrossRef] [PubMed]
- Lambert, P.F. Transgenic Mouse Models of Tumor Virus Action. Annu. Rev. Virol. 2016, 3, 473–489. [Google Scholar] [CrossRef]
- Meyers, J.M.; Uberoi, A.; Grace, M.; Lambert, P.F.; Munger, K. Cutaneous HPV8 and MmuPV1 E6 Proteins Target the NOTCH and TGF-beta Tumor Suppressors to Inhibit Differentiation and Sustain Keratinocyte Proliferation. PLoS Pathog. 2017, 13, e1006171. [Google Scholar] [CrossRef]
- Santos, C.; Vilanova, M.; Medeiros, R.; Gil da Costa, R.M. HPV-transgenic mouse models: Tools for studying the cancer-associated immune response. Virus Res. 2017, 235, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Tuong, Z.K.; Noske, K.; Kuo, P.; Bashaw, A.A.; Teoh, S.M.; Frazer, I.H. Murine HPV16 E7-expressing transgenic skin effectively emulates the cellular and molecular features of human high-grade squamous intraepithelial lesions. Papillomavirus Res. 2018, 5, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Bodily, J.M.; Alam, S.; Meyers, C. Regulation of human papillomavirus type 31 late promoter activation and genome amplification by protein kinase C. Virology 2006, 348, 328–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyers, C.; Frattini, M.G.; Hudson, J.B.; Laimins, L.A. Biosynthesis of human papillomavirus from a continuous cell line upon epithelial differentiation. Science 1992, 257, 971–973. [Google Scholar] [CrossRef]
- De Martel, C.; Plummer, M.; Vignat, J.; Franceschi, S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer 2017, 141, 664–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz, N.; Bosch, F.X.; de Sanjose, S.; Herrero, R.; Castellsague, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J. International Agency for Research on Cancer Multicenter Cervical Cancer Study Group. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef]
- Van Dyne, E.A.; Henley, S.J.; Saraiya, M.; Thomas, C.C.; Markowitz, L.E.; Benard, V.B. Trends in Human Papillomavirus–Associated Cancers—United States, 1999–2015. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 918–924. [Google Scholar] [CrossRef]
- Klingelhutz, A.J.; Roman, A. Cellular transformation by human papillomaviruses: Lessons learned by comparing high- and low-risk viruses. Virology 2012, 424, 77–98. [Google Scholar] [CrossRef] [Green Version]
- Durst, M.; Gissmann, L.; Ikenberg, H.; zur Hausen, H. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc. Natl. Acad. Sci. USA 1983, 80, 3812–3815. [Google Scholar] [CrossRef]
- Boshart, M.; Gissmann, L.; Ikenberg, H.; Kleinheinz, A.; Scheurlen, W.; zur Hausen, H. A new type of papillomavirus DNA, its presence in genital cancer biopsies and in cell lines derived from cervical cancer. EMBO J. 1984, 3, 1151–1157. [Google Scholar] [CrossRef]
- Baker, C.C.; Phelps, W.C.; Lindgren, V.; Braun, M.J.; Gonda, M.A.; Howley, P.M. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J. Virol. 1987, 61, 962–971. [Google Scholar]
- Schneider-Gadicke, A.; Schwarz, E. Different human cervical carcinoma cell lines show similar transcription patterns of human papillomavirus type 18 early genes. EMBO J. 1986, 5, 2285–2292. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.; Freese, U.K.; Gissmann, L.; Mayer, W.; Roggenbuck, B.; Stremlau, A.; zur Hausen, H. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature 1985, 314, 111–114. [Google Scholar] [CrossRef]
- Smotkin, D.; Wettstein, F.O. Transcription of human papillomavirus type 16 early genes in a cervical cancer and a cancer-derived cell line and identification of the E7 protein. Proc. Natl. Acad. Sci. USA 1986, 83, 4680–4684. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.; Phelps, W.C.; Bubb, V.; Howley, P.M.; Schlegel, R. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J. Virol. 1989, 63, 4417–4421. [Google Scholar]
- Bedell, M.A.; Jones, K.H.; Laimins, L.A. The E6-E7 region of human papillomavirus type 18 is sufficient for transformation of NIH 3T3 and rat-1 cells. J. Virol. 1987, 61, 3635–3640. [Google Scholar]
- Bedell, M.A.; Jones, K.H.; Grossman, S.R.; Laimins, L.A. Identification of human papillomavirus type 18 transforming genes in immortalized and primary cells. J. Virol. 1989, 63, 1247–1255. [Google Scholar] [PubMed]
- Yee, C.; Krishnan-Hewlett, I.; Baker, C.C.; Schlegel, R.; Howley, P.M. Presence and expression of human papillomavirus sequences in human cervical carcinoma cell lines. Am. J. Pathol. 1985, 119, 361–366. [Google Scholar]
- Dyson, N.; Guida, P.; Munger, K.; Harlow, E. Homologous sequences in adenovirus E1A and human papillomavirus E7 proteins mediate interaction with the same set of cellular proteins. J. Virol. 1992, 66, 6893–6902. [Google Scholar] [PubMed]
- Dyson, N.; Howley, P.M.; Munger, K.; Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.; Werness, B.A.; Dyson, N.; Phelps, W.C.; Harlow, E.; Howley, P.M. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989, 8, 4099–4105. [Google Scholar] [CrossRef] [PubMed]
- Funk, J.O.; Waga, S.; Harry, J.B.; Espling, E.; Stillman, B.; Galloway, D.A. Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev. 1997, 11, 2090–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerfass-Thome, K.; Zwerschke, W.; Mannhardt, B.; Tindle, R.; Botz, J.W.; Jansen-Durr, P. Inactivation of the cdk inhibitor p27KIP1 by the human papillomavirus type 16 E7 oncoprotein. Oncogene 1996, 13, 2323–2330. [Google Scholar] [PubMed]
- Jones, D.L.; Alani, R.M.; Munger, K. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2. Genes Dev. 1997, 11, 2101–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demers, G.W.; Halbert, C.L.; Galloway, D.A. Elevated wild-type p53 protein levels in human epithelial cell lines immortalized by the human papillomavirus type 16 E7 gene. Virology 1994, 198, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Werness, B.A.; Levine, A.J.; Howley, P.M. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 1990, 248, 76–79. [Google Scholar] [CrossRef]
- Duensing, S.; Lee, L.Y.; Duensing, A.; Basile, J.; Piboonniyom, S.; Gonzalez, S.; Crum, C.P.; Munger, K. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc. Natl. Acad. Sci. USA 2000, 97, 10002–10007. [Google Scholar] [CrossRef] [Green Version]
- Duensing, S.; Munger, K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002, 62, 7075–7082. [Google Scholar]
- Ueno, T.; Sasaki, K.; Yoshida, S.; Kajitani, N.; Satsuka, A.; Nakamura, H.; Sakai, H. Molecular mechanisms of hyperplasia induction by human papillomavirus E7. Oncogene 2006, 25, 4155–4164. [Google Scholar] [CrossRef] [Green Version]
- Akagi, K.; Li, J.; Broutian, T.R.; Padilla-Nash, H.; Xiao, W.; Jiang, B.; Rocco, J.W.; Teknos, T.N.; Kumar, B.; Wangsa, D.; et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2014, 24, 185–199. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Munger, K. The human papillomavirus E7 oncoprotein. Virology 2009, 384, 335–344. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef]
- Munger, K.; Jones, D.L. Human papillomavirus carcinogenesis: An identity crisis in the retinoblastoma tumor suppressor pathway. J. Virol. 2015, 89, 4708–4711. [Google Scholar] [CrossRef] [PubMed]
- Storey, A.; Osborn, K.; Crawford, L. Co-transformation by human papillomavirus types 6 and 11. J. Gen. Virol. 1990, 71 Pt 1, 165–171. [Google Scholar] [CrossRef]
- Ciccolini, F.; Di Pasquale, G.; Carlotti, F.; Crawford, L.; Tommasino, M. Functional studies of E7 proteins from different HPV types. Oncogene 1994, 9, 2633–2638. [Google Scholar]
- Ibaraki, T.; Satake, M.; Kurai, N.; Ichijo, M.; Ito, Y. Transacting activities of the E7 genes of several types of human papillomavirus. Virus Genes 1993, 7, 187–196. [Google Scholar] [CrossRef]
- Phelps, W.C.; Munger, K.; Yee, C.L.; Barnes, J.A.; Howley, P.M. Structure-function analysis of the human papillomavirus type 16 E7 oncoprotein. J. Virol. 1992, 66, 2418–2427. [Google Scholar] [PubMed]
- Jewers, R.J.; Hildebrandt, P.; Ludlow, J.W.; Kell, B.; McCance, D.J. Regions of human papillomavirus type 16 E7 oncoprotein required for immortalization of human keratinocytes. J. Virol. 1992, 66, 1329–1335. [Google Scholar]
- White, E.A.; Kramer, R.E.; Hwang, J.H.; Pores Fernando, A.T.; Naetar, N.; Hahn, W.C.; Roberts, T.M.; Schaffhausen, B.S.; Livingston, D.M.; Howley, P.M. Papillomavirus e7 oncoproteins share functions with polyomavirus small T antigens. J. Virol. 2015, 89, 2857–2865. [Google Scholar] [CrossRef]
- Helt, A.M.; Galloway, D.A. Destabilization of the retinoblastoma tumor suppressor by human papillomavirus type 16 E7 is not sufficient to overcome cell cycle arrest in human keratinocytes. J. Virol. 2001, 75, 6737–6747. [Google Scholar] [CrossRef]
- Banks, L.; Edmonds, C.; Vousden, K.H. Ability of the HPV16 E7 protein to bind RB and induce DNA synthesis is not sufficient for efficient transforming activity in NIH3T3 cells. Oncogene 1990, 5, 1383–1389. [Google Scholar]
- Huh, K.W.; DeMasi, J.; Ogawa, H.; Nakatani, Y.; Howley, P.M.; Munger, K. Association of the human papillomavirus type 16 E7 oncoprotein with the 600-kDa retinoblastoma protein-associated factor, p600. Proc. Natl. Acad. Sci. USA 2005, 102, 11492–11497. [Google Scholar] [CrossRef] [Green Version]
- Strati, K.; Lambert, P.F. Role of Rb-dependent and Rb-independent functions of papillomavirus E7 oncogene in head and neck cancer. Cancer Res. 2007, 67, 11585–11593. [Google Scholar] [CrossRef] [PubMed]
- Balsitis, S.; Dick, F.; Dyson, N.; Lambert, P.F. Critical roles for non-pRb targets of human papillomavirus type 16 E7 in cervical carcinogenesis. Cancer Res. 2006, 66, 9393–9400. [Google Scholar] [CrossRef] [PubMed]
- Balsitis, S.; Dick, F.; Lee, D.; Farrell, L.; Hyde, R.K.; Griep, A.E.; Dyson, N.; Lambert, P.F. Examination of the pRb-dependent and pRb-independent functions of E7 in vivo. J. Virol. 2005, 79, 11392–11402. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Crum, C.P.; Munger, K. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc. Natl. Acad. Sci. USA 2011, 108, 2130–2135. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin-Drubin, M.E.; Park, D.; Munger, K. Tumor suppressor p16INK4A is necessary for survival of cervical carcinoma cell lines. Proc. Natl. Acad. Sci. USA 2013, 110, 16175–16180. [Google Scholar] [CrossRef] [Green Version]
- Soto, D.R.; Barton, C.; Munger, K.; McLaughlin-Drubin, M.E. KDM6A addiction of cervical carcinoma cell lines is triggered by E7 and mediated by p21CIP1 suppression of replication stress. PLoS Pathog. 2017, 13, e1006661. [Google Scholar] [CrossRef]
- Mirabello, L.; Yeager, M.; Yu, K.; Clifford, G.M.; Xiao, Y.; Zhu, B.; Cullen, M.; Boland, J.F.; Wentzensen, N.; Nelson, C.W.; et al. HPV16 E7 Genetic Conservation Is Critical to Carcinogenesis. Cell 2017, 170, 1164–1174.e6. [Google Scholar] [CrossRef]
- White, E.A.; Munger, K. Crowd Control: E7 Conservation Is the Key to Cancer. Cell 2017, 170, 1057–1059. [Google Scholar] [CrossRef] [PubMed]
- Riley, R.R.; Duensing, S.; Brake, T.; Munger, K.; Lambert, P.F.; Arbeit, J.M. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003, 63, 4862–4871. [Google Scholar]
- Mendelsohn, A.H.; Lai, C.K.; Shintaku, I.P.; Elashoff, D.A.; Dubinett, S.M.; Abemayor, E.; St John, M.A. Histopathologic findings of HPV and p16 positive HNSCC. Laryngoscope 2010, 120, 1788–1794. [Google Scholar] [CrossRef]
- Pai, S.I.; Westra, W.H. Molecular pathology of head and neck cancer: Implications for diagnosis, prognosis, and treatment. Annu. Rev. Pathol. 2009, 4, 49–70. [Google Scholar] [CrossRef]
- Hatterschide, J.; Bohidar, A.E.; Grace, M.; Nulton, T.J.; Kim, H.W.; Windle, B.; Morgan, I.M.; Munger, K.; White, E.A. PTPN14 degradation by high-risk human papillomavirus E7 limits keratinocyte differentiation and contributes to HPV-mediated oncogenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 7033–7042. [Google Scholar] [CrossRef]
- Harden, M.E.; Prasad, N.; Griffiths, A.; Munger, K. Modulation of microRNA-mRNA Target Pairs by Human Papillomavirus 16 Oncoproteins. mBio 2017, 8. [Google Scholar] [CrossRef]
- Roland, N.J.; Rowley, H.; Scraggs, M.; Johnson, P.; Jones, A.S. MIB-1 and involucrin expression in laryngeal squamous carcinoma: The relationship to host and tumour factors and survival. Clin. Otolaryngol. Allied Sci. 1996, 21, 429–438. [Google Scholar] [CrossRef]
- Keck, M.K.; Zuo, Z.; Khattri, A.; Stricker, T.P.; Brown, C.D.; Imanguli, M.; Rieke, D.; Endhardt, K.; Fang, P.; Bragelmann, J.; et al. Integrative analysis of head and neck cancer identifies two biologically distinct HPV and three non-HPV subtypes. Clin. Cancer Res. 2015, 21, 870–881. [Google Scholar] [CrossRef]
- Santoro, A.; Pannone, G.; Ninivaggi, R.; Petruzzi, M.; Santarelli, A.; Russo, G.M.; Lepore, S.; Pietrafesa, M.; Laurenzana, I.; Leonardi, R.; et al. Relationship between CK19 expression, deregulation of normal keratinocyte differentiation pattern and high risk-human papilloma virus infection in oral and oropharyngeal squamous cell carcinoma. Infect. Agents Cancer 2015, 10, 46. [Google Scholar] [CrossRef]
- Kopan, R.; Traska, G.; Fuchs, E. Retinoids as important regulators of terminal differentiation: Examining keratin expression in individual epidermal cells at various stages of keratinization. J. Cell Biol. 1987, 105, 427–440. [Google Scholar] [CrossRef]
- Wu, Y.J.; Rheinwald, J.G. A new small (40 kd) keratin filament protein made by some cultured human squamous cell carcinomas. Cell 1981, 25, 627–635. [Google Scholar] [CrossRef]
- McCance, D.J.; Kopan, R.; Fuchs, E.; Laimins, L.A. Human papillomavirus type 16 alters human epithelial cell differentiation in vitro. Proc. Natl. Acad. Sci. USA 1988, 85, 7169–7173. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Allen-Hoffmann, B.L.; Lee, D.; Lambert, P.F. The human papillomavirus type 16 E7 oncogene is required for the productive stage of the viral life cycle. J. Virol. 2000, 74, 6622–6631. [Google Scholar] [CrossRef] [PubMed]
- Halbert, C.L.; Demers, G.W.; Galloway, D.A. The E6 and E7 genes of human papillomavirus type 6 have weak immortalizing activity in human epithelial cells. J. Virol. 1992, 66, 2125–2134. [Google Scholar]
- Hudson, J.B.; Bedell, M.A.; McCance, D.J.; Laiminis, L.A. Immortalization and altered differentiation of human keratinocytes in vitro by the E6 and E7 open reading frames of human papillomavirus type 18. J. Virol. 1990, 64, 519–526. [Google Scholar]
- Woodworth, C.D.; Cheng, S.; Simpson, S.; Hamacher, L.; Chow, L.T.; Broker, T.R.; DiPaolo, J.A. Recombinant retroviruses encoding human papillomavirus type 18 E6 and E7 genes stimulate proliferation and delay differentiation of human keratinocytes early after infection. Oncogene 1992, 7, 619–626. [Google Scholar]
- Schutze, D.M.; Snijders, P.J.; Bosch, L.; Kramer, D.; Meijer, C.J.; Steenbergen, R.D. Differential in vitro immortalization capacity of eleven (probable) [corrected] high-risk human papillomavirus types. J. Virol. 2014, 88, 1714–1724. [Google Scholar] [CrossRef]
- Flores, E.R.; Allen-Hoffmann, B.L.; Lee, D.; Sattler, C.A.; Lambert, P.F. Establishment of the human papillomavirus type 16 (HPV-16) life cycle in an immortalized human foreskin keratinocyte cell line. Virology 1999, 262, 344–354. [Google Scholar] [CrossRef]
- Blanton, R.A.; Coltrera, M.D.; Gown, A.M.; Halbert, C.L.; McDougall, J.K. Expression of the HPV16 E7 gene generates proliferation in stratified squamous cell cultures which is independent of endogenous p53 levels. Cell Growth Differ. 1992, 3, 791–802. [Google Scholar]
- Bergner, S.; Halec, G.; Schmitt, M.; Aubin, F.; Alonso, A.; Auvinen, E. Individual and Complementary Effects of Human Papillomavirus Oncogenes on Epithelial Cell Proliferation and Differentiation. Cells Tissues Organs 2016, 201, 97–108. [Google Scholar] [CrossRef]
- Lee, C.; Laimins, L.A. Role of the PDZ domain-binding motif of the oncoprotein E6 in the pathogenesis of human papillomavirus type 31. J. Virol. 2004, 78, 12366–12377. [Google Scholar] [CrossRef] [PubMed]
- Zehbe, I.; Richard, C.; DeCarlo, C.A.; Shai, A.; Lambert, P.F.; Lichtig, H.; Tommasino, M.; Sherman, L. Human papillomavirus 16 E6 variants differ in their dysregulation of human keratinocyte differentiation and apoptosis. Virology 2009, 383, 69–77. [Google Scholar] [CrossRef]
- Nees, M.; Geoghegan, J.M.; Munson, P.; Prabhu, V.; Liu, Y.; Androphy, E.; Woodworth, C.D. Human papillomavirus type 16 E6 and E7 proteins inhibit differentiation-dependent expression of transforming growth factor-beta2 in cervical keratinocytes. Cancer Res. 2000, 60, 4289–4298. [Google Scholar] [PubMed]
- Duffy, C.L.; Phillips, S.L.; Klingelhutz, A.J. Microarray analysis identifies differentiation-associated genes regulated by human papillomavirus type 16 E6. Virology 2003, 314, 196–205. [Google Scholar] [CrossRef] [Green Version]
- Wan, F.; Miao, X.; Quraishi, I.; Kennedy, V.; Creek, K.E.; Pirisi, L. Gene expression changes during HPV-mediated carcinogenesis: A comparison between an in vitro cell model and cervical cancer. Int. J. Cancer 2008, 123, 32–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santin, A.D.; Zhan, F.; Bignotti, E.; Siegel, E.R.; Cane, S.; Bellone, S.; Palmieri, M.; Anfossi, S.; Thomas, M.; Burnett, A.; et al. Gene expression profiles of primary HPV16- and HPV18-infected early stage cervical cancers and normal cervical epithelium: Identification of novel candidate molecular markers for cervical cancer diagnosis and therapy. Virology 2005, 331, 269–291. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.F.; Cheung, T.H.; Tsao, G.S.; Lo, K.W.; Yim, S.F.; Wang, V.W.; Heung, M.M.; Chan, S.C.; Chan, L.K.; Ho, T.W.; et al. Genome-wide gene expression profiling of cervical cancer in Hong Kong women by oligonucleotide microarray. Int. J. Cancer 2006, 118, 2461–2469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyongyosi, E.; Szalmas, A.; Ferenczi, A.; Poliska, S.; Konya, J.; Veress, G. Transcriptional regulation of genes involved in keratinocyte differentiation by human papillomavirus 16 oncoproteins. Arch. Virol. 2015, 160, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Klymenko, T.; Gu, Q.; Herbert, I.; Stevenson, A.; Iliev, V.; Watkins, G.; Pollock, C.; Bhatia, R.; Cuschieri, K.; Herzyk, P.; et al. RNASeq analysis of differentiated keratinocytes reveals a massive response to late events during human papillomavirus type 16 infection, including loss of epithelial barrier function. J. Virol. 2017. [Google Scholar] [CrossRef]
- Gyongyosi, E.; Szalmas, A.; Ferenczi, A.; Konya, J.; Gergely, L.; Veress, G. Effects of human papillomavirus (HPV) type 16 oncoproteins on the expression of involucrin in human keratinocytes. Virol. J. 2012, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Howley, P.M.; Pfister, H.J. Beta genus papillomaviruses and skin cancer. Virology 2015, 479–480, 290–296. [Google Scholar] [CrossRef] [PubMed]
- White, E.A.; Kramer, R.E.; Tan, M.J.; Hayes, S.D.; Harper, J.W.; Howley, P.M. Comprehensive analysis of host cellular interactions with human papillomavirus E6 proteins identifies new E6 binding partners and reflects viral diversity. J. Virol. 2012, 86, 13174–13186. [Google Scholar] [CrossRef]
- Rozenblatt-Rosen, O.; Deo, R.C.; Padi, M.; Adelmant, G.; Calderwood, M.A.; Rolland, T.; Grace, M.; Dricot, A.; Askenazi, M.; Tavares, M.; et al. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 2012, 487, 491–495. [Google Scholar] [CrossRef]
- Tan, M.J.; White, E.A.; Sowa, M.E.; Harper, J.W.; Aster, J.C.; Howley, P.M. Cutaneous beta-human papillomavirus E6 proteins bind Mastermind-like coactivators and repress Notch signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1473–1480. [Google Scholar] [CrossRef]
- Brimer, N.; Lyons, C.; Wallberg, A.E.; Vande Pol, S.B. Cutaneous papillomavirus E6 oncoproteins associate with MAML1 to repress transactivation and NOTCH signaling. Oncogene 2012, 31, 4639–4646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neary, K.; DiMaio, D. Open reading frames E6 and E7 of bovine papillomavirus type 1 are both required for full transformation of mouse C127 cells. J. Virol. 1989, 63, 259–266. [Google Scholar] [PubMed]
- Sarver, N.; Rabson, M.S.; Yang, Y.C.; Byrne, J.C.; Howley, P.M. Localization and analysis of bovine papillomavirus type 1 transforming functions. J. Virol. 1984, 52, 377–388. [Google Scholar] [PubMed]
- Tong, X.; Howley, P.M. The bovine papillomavirus E6 oncoprotein interacts with paxillin and disrupts the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 1997, 94, 4412–4417. [Google Scholar] [CrossRef] [Green Version]
- Tong, X.; Salgia, R.; Li, J.L.; Griffin, J.D.; Howley, P.M. The bovine papillomavirus E6 protein binds to the LD motif repeats of paxillin and blocks its interaction with vinculin and the focal adhesion kinase. J. Biol. Chem. 1997, 272, 33373–33376. [Google Scholar] [CrossRef]
- White, E.A.; Walther, J.; Javanbakht, H.; Howley, P.M. Genus beta human papillomavirus E6 proteins vary in their effects on the transactivation of p53 target genes. J. Virol. 2014, 88, 8201–8212. [Google Scholar] [CrossRef]
- Martinez-Zapien, D.; Ruiz, F.X.; Poirson, J.; Mitschler, A.; Ramirez, J.; Forster, A.; Cousido-Siah, A.; Masson, M.; Vande Pol, S.; Podjarny, A.; et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 2016, 529, 541–545. [Google Scholar] [CrossRef] [Green Version]
- Nomine, Y.; Masson, M.; Charbonnier, S.; Zanier, K.; Ristriani, T.; Deryckere, F.; Sibler, A.P.; Desplancq, D.; Atkinson, R.A.; Weiss, E.; et al. Structural and functional analysis of E6 oncoprotein: Insights in the molecular pathways of human papillomavirus-mediated pathogenesis. Mol. Cell 2006, 21, 665–678. [Google Scholar] [CrossRef]
- Zanier, K.; Charbonnier, S.; Sidi, A.O.; McEwen, A.G.; Ferrario, M.G.; Poussin-Courmontagne, P.; Cura, V.; Brimer, N.; Babah, K.O.; Ansari, T.; et al. Structural basis for hijacking of cellular LxxLL motifs by papillomavirus E6 oncoproteins. Science 2013, 339, 694–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohl, J.; Das, K.; Dasgupta, B.; Vande Pol, S.B. Competitive binding to a charged leucine motif represses transformation by a papillomavirus E6 oncoprotein. Virology 2000, 271, 163–170. [Google Scholar] [CrossRef]
- Chen, J.J.; Hong, Y.; Rustamzadeh, E.; Baleja, J.D.; Androphy, E.J. Identification of an alpha helical motif sufficient for association with papillomavirus E6. J. Biol. Chem. 1998, 273, 13537–13544. [Google Scholar] [CrossRef]
- McCormack, S.J.; Brazinski, S.E.; Moore, J.L., Jr.; Werness, B.A.; Goldstein, D.J. Activation of the focal adhesion kinase signal transduction pathway in cervical carcinoma cell lines and human genital epithelial cells immortalized with human papillomavirus type 18. Oncogene 1997, 15, 265–274. [Google Scholar] [CrossRef] [Green Version]
- Vande Pol, S.B.; Brown, M.C.; Turner, C.E. Association of Bovine Papillomavirus Type 1 E6 oncoprotein with the focal adhesion protein paxillin through a conserved protein interaction motif. Oncogene 1998, 16, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brimer, N.; Drews, C.M.; Vande Pol, S.B. Association of papillomavirus E6 proteins with either MAML1 or E6AP clusters E6 proteins by structure, function, and evolutionary relatedness. PLoS Pathog. 2017, 13, e1006781. [Google Scholar] [CrossRef]
- McElhinny, A.S.; Li, J.L.; Wu, L. Mastermind-like transcriptional co-activators: Emerging roles in regulating cross talk among multiple signaling pathways. Oncogene 2008, 27, 5138–5147. [Google Scholar] [CrossRef] [PubMed]
- Saint Just Ribeiro, M.; Wallberg, A.E. Transcriptional mechanisms by the coregulator MAML1. Curr. Protein Pept. Sci. 2009, 10, 570–576. [Google Scholar] [CrossRef]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.T.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef]
- Rangarajan, A.; Talora, C.; Okuyama, R.; Nicolas, M.; Mammucari, C.; Oh, H.; Aster, J.C.; Krishna, S.; Metzger, D.; Chambon, P.; et al. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J. 2001, 20, 3427–3436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.E.; Macdonald, R.J. Notch-independent functions of CSL. Curr. Top. Dev. Biol. 2011, 97, 55–74. [Google Scholar] [CrossRef]
- Shao, H.; Huang, Q.; Liu, Z.J. Targeting Notch signaling for cancer therapeutic intervention. Adv. Pharmacol. 2012, 65, 191–234. [Google Scholar] [CrossRef]
- Meyers, J.M.; Spangle, J.M.; Munger, K. The human papillomavirus type 8 E6 protein interferes with NOTCH activation during keratinocyte differentiation. J. Virol. 2013, 87, 4762–4767. [Google Scholar] [CrossRef]
- Wu, L.; Aster, J.C.; Blacklow, S.C.; Lake, R.; Artavanis-Tsakonas, S.; Griffin, J.D. MAML1, a human homologue of Drosophila mastermind, is a transcriptional co-activator for NOTCH receptors. Nat. Genet. 2000, 26, 484–489. [Google Scholar] [CrossRef]
- Liang, Y.; Ganem, D. RBP-J (CSL) is essential for activation of the K14/vGPCR promoter of Kaposi’s sarcoma-associated herpesvirus by the lytic switch protein RTA. J. Virol. 2004, 78, 6818–6826. [Google Scholar] [CrossRef]
- Robertson, E.S.; Grossman, S.; Johannsen, E.; Miller, C.; Lin, J.; Tomkinson, B.; Kieff, E. Epstein-Barr virus nuclear protein 3C modulates transcription through interaction with the sequence-specific DNA-binding protein J kappa. J. Virol. 1995, 69, 3108–3116. [Google Scholar]
- Wang, L.; Grossman, S.R.; Kieff, E. Epstein-Barr virus nuclear protein 2 interacts with p300, CBP, and PCAF histone acetyltransferases in activation of the LMP1 promoter. Proc. Natl. Acad. Sci. USA 2000, 97, 430–435. [Google Scholar] [CrossRef] [Green Version]
- Grossman, S.R.; Johannsen, E.; Tong, X.; Yalamanchili, R.; Kieff, E. The Epstein-Barr virus nuclear antigen 2 transactivator is directed to response elements by the J kappa recombination signal binding protein. Proc. Natl. Acad. Sci. USA 1994, 91, 7568–7572. [Google Scholar] [CrossRef]
- Zimber-Strobl, U.; Strobl, L.J.; Meitinger, C.; Hinrichs, R.; Sakai, T.; Furukawa, T.; Honjo, T.; Bornkamm, G.W. Epstein-Barr virus nuclear antigen 2 exerts its transactivating function through interaction with recombination signal binding protein RBP-J kappa, the homologue of Drosophila Suppressor of Hairless. EMBO J. 1994, 13, 4973–4982. [Google Scholar] [CrossRef]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The Varied Roles of Notch in Cancer. Annu. Rev. Pathol. 2017, 12, 245–275. [Google Scholar] [CrossRef]
- Ansieau, S.; Strobl, L.J.; Leutz, A. Activation of the Notch-regulated transcription factor CBF1/RBP-Jkappa through the 13SE1A oncoprotein. Genes Dev. 2001, 15, 380–385. [Google Scholar] [CrossRef]
- Sherman, L.; Schlegel, R. Serum- and calcium-induced differentiation of human keratinocytes is inhibited by the E6 oncoprotein of human papillomavirus type 16. J. Virol. 1996, 70, 3269–3279. [Google Scholar]
- Pei, X.F.; Sherman, L.; Sun, Y.H.; Schlegel, R. HPV-16 E7 protein bypasses keratinocyte growth inhibition by serum and calcium. Carcinogenesis 1998, 19, 1481–1486. [Google Scholar] [CrossRef] [Green Version]
- Lichtig, H.; Algrisi, M.; Botzer, L.E.; Abadi, T.; Verbitzky, Y.; Jackman, A.; Tommasino, M.; Zehbe, I.; Sherman, L. HPV16 E6 natural variants exhibit different activities in functional assays relevant to the carcinogenic potential of E6. Virology 2006, 350, 216–227. [Google Scholar] [CrossRef] [Green Version]
- Lefort, K.; Mandinova, A.; Ostano, P.; Kolev, V.; Calpini, V.; Kolfschoten, I.; Devgan, V.; Lieb, J.; Raffoul, W.; Hohl, D.; et al. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev. 2007, 21, 562–577. [Google Scholar] [CrossRef]
- Dotto, G.P. Crosstalk of Notch with p53 and p63 in cancer growth control. Nat. Rev. Cancer 2009, 9, 587–595. [Google Scholar] [CrossRef]
- Ben Khalifa, Y.; Teissier, S.; Tan, M.K.; Phan, Q.T.; Daynac, M.; Wong, W.Q.; Thierry, F. The human papillomavirus E6 oncogene represses a cell adhesion pathway and disrupts focal adhesion through degradation of TAp63beta upon transformation. PLoS Pathog. 2011, 7, e1002256. [Google Scholar] [CrossRef]
- Yugawa, T.; Handa, K.; Narisawa-Saito, M.; Ohno, S.; Fujita, M.; Kiyono, T. Regulation of Notch1 gene expression by p53 in epithelial cells. Mol. Cell. Biol. 2007, 27, 3732–3742. [Google Scholar] [CrossRef]
- Kranjec, C.; Holleywood, C.; Libert, D.; Griffin, H.; Mahmood, R.; Isaacson, E.; Doorbar, J. Modulation of basal cell fate during productive and transforming HPV-16 infection is mediated by progressive E6-driven depletion of Notch. J. Pathol. 2017, 242, 448–462. [Google Scholar] [CrossRef] [PubMed]
- Proweller, A.; Tu, L.; Lepore, J.J.; Cheng, L.; Lu, M.M.; Seykora, J.; Millar, S.E.; Pear, W.S.; Parmacek, M.S. Impaired notch signaling promotes de novo squamous cell carcinoma formation. Cancer Res. 2006, 66, 7438–7444. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, M.; Wolfer, A.; Raj, K.; Kummer, J.A.; Mill, P.; van Noort, M.; Hui, C.C.; Clevers, H.; Dotto, G.P.; Radtke, F. Notch1 functions as a tumor suppressor in mouse skin. Nat. Genet. 2003, 33, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Kuncharin, Y.; Sangphech, N.; Kueanjinda, P.; Bhattarakosol, P.; Palaga, T. MAML1 regulates cell viability via the NF-kappaB pathway in cervical cancer cell lines. Exp. Cell Res. 2011, 317, 1830–1840. [Google Scholar] [CrossRef]
- Ramdass, B.; Maliekal, T.T.; Lakshmi, S.; Rehman, M.; Rema, P.; Nair, P.; Mukherjee, G.; Reddy, B.K.; Krishna, S.; Radhakrishna Pillai, M. Coexpression of Notch1 and NF-kappaB signaling pathway components in human cervical cancer progression. Gynecol. Oncol. 2007, 104, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Rong, C.; Feng, Y.; Ye, Z. Notch is a critical regulator in cervical cancer by regulating Numb splicing. Oncol. Lett. 2017, 13, 2465–2470. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, R.; Rath, G.; Hussain, S.; Jawanjal, P.; Bandil, K.; Sharma, V.; Bharadwaj, M.; Mehrotra, R. Jagged-1 induced molecular alterations in HPV associated invasive squamous cell and adenocarcinoma of the human uterine cervix. Sci. Rep. 2018, 8, 9359. [Google Scholar] [CrossRef]
- Vazquez-Ulloa, E.; Ramos-Cruz, A.C.; Prada, D.; Aviles-Salas, A.; Chavez-Blanco, A.D.; Herrera, L.A.; Lizano, M.; Contreras-Paredes, A. Loss of nuclear NOTCH1, but not its negative regulator NUMB, is an independent predictor of cervical malignancy. Oncotarget 2018, 9, 18916–18928. [Google Scholar] [CrossRef] [Green Version]
- Zagouras, P.; Stifani, S.; Blaumueller, C.M.; Carcangiu, M.L.; Artavanis-Tsakonas, S. Alterations in Notch signaling in neoplastic lesions of the human cervix. Proc. Natl. Acad. Sci. USA 1995, 92, 6414–6418. [Google Scholar] [CrossRef]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.X.; Zhang, J.; Wang, J.; et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef]
- Fukusumi, T.; Califano, J.A. The NOTCH Pathway in Head and Neck Squamous Cell Carcinoma. J. Dent. Res. 2018, 97, 645–653. [Google Scholar] [CrossRef] [Green Version]
- Rettig, E.M.; Chung, C.H.; Bishop, J.A.; Howard, J.D.; Sharma, R.; Li, R.J.; Douville, C.; Karchin, R.; Izumchenko, E.; Sidransky, D.; et al. Cleaved NOTCH1 Expression Pattern in Head and Neck Squamous Cell Carcinoma Is Associated with NOTCH1 Mutation, HPV Status, and High-Risk Features. Cancer Prev. Res. 2015, 8, 287–295. [Google Scholar] [CrossRef]
- Meyers, J.M.; Grace, M.; Uberoi, A.; Lambert, P.F.; Munger, K. Inhibition of TGF-beta and NOTCH Signaling by Cutaneous Papillomaviruses. Front. Microbiol. 2018, 9, 389. [Google Scholar] [CrossRef] [PubMed]
- Fleming, N.I.; Jorissen, R.N.; Mouradov, D.; Christie, M.; Sakthianandeswaren, A.; Palmieri, M.; Day, F.; Li, S.; Tsui, C.; Lipton, L.; et al. SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Res. 2013, 73, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Schonleben, F.; Li, X.; Su, G.H. Disruption of transforming growth factor beta-Smad signaling pathway in head and neck squamous cell carcinoma as evidenced by mutations of SMAD2 and SMAD4. Cancer Lett. 2007, 245, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Kanuma, T.; Mizunuma, H.; Takama, F.; Ibuki, Y.; Wake, N.; Mogi, A.; Shitara, Y.; Takenoshita, S. Analysis of specific gene mutations in the transforming growth factor-beta signal transduction pathway in human ovarian cancer. Cancer Res. 2000, 60, 4507–4512. [Google Scholar] [PubMed]
- Valcourt, U.; Carthy, J.; Okita, Y.; Alcaraz, L.; Kato, M.; Thuault, S.; Bartholin, L.; Moustakas, A. Analysis of Epithelial-Mesenchymal Transition Induced by Transforming Growth Factor beta. Methods Mol. Biol. 2016, 1344, 147–181. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, J.A.; Jacob, Y.; Cassonnet, P.; Favre, M. Human papillomavirus type 5 E6 oncoprotein represses the transforming growth factor beta signaling pathway by binding to SMAD3. J. Virol. 2006, 80, 12420–12424. [Google Scholar] [CrossRef]
- Roman, A.; Munger, K. The papillomavirus E7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef] [Green Version]
- Collins, A.S.; Nakahara, T.; Do, A.; Lambert, P.F. Interactions with pocket proteins contribute to the role of human papillomavirus type 16 E7 in the papillomavirus life cycle. J. Virol. 2005, 79, 14769–14780. [Google Scholar] [CrossRef]
- Westphal, K.; Akgul, B.; Storey, A.; Nindl, I. Cutaneous human papillomavirus E7 type-specific effects on differentiation and proliferation of organotypic skin cultures. Anal. Cell. Pathol. 2009, 31, 213–226. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, W.; Roman, A. The E7 proteins of low- and high-risk human papillomaviruses share the ability to target the pRB family member p130 for degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 437–442. [Google Scholar] [CrossRef]
- Balsitis, S.J.; Sage, J.; Duensing, S.; Munger, K.; Jacks, T.; Lambert, P.F. Recapitulation of the effects of the human papillomavirus type 16 E7 oncogene on mouse epithelium by somatic Rb deletion and detection of pRb-independent effects of E7 in vivo. Mol. Cell. Biol. 2003, 23, 9094–9103. [Google Scholar] [CrossRef]
- Pietenpol, J.A.; Stein, R.W.; Moran, E.; Yaciuk, P.; Schlegel, R.; Lyons, R.M.; Pittelkow, M.R.; Munger, K.; Howley, P.M.; Moses, H.L. TGF-beta 1 inhibition of c-myc transcription and growth in keratinocytes is abrogated by viral transforming proteins with pRB binding domains. Cell 1990, 61, 777–785. [Google Scholar] [CrossRef]
- Lee, D.K.; Kim, B.C.; Kim, I.Y.; Cho, E.A.; Satterwhite, D.J.; Kim, S.J. The human papilloma virus E7 oncoprotein inhibits transforming growth factor-beta signaling by blocking binding of the Smad complex to its target sequence. J. Biol. Chem. 2002, 277, 38557–38564. [Google Scholar] [CrossRef]
- Soares, E.; Zhou, H. Master regulatory role of p63 in epidermal development and disease. Cell. Mol. Life Sci. 2017. [Google Scholar] [CrossRef]
- Melar-New, M.; Laimins, L.A. Human papillomaviruses modulate expression of microRNA 203 upon epithelial differentiation to control levels of p63 proteins. J. Virol. 2010, 84, 5212–5221. [Google Scholar] [CrossRef]
- Mighty, K.K.; Laimins, L.A. p63 is necessary for the activation of human papillomavirus late viral functions upon epithelial differentiation. J. Virol. 2011, 85, 8863–8869. [Google Scholar] [CrossRef]
- Eldakhakhny, S.; Zhou, Q.; Crosbie, E.J.; Sayan, B.S. Human papillomavirus E7 induces p63 expression to modulate DNA damage response. Cell Death Dis. 2018, 9, 127. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.Y.; Chen, B.F.; Yang, Y.C.; Chen, H.; Wang, Y.; Cviko, A.; Quade, B.J.; Sun, D.; Yang, A.; McKeon, F.D.; et al. Histologic and immunophenotypic classification of cervical carcinomas by expression of the p53 homologue p63: A study of 250 cases. Hum. Pathol. 2001, 32, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Szalmas, A.; Tomaic, V.; Basukala, O.; Massimi, P.; Mittal, S.; Konya, J.; Banks, L. The PTPN14 Tumor Suppressor Is a Degradation Target of Human Papillomavirus E7. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- White, E.A.; Munger, K.; Howley, P.M. High-Risk Human Papillomavirus E7 Proteins Target PTPN14 for Degradation. mBio 2016, 7. [Google Scholar] [CrossRef]
- Hansen, C.G.; Moroishi, T.; Guan, K.L. YAP and TAZ: A nexus for Hippo signaling and beyond. Trends Cell Biol. 2015, 25, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.M.; Nagatomo, I.; Suzuki, E.; Mizuno, T.; Kumagai, T.; Berezov, A.; Zhang, H.; Karlan, B.; Greene, M.I.; Wang, Q. YAP modifies cancer cell sensitivity to EGFR and survivin inhibitors and is negatively regulated by the non-receptor type protein tyrosine phosphatase 14. Oncogene 2013, 32, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, N.; Figel, S.A.; Wilson, K.E.; Morrison, C.D.; Gelman, I.H.; Zhang, J. PTPN14 interacts with and negatively regulates the oncogenic function of YAP. Oncogene 2013, 32, 1266–1273. [Google Scholar] [CrossRef]
- Michaloglou, C.; Lehmann, W.; Martin, T.; Delaunay, C.; Hueber, A.; Barys, L.; Niu, H.; Billy, E.; Wartmann, M.; Ito, M.; et al. The tyrosine phosphatase PTPN14 is a negative regulator of YAP activity. PLoS ONE 2013, 8, e61916. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Huang, J.; Wang, X.; Yuan, J.; Li, X.; Feng, L.; Park, J.I.; Chen, J. PTPN14 is required for the density-dependent control of YAP1. Genes Dev. 2012, 26, 1959–1971. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Pasolli, H.A.; Fuchs, E. Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc. Natl. Acad. Sci. USA 2011, 108, 2270–2275. [Google Scholar] [CrossRef] [Green Version]
- Wasson, C.W.; Morgan, E.L.; Muller, M.; Ross, R.L.; Hartley, M.; Roberts, S.; Macdonald, A. Human papillomavirus type 18 E5 oncogene supports cell cycle progression and impairs epithelial differentiation by modulating growth factor receptor signalling during the virus life cycle. Oncotarget 2017, 8, 103581–103600. [Google Scholar] [CrossRef] [Green Version]
- De Thé, H. Differentiation therapy revisited. Nat. Rev. Cancer 2018, 18, 117–127. [Google Scholar] [CrossRef]
- Ablain, J.; de Thé, H. Retinoic acid signaling in cancer: The parable of acute promyelocytic leukemia. Int. J. Cancer 2014, 135, 2262–2272. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Liu, Q. Differentiation therapy: A promising strategy for cancer treatment. Chin. J. Cancer 2016, 35, 3. [Google Scholar] [CrossRef]
- Cheepala, S.B.; Yin, W.; Syed, Z.; Gill, J.N.; McMillian, A.; Kleiner, H.E.; Lynch, M.; Loganantharaj, R.; Trutschl, M.; Cvek, U.; et al. Identification of the B-Raf/Mek/Erk MAP kinase pathway as a target for all-trans retinoic acid during skin cancer promotion. Mol. Cancer 2009, 8, 27. [Google Scholar] [CrossRef]
- Verma, A.K. Inhibition of both stage I and stage II mouse skin tumour promotion by retinoic acid and the dependence of inhibition of tumor promotion on the duration of retinoic acid treatment. Cancer Res. 1987, 47, 5097–5101. [Google Scholar]
- Zhang, M.L.; Tao, Y.; Zhou, W.Q.; Ma, P.C.; Cao, Y.P.; He, C.D.; Wei, J.; Li, L.J. All-trans retinoic acid induces cell-cycle arrest in human cutaneous squamous carcinoma cells by inhibiting the mitogen-activated protein kinase-activated protein 1 pathway. Clin. Exp. Dermatol. 2014, 39, 354–360. [Google Scholar] [CrossRef]
- Arany, I.; Ember, I.A.; Tyring, S.K. All-trans-retinoic acid activates caspase-1 in a dose-dependent manner in cervical squamous carcinoma cells. Anticancer Res. 2003, 23, 471–473. [Google Scholar]
- Arany, I.; Whitehead, W.E.; Ember, I.A.; Tyring, S.K. Dose-dependent activation of p21WAF1 transcription by all-trans-acid in cervical squamous carcinoma cells. Anticancer Res. 2003, 23, 495–497. [Google Scholar]
- Ruffin, M.T.; Bailey, J.M.; Normolle, D.P.; Michael, C.W.; Bieniasz, M.E.; Kmak, D.C.; Unger, E.R.; Brenner, D.E. Low-dose topical delivery of all-trans retinoic acid for cervical intraepithelial neoplasia II and III. Cancer Epidemiol. Biomark. Prev. 2004, 13, 2148–2152. [Google Scholar]
- Wahlberg, P.; Einarsdottir, M.; Fex, G.; Rydell, R. The additive antiproliferative effect of all-trans retinoic acid and interferon-alpha2a on human cervical carcinoma cell lines is not associated with increased expression of retinoid receptors. Anti-Cancer Drugs 1997, 8, 522–528. [Google Scholar] [CrossRef]
- Weiss, G.R.; Liu, P.Y.; Alberts, D.S.; Peng, Y.M.; Fisher, E.; Xu, M.J.; Scudder, S.A.; Baker, L.H., Jr.; Moore, D.F.; Lippman, S.M. 13-cis-retinoic acid or all-trans-retinoic acid plus interferon-alpha in recurrent cervical cancer: A Southwest Oncology Group phase II randomized trial. Gynecol. Oncol. 1998, 71, 386–390. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.K.; Li, Y.; Hafner, M.; Banerjee, N.S.; Tang, S.; Briskin, D.; Meyers, C.; Chow, L.T.; Xie, X.; et al. microRNAs are biomarkers of oncogenic human papillomavirus infections. Proc. Natl. Acad. Sci. USA 2014, 111, 4262–4267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freije, A.; Molinuevo, R.; Ceballos, L.; Cagigas, M.; Alonso-Lecue, P.; Rodriguez, R.; Menendez, P.; Aberdam, D.; De Diego, E.; Gandarillas, A. Inactivation of p53 in Human Keratinocytes Leads to Squamous Differentiation and Shedding via Replication Stress and Mitotic Slippage. Cell Rep. 2014, 9, 1349–1360. [Google Scholar] [CrossRef] [Green Version]
- Ying, Z.; Sandoval, M.; Beronja, S. Oncogenic activation of PI3K induces progenitor cell differentiation to suppress epidermal growth. Nat. Cell Biol. 2018, 20, 1256–1266. [Google Scholar] [CrossRef] [PubMed]
- Gandarillas, A.; Watt, F.M. c-Myc promotes differentiation of human epidermal stem cells. Genes Dev. 1997, 11, 2869–2882. [Google Scholar] [CrossRef] [Green Version]
- Molinuevo, R.; Freije, A.; de Pedro, I.; Stoll, S.W.; Elder, J.T.; Gandarillas, A. FOXM1 allows human keratinocytes to bypass the oncogene-induced differentiation checkpoint in response to gain of MYC or loss of p53. Oncogene 2017, 36, 956–965. [Google Scholar] [CrossRef]
- Waikel, R.L.; Wang, X.J.; Roop, D.R. Targeted expression of c-Myc in the epidermis alters normal proliferation, differentiation and UV-B induced apoptosis. Oncogene 1999, 18, 4870–4878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watt, F.M.; Frye, M.; Benitah, S.A. MYC in mammalian epidermis: How can an oncogene stimulate differentiation? Nat. Rev. Cancer 2008, 8, 234–242. [Google Scholar] [CrossRef]
- Gebhardt, A.; Frye, M.; Herold, S.; Benitah, S.A.; Braun, K.; Samans, B.; Watt, F.M.; Elsasser, H.P.; Eilers, M. Myc regulates keratinocyte adhesion and differentiation via complex formation with Miz1. J. Cell Biol. 2006, 172, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemp, C.J.; Donehower, L.A.; Bradley, A.; Balmain, A. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell 1993, 74, 813–822. [Google Scholar] [CrossRef]
Figure 1.
Human papillomavirus (HPV) replication requires and impairs epithelial differentiation. A schematic of epithelial differentiation in the absence (A) and presence (B) of HPV infection. In uninfected cells, self-renewing basal keratinocytes attach to the basement membrane. When keratin 5 and keratin 14-positive basal cells divide, one daughter cell can progress into the spinous layers and express keratins 1 and 10. Terminal differentiation and progression into the granular and cornified layers is marked by the expression of fillagrin and involucrin. Suprabasal epithelial cells are generally not proliferative. In an HPV-infected epithelium, proliferation is uncoupled from differentiation. Markers of proliferation are expressed and differentiation-related gene expression is impaired in suprabasal cells.
Figure 1.
Human papillomavirus (HPV) replication requires and impairs epithelial differentiation. A schematic of epithelial differentiation in the absence (A) and presence (B) of HPV infection. In uninfected cells, self-renewing basal keratinocytes attach to the basement membrane. When keratin 5 and keratin 14-positive basal cells divide, one daughter cell can progress into the spinous layers and express keratins 1 and 10. Terminal differentiation and progression into the granular and cornified layers is marked by the expression of fillagrin and involucrin. Suprabasal epithelial cells are generally not proliferative. In an HPV-infected epithelium, proliferation is uncoupled from differentiation. Markers of proliferation are expressed and differentiation-related gene expression is impaired in suprabasal cells.

Figure 2.
Genus beta HPV E6 bind MAML1 to inhibit NOTCH signaling. A schematic of activated NOTCH signaling in the absence (A) and presence (B) of genus beta HPV E6. Upon activation of the NOTCH signaling pathway, proteolytic cleavage of NOTCH releases the NOTCH intracellular domain (ICD), which can translocate to the nucleus. It displaces transcriptional repressors and forms a transcriptionally active complex including NOTCH, MAML1, and RBP-J. Genus beta HPV E6 bind to an LxxLL motif present in the MAML1 C-terminus. The oncoprotein does not disrupt formation of the transcription complex but does inhibit NOTCH signaling.
Figure 2.
Genus beta HPV E6 bind MAML1 to inhibit NOTCH signaling. A schematic of activated NOTCH signaling in the absence (A) and presence (B) of genus beta HPV E6. Upon activation of the NOTCH signaling pathway, proteolytic cleavage of NOTCH releases the NOTCH intracellular domain (ICD), which can translocate to the nucleus. It displaces transcriptional repressors and forms a transcriptionally active complex including NOTCH, MAML1, and RBP-J. Genus beta HPV E6 bind to an LxxLL motif present in the MAML1 C-terminus. The oncoprotein does not disrupt formation of the transcription complex but does inhibit NOTCH signaling.

© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
White, E.A. Manipulation of Epithelial Differentiation by HPV Oncoproteins. Viruses 2019, 11, 369. https://doi.org/10.3390/v11040369
AMA Style
White EA. Manipulation of Epithelial Differentiation by HPV Oncoproteins. Viruses. 2019; 11(4):369. https://doi.org/10.3390/v11040369
Chicago/Turabian StyleWhite, Elizabeth A. 2019. "Manipulation of Epithelial Differentiation by HPV Oncoproteins" Viruses 11, no. 4: 369. https://doi.org/10.3390/v11040369
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.