1. Introduction
The AIDS (Acquired Immunodeficiency Syndrome) pandemic that is caused by Human Immunodeficiency Virus (HIV) represents a global health problem with enormous disease control dimensions. Thanks to the efforts in strengthening HIV/AIDS prevention programs and the increasing numbers of HIV-infected individuals that have access to the highly effective antiretroviral therapy (HAART), the annual number of new HIV infections has declined by 16% since 2010 (
http://www.unaids.org). Nonetheless, at the end of 2016, over 36 million people worldwide were living with HIV/AIDS. As there is no functional cure for HIV infection thus far, an effective HIV vaccine remains the best long-term strategy for preventing viral infection and AIDS.
We have previously reported that poxvirus-based viral vectors are potential prophylactic vaccine candidates against HIV infection [
1,
2,
3,
4]. The modified vaccinia virus Ankara-B (MVA-B) vaccine simultaneously co-expressing the HIV-1 (clade B) monomeric gp120 envelope (Env) protein as a cell-released product and Gag-Pol-Nef (GPN) antigens as an intracellular polyprotein unable to form VLPs was safe, well tolerated, and elicited moderate and durable HIV-1-specific T cell and antibody responses when it was assayed in healthy volunteers in homologous regimen [
1,
2]. The same HIV-1 antigens, but from clade C (MVA-C), were also safe and triggered HIV-1 specific immune responses when used in a prophylactic phase I clinical trial in combination with a DNA vector and a gp140 protein component [
5]. Moreover, when these clade C HIV-1 antigens were vectored by the DNA and NYVAC strain, induced strong, broad, and polyfunctional T cell responses in humans when combined in the heterologous DNA prime/NYVAC boost regimen [
6]. Commonly, in these studies, the induced T cell responses were predominantly directed against Env, whereas GPN-specific responses were lower and less frequent.
In order to increase the expression levels of the encoded HIV-1 antigens and to achieve more balanced immune responses, a new generation of optimized HIV-1
env and
gag-pol-nef genes were designed and then inserted independently into different backbones, such as DNA vectors and attenuated poxvirus strains (NYVAC and ALVAC) [
7,
8,
9,
10,
11]. The improved antigens belong to the HIV-1 clade C, which is responsible for approximately 50% of all new infections worldwide. The original GPN polyprotein was further refined to allow for the efficient production and release of virus-like particles and to better balance the relative expression of Gag and Pol-Nef antigens and a trimeric soluble gp140 form was used instead of the monomeric gp120 to more closely resemble the native envelope structure. The new generation of recombinant vectors demonstrated an inducement of an enhanced HIV-1-specific immunogenicity profile in mice [
11] and non-human primates (NHPs) [
8,
9,
10,
12,
13] when combined in homologous or heterologous combination.
Since vaccine-induced protective immunity is critically determined by the HIV-1 Env conformation and Gag-specific cellular response, significant efforts are directed towards generating trimeric Env immunogens that assume native structures and Gag-induced VLPs with enhanced immunogenicity. Here, we generated and characterized single and double MVA-based vectors that expressed the HIV-1 clade C gp145(ZM96) Env as a membrane-bound gp145 trimeric protein and/or the improved Gag(ZM96)-Pol-Nef(CN54) (GPN) polyprotein, which is processed in a way that produces a 55 kDa Gag protein that is able to induce the formation of virus-like particles (VLPs) [
11]. The immunogenicity of the double MVA-gp145-GPN virus was evaluated in mice in comparison with single recombinants that individually expressed either gp145(ZM96) Env (MVA-gp145) or Gag(ZM96)-Pol-Nef(CN54) (GPN) polyprotein (MVA-GPN). Based on the broad capacity of membrane-bound gp145 to react with bNAbs and on the balanced HIV-1-specific immune responses that are induced by the double recombinant MVA vector (CD4, Tfh, GC B cells, and IgG2a/IgG1 ratio), our findings suggest a potential role of MVA-gp145-GPN as a relevant vaccine against HIV.
2. Materials and Methods
2.1. Cells and Viruses
Primary chicken embryo fibroblast (CEF) cells (obtained from pathogen-free 11-day-old eggs; MSD, Salamanca, Spain), DF-1 cells (a spontaneously immortalized CEF cell line) and HeLa cells (human epithelial cervix adenocarcinoma cells) were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 100 U/mL penicillin/100 µg/mL streptomycin (SIGMA, St. Louis, MO, USA), 2 mM l-glutamine (Merck, Kenilworth, NJ, USA), 0.1 mM non-essential amino acids (SIGMA), 0.5 μg/mL amphotericin B (Fungizone; Gibco-Life Technologies, Waltham, MA, USA) and 10% heat-inactivated fetal calf serum (FCS; SIGMA) for CEF and DF-1 cells or 10% newborn calf serum (NCS; SIGMA) for HeLa cells. The cells were maintained in a humidified air 5% CO2 atmosphere at 37 °C.
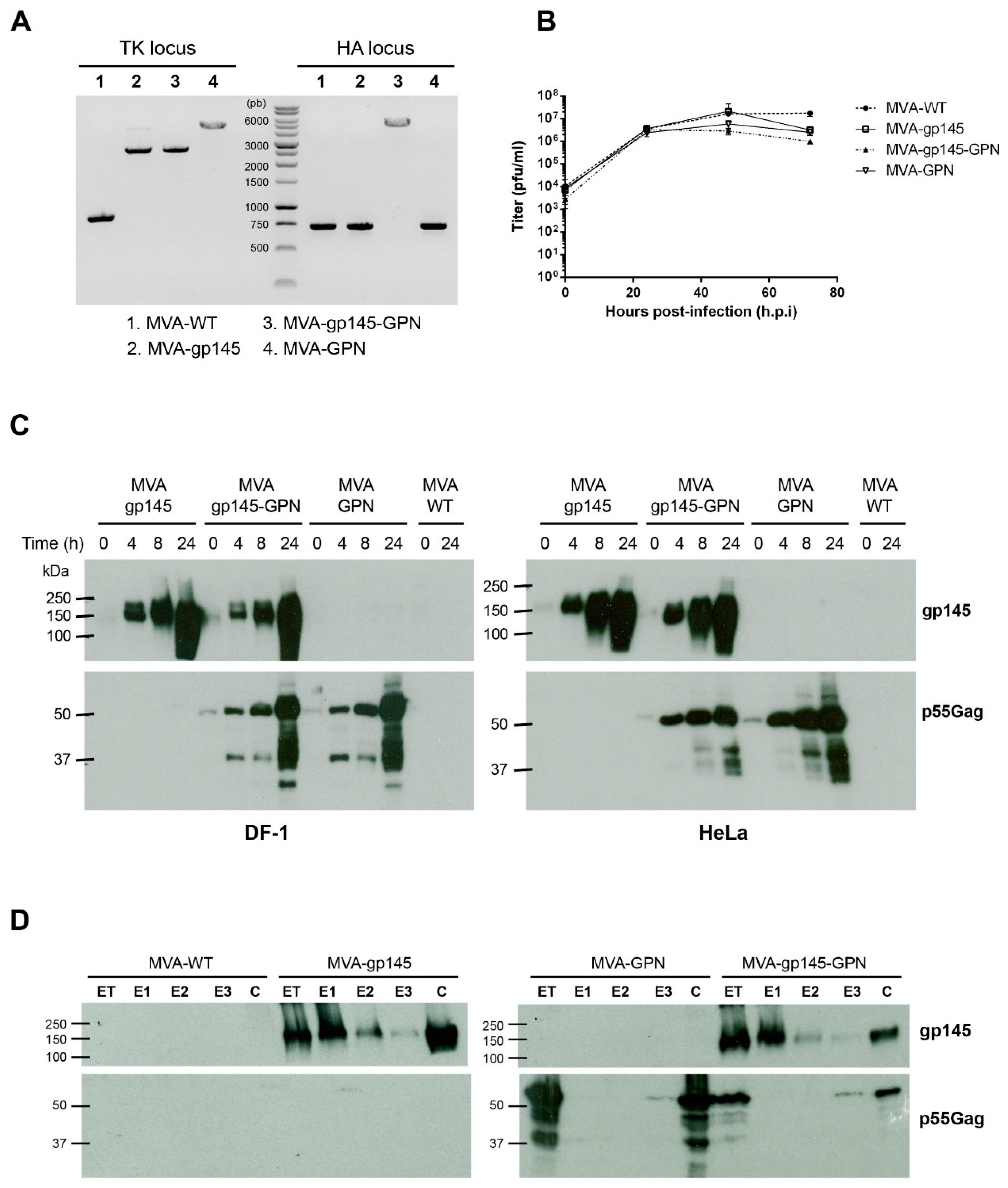
The viruses that were used in this work included: the attenuated wild-type modified vaccinia virus Ankara (MVA-WT) that was obtained from the Ankara strain after 586 serial passages in CEF cells (kindly provided by G. Sutter); the recombinant MVA-gp145(ZM96) expressing a membrane-bound trimeric HIV-1 clade C ZM96 gp145 protein from the viral thymidine kinase (TK) locus (shortly MVA-gp145); the recombinant MVA-Gag(ZM96)-Pol-Nef(CN54) expressing the optimized Gag(ZM96)-Pol-Nef(CN54) polyprotein, which is processed to produce a 55 kDa Gag protein that is able to induce the formation of VLPs from the viral TK locus (shortly MVA-GPN); and, the recombinant MVA-gp145(ZM96)-Gag(ZM96)-Pol-Nef(CN54) expressing gp145(ZM96) from the viral TK locus and Gag(ZM96)-Pol-Nef(CN54) polyprotein from the viral haemagglutinin (HA) locus (shortly MVA-gp145-GPN). In both of the GPN-expressing vectors, the natural ribosomal (−1) frameshift between Gag and Pol was restored to skew Gag:PolNef expression to approximately 10:1, and the N-terminal myristoylation signal was reintroduced to enable the release of GagPolNef virus-like particles from infected cells [
9]. Virus infections were performed with 2% FCS or NCS.
2.2. Construction of the Plasmid Transfer Vectors
2.2.1. Construction of the Plasmid Transfer Vector pCyA-gp145(ZM96)
The plasmid transfer vector pCyA-gp145(ZM96) (shortly pCyA-gp145), which was used for the insertion of gp145 antigen into the viral TK locus of MVA-WT, was obtained by standard cloning procedures. The codon optimized
env gen was amplified by PCR from plasmid plZAW1-gp145-ZM96-DeltaC6 (provided by Ralf Wagner, University of Regensburg) with oligonucleotides gp145TM-U1: (5´-GA
CTCGAGGCCACCATGGGAGTG-3´) (
XhoI site underlined) and gp145TM-L1: (5´-TA
GCGGCCGCTCAGTAGCCCTG-3´) (
NotI site underlined) (gp145 PCR product: 2178 bp), digested with
XhoI and
NotI, and then cloned into plasmid pCyA (7582 bp) [
14] that was previously digested with the same restriction enzymes to generate pCyA-gp145 (9654 bp). The resulting plasmid pCyA-gp145 was confirmed by DNA sequence analysis and it directs the insertion of gp145 antigen into the TK locus of MVA-WT.
2.2.2. Construction of the Plasmid Transfer Vector pHA-Gag(ZM96)-Pol-Nef(CN54)
The plasmid transfer vector pHA-Gag(ZM96)-Pol-Nef(CN54) (shortly pHA-GPN), which was used for the insertion of GPN antigen into the viral haemagglutinin (HA) locus of MVA-gp145 recombinant virus, was obtained by standard cloning procedures. The codon optimized GPN gen was amplified by PCR from plasmid plZAW1-Gag(ZM96)-Pol-Nef(CN54) [
11] with oligonucleotides GPN-
NotI (5’-AATT
GCGGCCGCTTACTTGGTCCTGTG-3’) (
NotI site underlined) and GPN-
KpnI (5’-AGA
GGTACCGCCACCATGGGAGCCAGAG-3’) (
KpnI site underlined) (GPN PCR product: 4070 bp), digested with
NotI and
KpnI, and then cloned into plasmid pHA (6639 bp) that was previously digested with the same restriction enzymes to generate pHA-GPN (10,666 bp). The plasmid pHA has been previously described [
15] and it comprises the viral sE/L promoter, a multiple-cloning site, HA flanking regions of MVA genome, and the selectable marker genes for ampicillin and β-glucuronidase. The resulting plasmid pHA-GPN was confirmed by DNA sequence analysis and it directs the insertion of the GPN antigen into the HA locus of MVA-gp145-GPN.
2.3. Construction of MVA-Based Recombinant Viruses
For the construction of MVA-gp145 and MVA-GPN recombinant viruses, 3 × 10
6 DF-1 cells were infected with MVA-WT at a multiplicity of infection (MOI) of 0.05 pfu/cell and then transfected one hour later with 6 µg DNA of pCyA-gp145 or plZAW1-Gag(ZM96)-Pol-Nef(CN54) [
11], respectively, using Lipofectamine-2000 (Invitrogen, Carlsbad, CA, USA) and following the manufacturer’s instructions. After 72 h post-infection (h.p.i.), the infected cells were collected, lysed by freeze-thaw cycling, sonicated, and then used for the screening of the MVA-based recombinant viruses. MVA viruses containing gp145 or Gag-Pol-Nef genes and transiently co-expressing the β-gal marker gene were isolated by three consecutive rounds of plaque purification steps in monolayers of DF-1 cells that were stained with 5-bromo-4-chloro-3-indolyl β-D-galactopyranoside (X-Gal; 1.2 mg/mL). After the recombinant viruses expressing gp145 or Gag-Pol-Nef and the β-gal marker have been isolated, further propagation of these MVA-based recombinant viruses leads to the self-deletion of β-gal by homologous recombination events between the short TK left arm repeat and the TK left arm that are flanking the marker. Therefore, in the subsequent three rounds, MVA-based recombinant viruses containing gp145 or Gag-Pol-Nef genes and having deleted the β-gal marker gene were selected by plaque purification screening for non-stained viral plaques in DF-1 cells in the presence of 5-bromo-4-chloro-3-indolyl β-D-galactopyranoside (1.2 mg/mL).
For the construction of MVA-gp145-GPN recombinant virus, 3 × 106 DF-1 cells were infected with MVA-gp145 recombinant virus at 0.05 pfu/cell and then transfected one hour later with 6 µg of plasmid pHA-GPN using Lipofectamine-2000 (Invitrogen). At 72 h post-infection (h.p.i.), the cells were harvested, lysed by freeze-thaw cycling, sonicated, and used for recombinant virus screening using X-Gluc (5-bromo-4-chloro-3-indolyl-β-d-glucuronic acid; 800 μg/mL) as substrate for the product of the β-glucuronidase (β-GUS) marker gene. As described above for the MVA-gp145 and MVA-GPN viruses, during the purification steps, the β-GUS marker gene was deleted by homologous recombination.
The resulting MVA-based recombinant viruses were expanded in CEF cells and the crude preparations that were obtained were used for the propagation of the viruses in large cultures of CEF cells, followed by virus purification through two 36% (
w/
v) sucrose cushions and the virus titers were determined by the immunostaining plaque assay in DF-1 cells, as previously described [
16]. The titer determinations of the different viruses were performed at least three times.
2.4. PCR Analysis of MVA-Based Recombinant Viruses
To test the identity and purity of the different MVA-based recombinant viruses, viral DNA was extracted from DF-1 cells that were infected at 5 pfu/cell with MVA-WT, MVA-gp145, MVA-GPN or MVA-gp145-GPN for 24 h. The cell membranes were disrupted by proteinase K treatment (0.2 mg/mL proteinase K in 50 mM Tris-HCl pH 8, 100 mM EDTA pH 8, 100 mM NaCl, 1% SDS; 1 h, 55 °C), followed by incubation with RNase A (80 µg/mL). Viral DNA was precipitated using 2-propanol. Primers TK-L: 5’-TGATTAGTTTGATGCGATTC-3’ and TK-R: 5’-CTGCCGTATCAAGGACA-3’ spanning TK flanking regions and primers HA-MVA: 5’-TGACACGATTACCAATAC-3’ and HA-II: 5’-GATCCGCATCATCGGTGG-3’ spanning HA flanking regions were used for PCR analysis of TK and HA loci, respectively. The amplification reactions were carried out with Phusion High-Fidelity DNA polymerase (BioLabs, Ipswich, MA, USA), according to the manufacturer´s recommendations.
2.5. Analysis of Virus Growth
To determine the growth profile of the different MVA-based recombinant viruses, monolayers of primary CEF cells that were grown in 12-well plates were infected at 0.1 pfu/cell with MVA-WT, MVA-gp145, MVA-GPN or MVA-gp145-GPN. Following virus adsorption for 60 min at 37 °C, the inoculum was removed and the infected cells were incubated with fresh DMEM containing 2% FCS at 37 °C in a 5% CO2 atmosphere. At different times post-infection (0, 24, 48 and 72 h), the cells were harvested by scraping, centrifuged for 5 min at 3000 rpm, supernatant removed, 0.1 mL of complete DMEM added to the cellular pellet, freeze-thawed three times, and briefly sonicated. Virus titers in cell lysates were determined by immunostaining plaque assay in DF-1 cells using rabbit polyclonal anti-VACV strain WR (1:1000; CNB), followed by anti-rabbit-horseradish peroxidase (HRP) (1:1000; SIGMA).
2.6. Time-Course Expression of HIV-1 Proteins gp145 and GPN by Western-Blot Analysis
Time-course expression of gp145 and GPN proteins from single and double recombinant viruses was performed by Western-blot. Monolayers of DF-1 or HeLa cells that were grown in 24-well plates were infected at 5 pfu/cell with MVA-WT, MVA-gp145, MVA-GPN or MVA-gp145-GPN viruses. At different times post-infection (0, 4, 8 and 24 h), infected cells were collected, and the cells extracts were fractionated by 10% SDS-PAGE and then analyzed by Western-blot using rabbit anti-gp120 antibody (1:3000; CNB) to evaluate the expression of gp145 protein, or rabbit anti-gag p24 antibody (1:1000; NIBSC) to determine the expression of GPN protein. Goat anti-rabbit-HRP (1:5000; SIGMA) was used as secondary antibody. The immunocomplexes were detected by an enhanced chemiluminescence system (ECL, GE Healthcare, Chicago, IL, USA).
2.7. Genetic Stability of MVA-Based Recombinant Viruses
The stability of HIV-1 antigens that were expressed by the different recombinant viruses was analyzed by serial passages in CEF cells grown in T25 tissue culture flasks. Monolayers of CEF cells were infected with MVA-gp145, MVA-GPN or MVA-gp145-GPN at 0.05 pfu/cell, and after 48–72 h the infected cells were collected by scrapping, freeze-thawed three times, and cellular extract was used to infect a monolayer of CEF cells for a new round of infection. This same procedure was repeated until seven serial passages were performed. Additionally, monolayers of DF-1 cells that were grown in 6-well culture plates were infected with serial dilutions of the virus stock from passage 7. After 1 h of viral adsorption, the inoculum was removed and the infected cells were overlayed with 1.9% Agar:DMEM 2X:2% FCS. When lysis plaques were visible, 20–25 isolated plaques were picked up. The correct expression of gp145 and GPN antigens both in passages P1 to P7 and in isolated plaques from P7 was analyzed by Western-blot using rabbit anti-gp120 or anti-gag p24 antibodies.
2.8. Fractionation of HIV-1 gp145 and Gag Antigens into Different Compartments after Detergent Treatment of Purified Recombinant MVA Particles
The localization of HIV-1 gp145 and Gag antigens in MVA recombinants was analyzed by sequential detergent treatment, as previously described [
17]. Briefly, virions that were purified by sucrose gradients were resuspended by sonication in 0.1 mL of Tris-buffer (50 mM Tris-HCl pH 8.5, 10 mM MgCl
2) containing the nonionic detergent NP-40 (1%). This and the following treatments were performed for 30 min at 37 °C. The E1 fraction (soluble lipid envelopes) was discarded by centrifugation, and the remaining pellet fraction was resuspended in 0.1 mL of Tris-buffer containing 1% NP-40 plus 50 mM DTT. The E2 fraction (soluble protein matrix-like membranes) was collected after centrifugation and the pellet was resuspended in 0.1 mL of the previous buffer with the addition of 0.5% DOC and 0.1% SDS. The soluble core proteins (E3 fraction) were removed by centrifugation, and the pellet fraction containing the remaining cores (C fraction) was resuspended in 0.1 mL of milliQ H
2O. The collected fractions were resolved by SDS-PAGE under reducing conditions and the HIV-1 gp145 and Gag proteins were identified by Western-blot with specific antibodies.
2.9. Detection of Gag-Induced VLPs by Electron Microscopy Analysis
To detect HIV-1 Gag-induced VLPs in the supernatant of infected cells, 150 × 106 HeLa cells were infected with MVA-GPN or MVA-gp145-GPN viruses at 5 pfu/cell for 24 hours. The VLPs were pelleted from infected cell culture supernatants by ultracentrifugation through a 20% sucrose cushion at 25,000 rpm in a Beckman SW28 rotor for 3 h. The VLP-containing pellet was resuspended in PBS buffer, loaded onto a 20%–60% w/v sucrose gradient, and then centrifuged at 35,000 rpm in a Beckmann SW41 rotor for 18 h. The gradient was fractionated from the top in 500 μL aliquots and then analysed by Western-blot to detect the presence of HIV-1 Gag specific p24 protein. The fraction of each virus that exhibited the highest Gag expression level was dialyzed to eliminate sucrose and salts through a 0.025 μm pore size membrane filter (Merck Millipore, Burlington, MA, USA) overnight at 4 °C. After dialysis, 100 μL of each sample was fixed with 4% paraformaldehyde for 30 min at 4 °C. For the negative staining of VLPs, 20 µL of each sample was adsorbed to carbon-coated collodion films that were mounted on 400-mesh/inch nickel grids (Aname, Madrid, Spain), floated two times in NH4Cl drops, washed several times with PBS, and stained with 2% uranyl acetate (Aname) for 1 min. For the immunogold labelling to detect gp145 that is associated to the VLP, the samples were incubated with a rabbit polyclonal anti-gp120 primary antibody (1:20; CNB), followed by an anti-IgG secondary antibody that was coupled to 10-nm colloidal gold beads (1:40; BBI Solutions, Crumlin, UK) and then stained with 2% uranyl acetate. Finally, VLP samples from negative staining or immunogold labelling were analysed in a transmission electron microscope (JEOL JEM-1011; CNB-CSIC Electron Microscopy Service, Madrid, Spain) that was equipped with an ES1000W Erlangshen charge-coupled-device (CCD) camera (Gatan Inc., Pleasanton, CA, USA) at an acceleration voltage of 40 to 100 kV.
2.10. Detection of HIV-1 Clade C ZM96 gp145 Protein on the Surface of Infected Cells
The presence of membrane-bound trimeric gp145 protein on the surface of non-permeabilized infected cells was assessed by flow cytometry using a panel of human broadly neutralizing antibodies (bNAbs) targeting quaternary V1/V2 N-glycans (PG9, PGT145 and PG16), V3 N-glycans (10-1074 and PGT121), outer domain (OD)-glycans (2G12), CD4 binding site (VRC01, VRC03 and b12), or gp120/gp41ECTO interface (PGT151 and 35O22) epitopes on the native Env protein. bNAbs were obtained through the AIDS Reagent Program, Division of AIDS, NIAID. HeLa cells were infected with MVA-gp145, MVA-gp145-GPN or MVA-GPN recombinant viruses at 3 pfu/cell. At 16 h.p.i., the cells were rinsed with PBS (no calcium/magnesium), dissociated with 2 mM EDTA in 1X PBS, washed with FACS buffer (1% BSA, 2 mM EDTA in 1X PBS), and pelleted at 1500 rpm for 5 min. Cells were then stained with live/death fixable red dye (1:200, Invitrogen) for 30 min at 4 °C in the dark, washed twice with FACS buffer, and then blocked with 3% BSA for 30 min at 4 °C. 10 µg/mL in 50 µL FACS buffer of each primary human IgG anti-Env bNAb were used to stain 106 cells for 30 min at 4 °C in the dark. The cells were then washed twice with FACS buffer and secondary F(ab’)2-goat anti-human IgG (H + L)-PE antibody (1:200, Beckman Coulter, Brea, CA, USA) in 50 µL FACS buffer was added onto the cells. After 30 min incubation at 4 °C in the dark, the cells were washed twice with FACS buffer and fixed with 0.5% formaldehyde. The samples were acquired in a GALLIOS flow cytometer (Beckman Coulter) and data analyses were performed using FlowJo software (Version 10.4.2; Tree Star, Ashland, OR, USA). Geometric Mean Fluorescence Intensity (gMFI) values on the “live cells” gate were used to analyse the results.
2.11. Peptides
The HIV-1 clade C ZM96 gp140 peptides were provided by the Centralised Facility for AIDS Reagents, NIBSC, UK. They spanned the HIV-1 gp140 from clade C (ZM96) that was included in the recombinant viruses MVA-gp145 and MVA-gp145-GPN as consecutive 15-mers overlapping by 11 amino acids. They were pooled in three different Env peptide pools: Env-1 (61 peptides), Env-2 (65 peptides), and Env-3 (40 peptides). The HIV-1 clade C ZM96 Gag peptide pool was provided by the NIH AIDS Reagent Program (Maryland, USA) and it spanned the HIV-1 Gag region from clade C (ZM96) included in MVA-GPN and MVA-gp145-GPN viruses as consecutive 20-mers overlapping by 10 amino acids. The HIV-1 clade C CN54 Pol and Nef peptide pools were provided by the EuroVacc Foundation (Lausanne, Switzerland) and spanned the HIV-1 Pol and Nef antigens from clade C (CN54) included in MVA-GPN and MVA-gp145-GPN viruses as consecutive 15-mers overlapping by 11 amino acids. For the analysis of the HIV-1 Pol-Nef-specific cellular immune responses, the following peptide pools were used: Gag-Pol, Pol-1, Pol-2 and Nef. The vaccinia virus (VACV) E3
140–148 peptide (VGPSNSPTF; CNB-CSIC Proteomics Service), described as an immunodominant epitope in BALB/c mice [
18], was used to detect the VACV-specific CD8 T cell responses.
2.12. Ethics Statement
The Ethical Committee of Animal Experimentation (CEEA) of Centro Nacional de Biotecnología (CNB-CSIC, Madrid, Spain) approved the animal experimental protocols, according to International EU Guidelines 2010/63/UE on protection of animals used for experimentation and other scientific purposes, Spanish National Royal Decree RD 1201/2005 and Spanish National Law 32/2007 on animal welfare, exploitation, transport and sacrifice (permit number PROEX 281/16).
2.13. Mouse Immunization Schedule
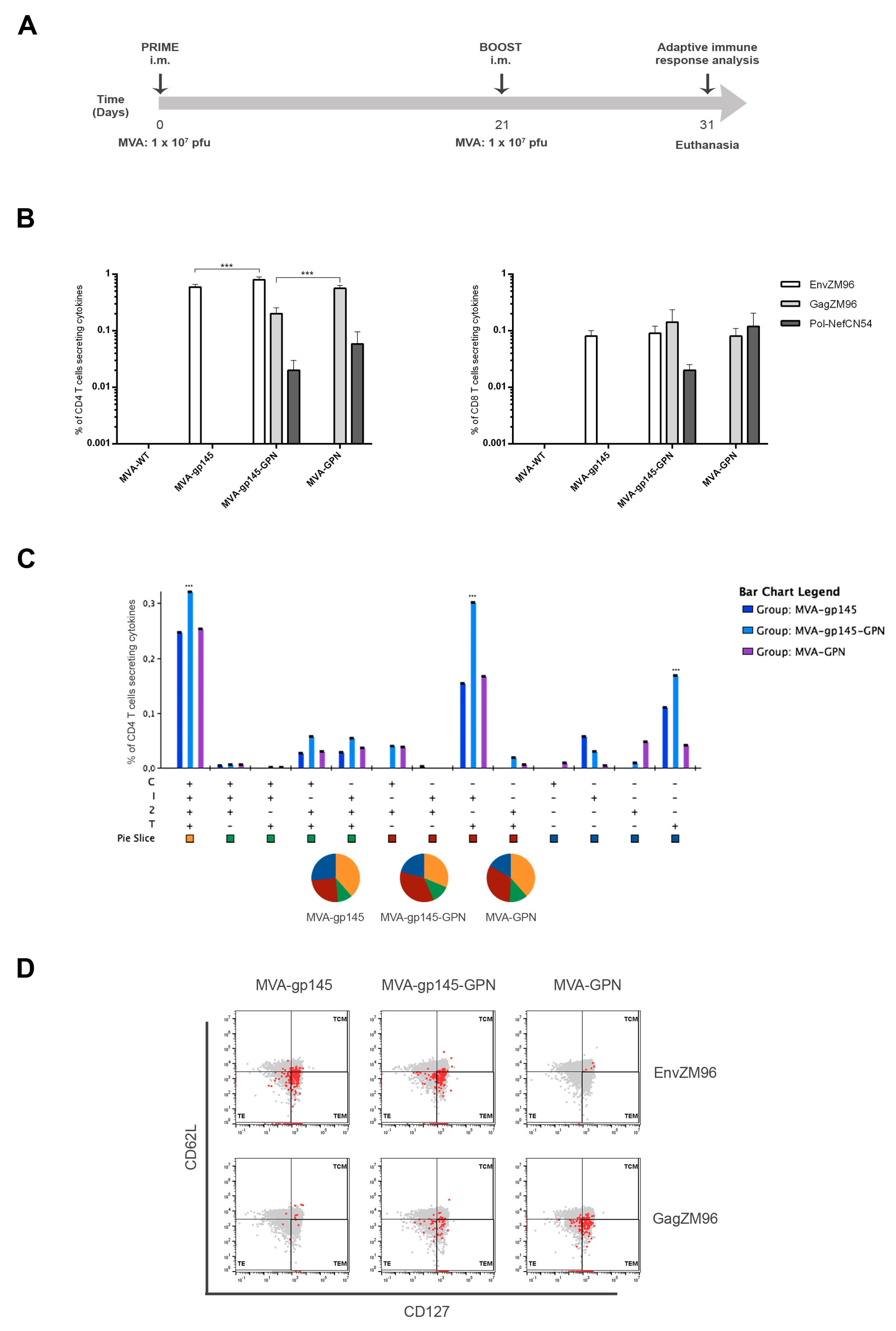
Groups of female BALB/c mice (6–8 week-old) purchased from ENVIGO (n = 5) were immunized with 1 × 107 pfu of MVA-WT, MVA-gp145, MVA-GPN or MVA-gp145-GPN by bilateral intramuscular (i.m.) route. Three weeks later, the animals were immunized with MVA constructions as in the prime and 10 days after the last immunization, mice were sacrificed, and the spleens and draining lymph nodes (DLNs) were processed for Intracellular Cytokine Staining (ICS) assay and sera harvested for Enzyme-Linked Immunosorbent Assay (ELISA) to measure the cellular and humoral adaptive immune responses against HIV-1 or VACV antigens, respectively.
2.14. Analysis of the Cellular Immune Responses by Intracellular Cytokine Staining (ICS) Assay
2.14.1. Analysis of HIV-1- and VACV-Specific CD4 and CD8 T Cell Responses
To determine the magnitude and phenotype of the HIV-1- or VACV-specific CD4 and CD8 T cell immune responses, 2 × 106 splenocytes (erythrocyte-depleted) were seeded on 96-well plates and stimulated for 6 h in supplemented RPMI 1640 medium with 10% FCS, 1 µL/mL Golgiplug (BD Biosciences, Franklin Lakes, NJ, USA), anti-CD107a-FITC (BD Biosciences) and 5 µg/mL of HIV-1 clade C ZM96 gp140 (Env-1 + Env-2 + Env-3) or Gag peptide pools or 1 µg/mL of HIV-1 clade C CN54 Pol-Nef (Gag-Pol + Pol-1 + Pol-2 + Nef) peptide pools or 10 µg/mL of VACV E3 peptide. After stimulation, the splenocytes were washed, stained for surface markers, permeabilized (Cytofix/Cytoperm kit; BD Biosciences), and stained intracellularly with appropriate fluorochromes. For the analysis of HIV-1- and VACV-specific CD4 and CD8 T cell responses, the following fluorochrome-conjugated antibodies were used: CD3-PECF594, CD4-APCCy7, CD8-V500, CD107a-FITC, IL-2-APC, IFN-γ-PeCy7 and TNF-α-PE for functional analyses and CD127-PerCPCy5.5 and CD62L-Alexa700 for the phenotypic analyses. All of the antibodies were from BD Biosciences. The dead cells were excluded using the violet LIVE/DEAD stain kit (Invitrogen).
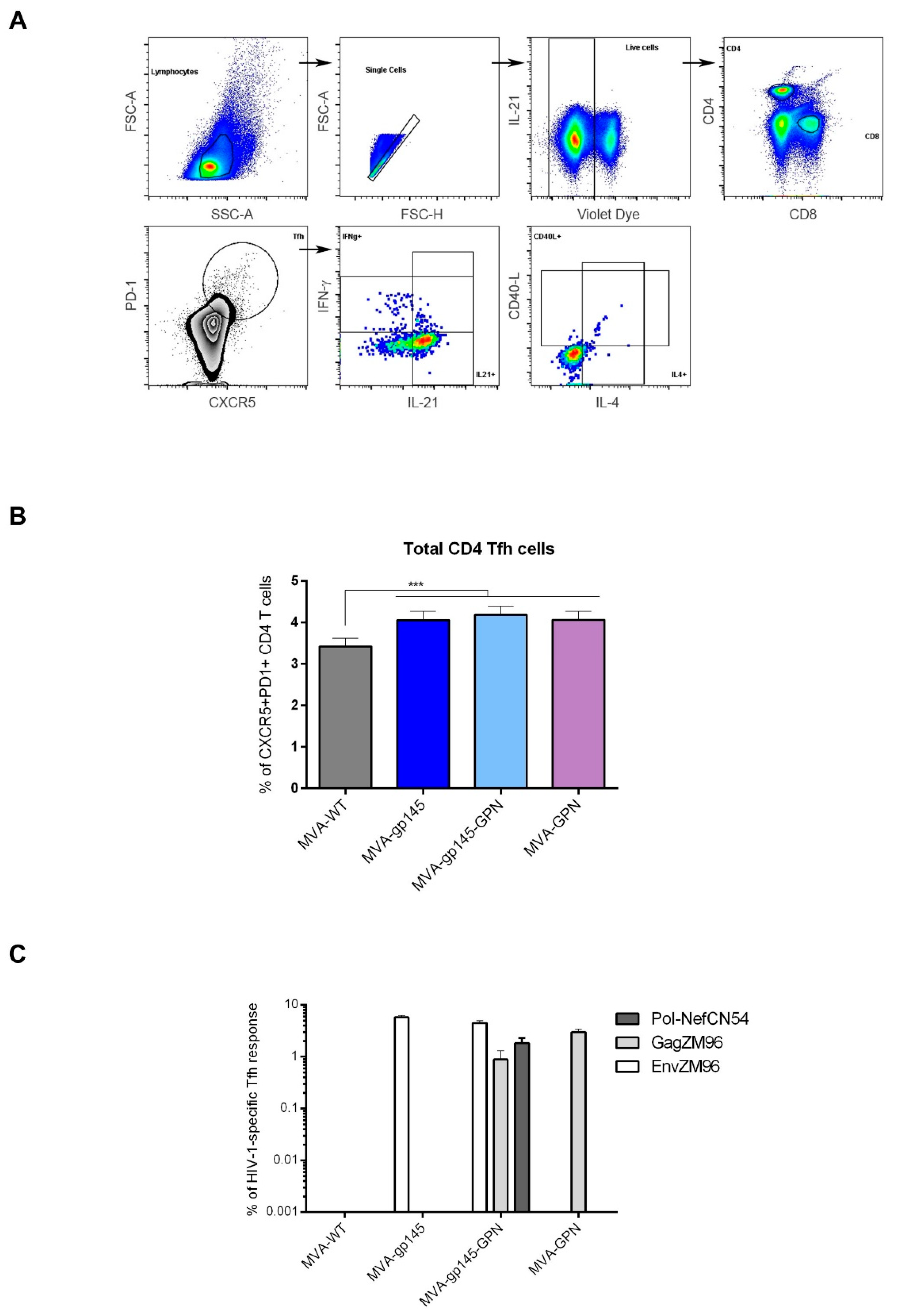
2.14.2. Analysis of HIV-1-Specific Tfh Cell Responses
To analyze the magnitude and phenotype of the HIV-1-specific Tfh cell immune responses, 2 × 106 splenocytes (erythrocyte-depleted) were seeded on 96-well plates and stimulated for 6 h in supplemented RPMI 1640 medium with 10% FCS, 1 µL/mL Golgiplug (BD Biosciences), anti-CD154 (CD40L)-Biotin (BD Biosciences) and 5 µg/mL of HIV-1 clade C ZM96 gp140 (Env-1 + Env-2 + Env-3) or Gag peptide pools or 1 µg/mL of HIV-1 clade C CN54 Pol-Nef (Gag-Pol + Pol-1 + Pol-2 + Nef) peptide pools. After stimulation, the splenocytes were washed, stained for surface markers, permeabilized (Cytofix/Cytoperm kit; BD Biosciences), and stained intracellularly with appropriate fluorochromes. For the analysis of HIV-1-specific Tfh cell responses, the following fluorochrome-conjugated antibodies were used: CD4-Alexa700, CD8-V500, CD154 (CD40L)-Biotin/Avidin-PE, IL-4-Alexa488, IFN-γ-PeCy7 and IL-21-APC for functional analyses and CXCR5-PECF594, PD1 (CD279)-APCefluor780 and CD44-PeCy5 (SPRD) for phenotypic analyses. All of the antibodies were from BD Biosciences. The dead cells were excluded using the violet LIVE/DEAD stain kit (Invitrogen).
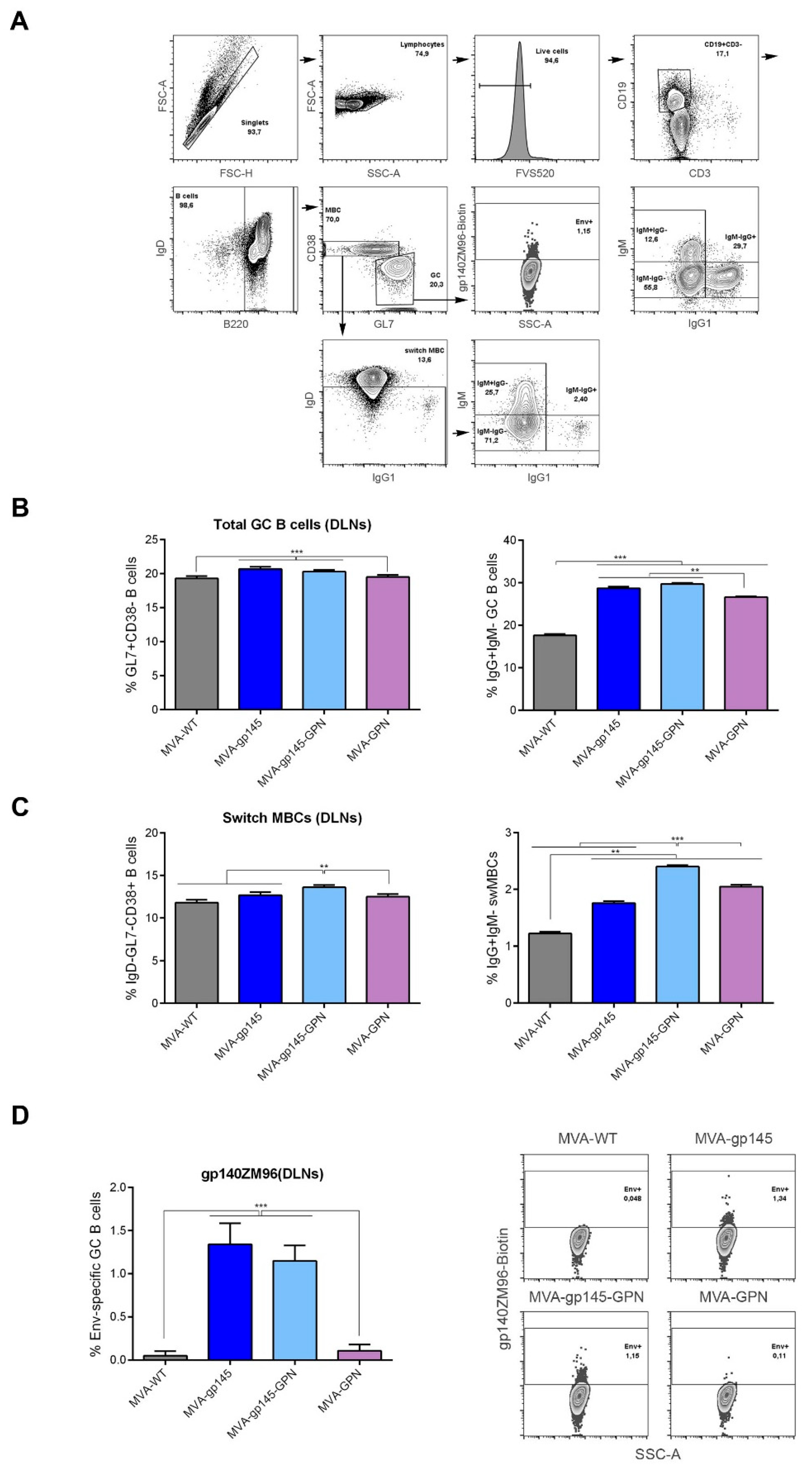
2.14.3. Analysis of gp140-Specific Germinal Center (GC) B Cell Responses
For the analysis of the magnitude and phenotype of the HIV-1 gp140-specific B cell immune responses, 2 × 106 cells from the draining lymph nodes were seeded on 96-well plates, centrifuged, and the dead cells were stained by incubation with Fixable Viability Stain 520 (FVS 520) (BD Biosciences). After blocking the Fc receptors with anti-CD16/CD32 (BD Biosciences), the cells were incubated with 0.3 µg/106 cells of biotinylated ZM96gp140 protein (University of Regensburg, Regensburg, Germany; Prof. Dr. Ralf Wagner) for 30 min. at 4 °C in the dark. After washing, the cells were stained with the following fluorochrome-conjugated antibodies for surface markers: CD3-FITC, B220-PECy7, IgD-APCH7, CD38-PerCPCy5.5, IgG1-BV421, GL7-Alexa647, IgM-PECF594 and CD19-Alexa700 (all from BD Biosciences).
For the analysis of the HIV-1- and VACV-specific T and B cell responses, cells from immunized animals were acquired in a GALLIOS flow cytometer (Beckman Coulter) and analyses of the data were performed using FlowJo software (Version 10.4.2; Tree Star, Ashland, OR, USA). Lymphocyte-gated events ranged between 105 and 5 × 105. After lymphocyte gating, Boolean combinations of single functional gates were generated using FlowJo to quantify the frequency of each response based on all the possible combinations of differentiation markers or cytokine expression. For each specific functional combination, background responses in the non-stimulated controls (RPMI) were subtracted from those that were obtained in stimulated samples, and the percentages of cytokine-producing cells in the control groups were also subtracted from the rest of the groups to eliminate the non-specific responses.
2.15. Antibody Measurement by Enzyme-Linked ImmunoSorbent Assay (ELISA)
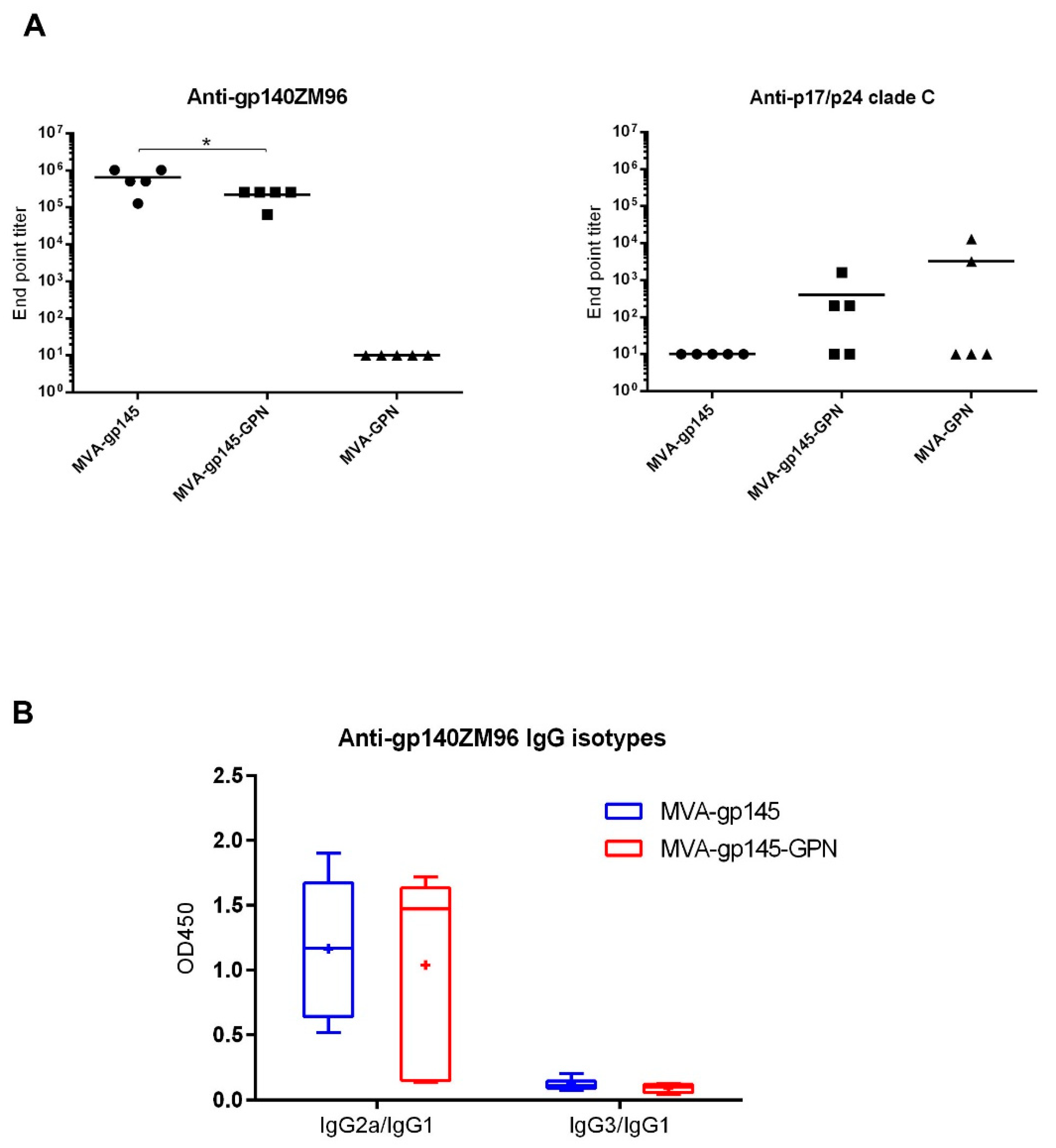
As previously described, ELISA was used to assess antibody binding to gp140 and GPN proteins in serum [
19]. Individual sera from immunized mice were two-fold serially diluted in duplicate and then incubated with 0.9 µg/mL of recombinant clade C ZM96gp140 purified protein (University of Regensburg, Regensburg, Germany; Prof. Dr. Ralf Wagner) or 1 μg/mL of the recombinant p17/p24 protein (C-clade consensus sequence; ARP695.1; NIBSC, Centralised Facility for AIDS Reagents, United Kingdom). The levels of binding anti-HIV-1 total IgG antibodies were defined as the last serum dilution that gave three times the mean OD
450 value of the control group (end point titer). The IgG1, IgG2a or IgG3 antibody isotypes against Env were determined for each individual serum diluted at 1:64,000.
2.16. Data Analysis and Statistics
For the statistical analysis of flow cytometry data using a panel of human bNAbs, we report the geometric mean of the fluorescence and compute the 2.5% and 97.5% percentiles of the fluorescence distribution.
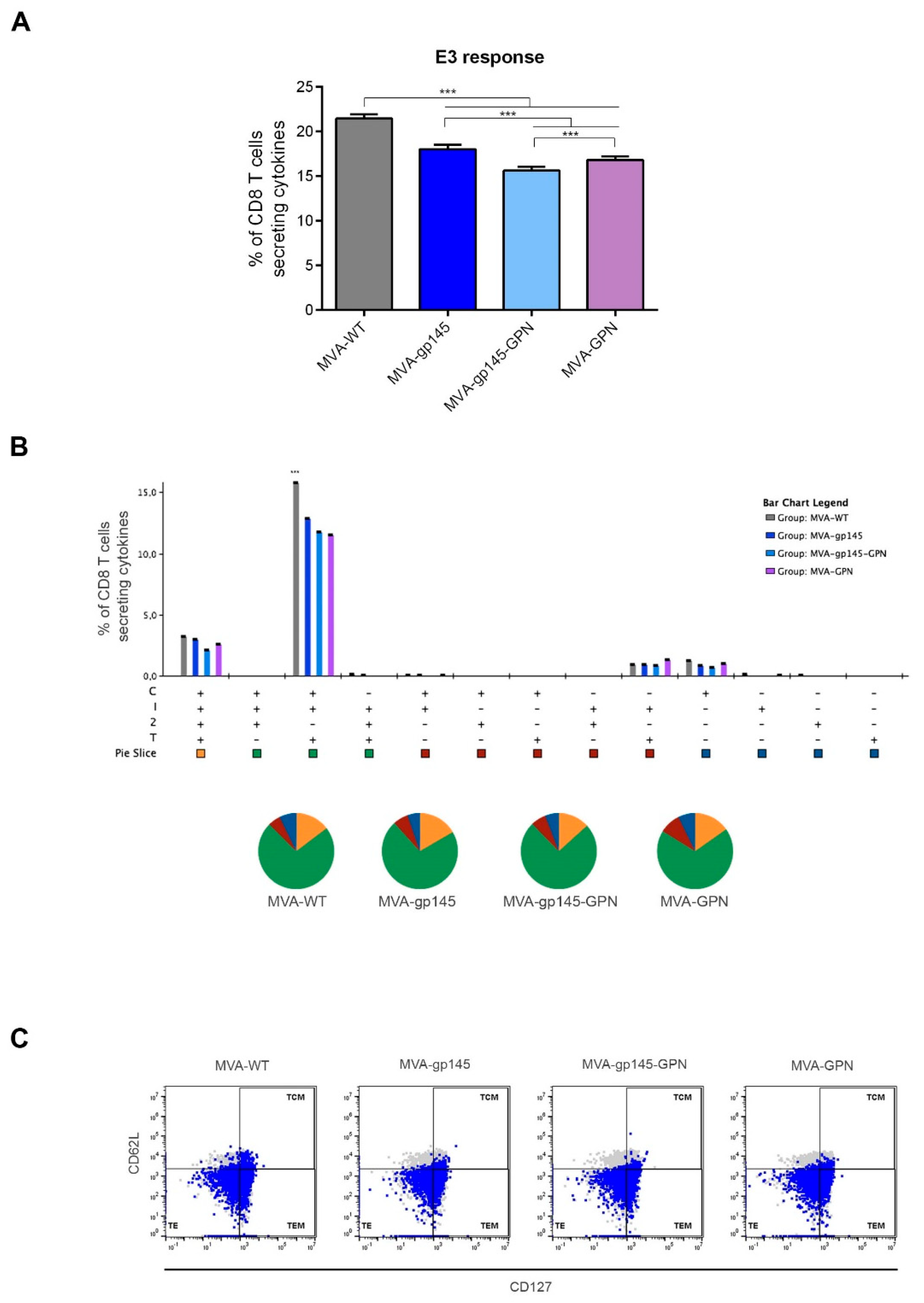
For the statistical analysis of the data obtained by ICS, an approach that corrects the values for the unstimulated controls (RPMI) and permits the calculation of confidence intervals and
p values of hypothesis tests was applied [
20]. Only antigen responses that were significantly higher than the corresponding RPMI values are depicted and the background that was obtained for the different cytokines in the non-stimulated controls never exceeded 0.05%. Analysis and presentation of the distribution of the polyfunctional responses were performed using SPICE version 5.1, which was downloaded from
http://exon.niaid.nih.gov [
21]. A comparison of distributions was performed using a Student’s T test and a partial permutation test, as previously described [
21]. All of the values used for analyzing the proportionate representation of responses are background-subtracted.
For the statistical analysis of ELISA data, a one-way ANOVA test that was followed by Tukey’s honest significant difference criterion was performed.
4. Discussion
Over three decades since the discovery of HIV virus as the AIDS causal agent, the development of an effective vaccine remains one of the major public health challenges of our society. Even though life expectancy of HIV-infected individuals has significantly improved due to the implementation of antiretroviral therapies, the development of a highly effective HIV/AIDS vaccine is essential in order to ultimately eradicate the HIV. Recombinant poxvirus vectors (including MVA, ALVAC or NYVAC viruses) have been extensively used in preclinical and clinical trials as vaccine candidates against HIV-1, being demonstrated to be safe and immunogenic. However, an ideal immunogen that is capable of inducing high protection by immunization has yet to be identified.
In the present study, we described the generation and preclinical evaluation in mice of new single and double MVA-based vaccine candidates, which expressed optimized HIV-1 clade C membrane-bound gp145(ZM96) trimeric protein and/or Gag(ZM96)-Pol-Nef(CN54) (GPN) polyprotein processed to form Gag-induced virus-like particles (VLPs). The HIV-1 clade C is highly prevalent in developing countries, like India and Southern Africa, where eradication efforts are further complicated by economic and cultural challenges.
Since vaccine-induced protective immunity is critically determined by HIV-1 Env conformation, the gp145 antigen was designed to be expressed as a membrane-bound trimeric protein resembling the native conformation. It has been shown that soluble recombinant native-like Env trimeric proteins are able to induce strong HIV-1-specific B cell and antibody responses, enhancing the development of broadly neutralizing antibodies (bNAbs) [
30]. Additionally, Gag, Pol and Nef antigens were designed to be expressed as Gag-induced VLPs with the aim of favouring both the B and T cell immune responses, as it has been previously described by Böckl and colleagues [
31]. The inclusion of both HIV-1 antigens in the same vector aims to potentiate and broaden the HIV-1-specific immune responses and to avoid the use of two different recombinant viruses expressing each protein individually, which represents a significant advantage regarding production cost.
All of the recombinant viruses were highly stable in cell culture and expressed high levels of HIV-1 gp145 and/or Gag as VLPs. Furthermore, we demonstrated that the insertion of foreign genes into the viral genome did not affect the virus replication of the singles MVA-gp145 and MVA-GPN or the double MVA-gp145-GPN viruses, since they grew as efficiently as the parental MVA-WT in CEF cells. In addition, the VLPs contain Env bound to the particles, as visualized by immunogold electron microscopy of purified VLPs from MVA-gp145-GPN-infected cells. Moreover, gp145 and Gag antigens were also detected in purified MVA virions and they were localized in different compartments, with gp145 being mainly found in the virus-membrane fraction and Gag in the insoluble Core fraction. The HIV-1 antigens that were incorporated within the recombinant MVA virus particles is an added advantage, as they could also contribute to promote immune responses to these antigens soon after virus infection.
The presence of properly processed and folded Env on the cell surface of the infected cells was demonstrated by flow cytometry using a panel of bNAbs that bind to the relatively conserved and functionally relevant regions of Env. These epitopes, called vulnerability sites, were exposed on the membrane-bound gp145 protein that was expressed by the single MVA-gp145 or the double MVA-gp145-GPN viruses, as indicated by specific binding of almost all of the bNAbs assayed. A membrane-bound conformation of gp145 at the cell surface would likely induce more bNAbs than a cell-released soluble trimeric gp140 form.
In the BALB/c mouse model, we observed that MVA-gp145-GPN was highly immunogenic, inducing high and polyfunctional Env- and Gag-specific CD4 T cell immune responses. Similar results were obtained in an immunization study that was performed in rhesus macaques, where a mix of single recombinant NYVAC vectors expressing Env as a trimeric soluble protein or Gag-induced VLPs elicited large CD4
+ T cell responses [
10]. Interestingly, we observed that the co-expression of Env and GPN antigens by the double MVA-gp145-GPN virus enhanced the Env-specific CD4 T cell responses when compared with the single MVA-gp145 recombinant, which is possibly due to the additional incorporation of gp145 antigen into Gag-induced VLPs, but did not abrogate the specific responses against Gag, although the magnitude was significantly reduced as compared with the single MVA-GPN virus. Lower levels of HIV-1-specific CD8 T cell immune responses were detected in immunized animals, which is consistent with the inefficiency of protein antigens in engaging the class I cellular machinery that is needed to elicit effective CD8 responses. In the case of VACV-specific CD8 T cells, we observed a reduction of this type of cells in the double MVA recombinant vector as compared to MVA-WT and the single recombinant vectors, which should be advantageous in vaccination when booster doses with the same vector are needed.
In terms of humoral immune responses, MVA-gp145-GPN recombinant virus induced titers of anti-gp140 binding antibodies in the order of 10
5. Remarkably, similar levels were recently described combining MVA and simian adenoviruses expressing native-like Env soluble trimers [
32]. Another study in rabbits using a soluble recombinant HIV-1 Env protein trimer reported end point titers that were in the order of 10
4–10
5, depending of the immunization regimen used [
33]. In both studies, animals developed NAbs against tier-1 viruses and, importantly, some immunization regimens also induced autologous tier-2 NAbs responses. Moreover, we have previously described that, in non-human primates, the combination of NYVAC vectors expressing cell-released gp140 and Gag-derived VLPs as a prime, with gp140 protein as a boost, induced high titers of Env-specific binding antibodies, HIV-1 neutralizing antibodies, and ADCC responses [
10]. These studies demonstrated that immunization with trimeric Env glycoprotein could induce the generation of protective responses. Therefore, it is expected that the MVA vector co-expressing gp145 and Gag-derived VLPs would trigger similar protective responses to HIV-1 antigens, although further studies in rabbits or non-human primates should be conducted.
Follicular helper T cells (Tfh) were identified in year 2000 as the key helper CD4 T cell population that was responsible for providing help to B cells [
24,
34]. During the last years, this cellular subset has been extensively studied in the settings of vaccination or chronic HIV infection. HIV-1-specific circulating Tfh (cTfh), defined by IL-21 production, were induced at higher levels by the partly efficacious RV144 HIV vaccine than by other HIV-1 vaccine candidates that had shown no efficacy [
35]. An association between the proportion of PD-1
+ cTfh or PD-1
+CXCR3
− cTfh, and the induction of broadly neutralizing antibodies was also reported in HIV-1-infected patients [
36]. Furthermore, a high frequency of HIV-1-specific cTfh was recently associated with preserved memory B cell responses in HIV controllers [
37].
Since all of these findings suggested a key role for the Tfh response in inducing protective responses against HIV, we decided to characterize, in detail, the frequencies of total and antigen-specific Tfh cells that were elicited by the double MVA-gp145-GPN vector in comparison with the singles MVA-gp145 and MVA-GPN viruses. We found that the homologous MVA/MVA immunization regimen was able to induce high numbers of CD4 T cells with Tfh phenotype (CXCR5
+PD-1
+), of which about 70% of Tfh cells stimulated or non-stimulated were positive for IL-21. Since this marker is critically involved in the Tfh cell help to GC B cells in their process of proliferation and mutation to increase affinity of B cell responses [
24], this result suggested that MVA-based vectors might represent an advantageous platform to potentially activate this subtype of CD4 T cells. A recent study involving patients from a cohort of individuals who naturally control HIV-1 replication, reported that cTfh help likely contributes to the persistence of controller memory B cell responses, as the frequency of HIV-1-specific cTfh is correlated with the induction of HIV-1-specific antibodies in functional assays. Moreover, a positive correlation between Gag-specific cTfh and the induction of Env-specific IgG was observed [
37]. MVA-gp145-GPN elicited HIV-1-specific Tfh responses with magnitudes that were similar to those that are induced by the singles MVA-gp145 or MVA-GPN viruses. These responses were primarily directed against Env pool, followed by Gag pool correlating with the HIV-1-specific humoral immunity elicited, since higher magnitudes of Env-specific than Gag-specific IgG antibodies were detected.
Since GC B cells are the target of Tfh help and also the main source of bNAbs, we analyzed this population using the different MVA-based vectors. High frequencies of total GC B cells were observed in the DLNs of animals that received parental or recombinant MVA viruses, and Env-specific GC responses were found in animals that were immunized with viruses expressing gp145. The simultaneous expression of gp145 and GPN by MVA-gp145-GPN virus did not affect the Env-specific GC responses, since similar levels were induced in MVA-gp145- or MVA-gp145-GPN-immunized animals. Our findings are promising, since it has been reported in a study with rhesus macaques that the frequency of Env-specific Tfh cells and GC B cells correlated with the subsequent development of NAbs after infecting the monkeys. Moreover, broader antibody neutralization was associated with greater affinity maturation in memory B cells, a process where Tfh cells are a key element [
38].
The MVA vectors that are described here have added immune advantages over vectors that were derived from the NYVAC strain [
9,
11,
13]. As previously described, both MVA and NYVAC vectors differ in the extent of activation of host innate immune signals, with higher induction of the cytokine/chemokine pattern and of other gene signatures by MVA over NYVAC [
39,
40]. In addition, the replication capacity of NYVAC in non-permissive human cells is more restricted than that of MVA [
41]. Additionally, the MVA backbone has permitted, in a stable manner, the insertion of both HIV-1 antigens. Co-expression of a membrane-bound trimeric Env for the induction of bNAbs and Gag-induced VLPs to potentiate Gag-specific responses from MVA-gp145-GPN could play an active immune role in the control of viremia during acute or chronic HIV infection [
42,
43].
Overall, our study revealed that the MVA-gp145-GPN virus induced high and polyfunctional HIV-1-specific CD4 T cell responses and triggered antigen-specific Tfh and GC B cells that correlated with robust HIV-1-specific humoral responses. It represents a novel vector in the HIV vaccine field with significant immune features that are relevant for protection.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






