Mechanisms of Strain Diversity of Disease-Associated in-Register Parallel β-Sheet Amyloids and Implications About Prion Strains


1
Division of Cellular and Molecular Biology, Nagasaki University Graduate School of Biomedical Sciences, Nagasaki 852-8523, Japan
2
Center for Bioinformatics and Molecular Medicine, Nagasaki University Graduate School of Biomedical Sciences, Nagasaki 852-8523, Japan
*
Author to whom correspondence should be addressed.
†
These two authors equally contributed to the work.
Viruses 2019, 11(2), 110; https://doi.org/10.3390/v11020110
Submission received: 25 December 2018
/
Revised: 22 January 2019
/
Accepted: 23 January 2019
/
Published: 28 January 2019
(This article belongs to the Special Issue Deciphering the Molecular Targets of Prion and Prion-Like Strains)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The mechanism of prion strain diversity remains unsolved. Investigation of inheritance and diversification of protein-based pathogenic information demands the identification of the detailed structures of abnormal isoforms of the prion protein (PrPSc); however, achieving purification is difficult without affecting infectivity. Similar prion-like properties are recognized also in other disease-associated in-register parallel β-sheet amyloids including Tau and α-synuclein (αSyn) amyloids. Investigations into structures of those amyloids via solid-state nuclear magnetic resonance spectroscopy and cryo-electron microscopy recently made remarkable advances due to their relatively small sizes and lack of post-translational modifications. Herein, we review advances regarding pathogenic amyloids, particularly Tau and αSyn, and discuss implications about strain diversity mechanisms of prion/PrPSc from the perspective that PrPSc is an in-register parallel β-sheet amyloid. Additionally, we present our recent data of molecular dynamics simulations of αSyn amyloid, which suggest significance of compatibility between β-sheet propensities of the substrate and local structures of the template for stability of amyloid structures. Detailed structures of αSyn and Tau amyloids are excellent models of pathogenic amyloids, including PrPSc, to elucidate strain diversity and pathogenic mechanisms.
1. Introduction
Strain diversity is one of the most mysterious features of mammalian prions. The strain-specific traits of prions are enciphered in the structures of the abnormal isoform prion protein (PrPSc), and they are stably inherited over generations through template-directed refolding of the normal isoform prion protein (PrPC), where the template PrPSc imprints the structural details onto the substrate PrPC [1]. Moreover, the strain-specific pathogenic information encoded in the conformation of PrPSc is reproducibly “translated” into the strain-specific clinicopathological traits in manifested diseases [2,3]. This view is widely accepted as the protein-only hypothesis but detailed mechanisms, e.g., specifically what structures of PrPSc encode pathogenic information and how the information is translated, are unknown. Investigations of the storage, inheritance, and diversification of the protein-based pathogenic information warrants identification of structure–phenotype correlations; however, such a feat is difficult because detailed structures of the entire PrPSc is not available yet due to its incompatibility with high-resolution structural analyses, i.e., X-ray crystallography or nuclear magnetic resonance spectrometry (NMR). The incompatibility is attributable to difficulty in purifying without losing infectivity [4], in recapitulating bona fide PrPSc prions in-vitro with infectivity and toxicity, and relatively large sizes of PrP with post-translational modifications. Solid-state NMR (ssNMR) revealed in-register parallel β-sheet structures of in vitro-formed amyloids of the peptide corresponding to residues 23–144 [5], and ssNMR and electron paramagnetic resonance analyses have also indicated parallel in-register architectures for in vitro-formed amyloids of PrP residues 90–231 [6,7,8], but the structures of the whole molecule would be necessary for elucidation of strain diversity. Based on such data and other empirical constraints for PrPSc, Groveman et al. propounded in-register parallel β-sheet structure models of PrPSc encompassing residues 90–231 [8]. However, the validity of these models for PrPSc is yet to be determined. On the other hand, cryo-electron microscopy (cryo-EM) of PrPSc lead to the four-rung β-solenoid model [9], although the resolutions were not high enough for atomic-level modeling. Which model is more plausible is still controversial in the field. As for the translation of the strain-specific structures, interactions of PrPSc with environments and/or other proteins are essential because each strain recognizes preferable cell groups to manifest the strain-specific lesion profiles, which provide the environment and factors required for efficient propagation [10,11,12].
Many clinically important neurodegenerative diseases are associated with amyloids, e.g., Alzheimer’s disease (AD) with β-amyloid and Tau amyloid; Parkinson’s disease (PD) and dementia with Lewy bodies (DLB) with α-synuclein amyloids; and tauopathies, including Pick’s disease, with Tau amyloids. Interestingly, they show “prion-like properties,” including transmissibility to animals and even strain diversity [13,14,15]. Although they have no amino acid homology, those amyloids share a common basic architecture, in-register parallel β-sheet structure [14,16,17], which is characterized by a stack of β-loop-β motifs with identical groups of amino acids aligned along the fibril axis with narrow 4.8 Å intervals (Figure 1A). A given residue of the amyloid therefore can interact most frequently with the identical group counterparts of the adjacent layers. This structural feature may accentuate the characteristics of each residue of the peptide and greatly contribute to amyloid conformations, e.g., hydrophobic residues make extensive hydrophobic patches spanning along the entire fibril axis, while charged residues may disorder the local structures by repulsion between the layers and be preferentially exposed to the solvent. This feature also explains why a single mutation greatly affects properties and structures of the amyloid because it replaces the entire column (Figure 1B). Why do different proteins take the same in-register parallel β-sheet structures that can efficiently propagate? The in-register amyloids are hypothesized to have lower free energy than native conformations [18]. The hydrogen bonds between the backbones of the amyloid thermodynamically favor the in-register alignment [19]. Roterman et al. regarded the in-register amyloid as a “ribbon-like micelle” in which exposed hydrophobicity at the stacked ends enable endless elongation [20]; this would hold true even in the four-rung β-solenoid model of PrPSc. Generally proteins with hydrophobic β-sheets exposed on the molecular surface are prone to aggregation formation. Soluble physiologically β-sheet-rich proteins have structural features to protect the solvent-facing β-sheets from undesired interactions and aggregation with other molecules [21]. Lack of such built-in protection systems in misfolded proteins, either in-register parallel β-sheets or β-solenoids, would allow uninhibited propagation of amyloids. Efficient propagation of the amyloids may stem from those thermodynamic features of the in-register parallel β-sheet structures. In-register parallel β-sheet amyloids are suitable experimental objects for molecular dynamics (MD) simulation if the structures of the amyloid are autonomously determined depending on the primary structure and thermodynamic principles.
Recent progresses in the structural analyses of disease-associated amyloids are even greater than those of PrPSc prions, partly because of their relatively small size, lack of post-translational modification, and in-vitro reproducibility of the cytotoxicity and infectivity of the amyloids [22]. Resolutions of the structures of those in-register amyloids determined by ssNMR or cryo-EM [14,17,23,24,25] are high enough to allow analyses of structure–phenotype relations. Here, we review recent advances in investigations of the structure and strain diversity of disease-associated in-register parallel β-sheet amyloids. Moreover, we herein introduce our in-silico data on the αSyn amyloid and discuss whether obtained knowledge is applicable to PrPSc. To note, we particularly focus on relatively large pathogenic amyloids, such as α-synuclein and Tau.
2. Progress in Investigation of Tau Amyloids
The Tau protein is abundantly expressed in the nervous system, especially in nerve cell axons, and intrinsically disordered with a low content of secondary structures [26]. Tau has six isoforms depending on patterns of alternative splicing of exons 2, 3, and 10. Exon 10 corresponds to the second repeat of the four-repeat motif of the C-terminal microtubule-binding domain, and the absence or presence of exon 10 results in “three-repeat” (3R) or “four-repeat” (4R) Tau, respectively. The repeat motifs convert to an in-register parallel β-sheet structure common in tauopathies. Interestingly, which of the six isoforms preferentially converts to amyloids varies depending on disease types. For example, in Pick’s disease, the amyloids consist predominantly of the 3R-Tau, whereas AD amyloids contain both 3R- and 4R-Tau [27,28].
Recent work advanced our knowledge of the mechanism of strain diversity of amyloids, wherein cryo-EM detailed structures of two distinct Tau amyloids isolated from the brains of AD and Pick’s disease patients [14,25]. The distinct structures of the Tau amyloids demonstrated unequivocal examples of strain-specific structures. One of the notable differences between AD Tau and Pick’s Tau amyloids is the position of acute-angle β-arches, which was reminiscent of the mechanism of strain diversity of a yeast prion Sup35 postulated by Kajava et al. [29]. Interestingly, except for one β-sheet that is straight in the Pick’s fold but bent in the AD fold to make a new β-arch, many of the β-sheet regions in the AD amyloid also maintained β-sheet structure in the Pick’s amyloid, as if the positions of β-sheets are fixed, albeit with different facing partners for cross-β spine formation. The primary structures might be significant determinants of positions of β-sheets in in-register parallel amyloids, and cross-β-spine formation may be necessary to stabilize β-sheets by concealing hydrophobic residues from water [30]. Consistent with different patterns of the cross-β spines, Pick’s form and the AD form of Tau amyloids showed different proteolytic-fragment patterns on immunoblots after trypsin digestion [31].
Falcon and colleagues suggested mechanisms of strain diversity of Tau amyloids, which is applicable to other amyloids as well. They postulated that steric conflicts of the branching Cβ of Val300 of 4R-Tau with the omega-like structure formed by Pro270-Gly-Gly-Gly273 of the 3R-Tau of Pick’s amyloid makes the 4R-Tau an incompatible substrate, hampering cross-seeding of 4R-Tau by the Pick’s amyloid [27,32,33]. This type of strain barrier, due to incompatibility of local structures between the substrate monomer and the template amyloid, is discussed in detail regarding αSyn and PrPSc.
Shifts of cross-β spine pairs accompanied by alterations in the positions of β-arches enable generation of structural polymorphs, even by amyloids with relatively simple conformations like Tau amyloids. Therefore, the similar mechanisms may operate in the diversification of strains of other amyloids including PrPSc. For example, this may possibly explain the mechanism of how smaller proteolytic fragments of 12 or 13 kDa of PrPSc are produced, depending on the disease types [34,35,36]. If the fragments are produced by cleavage at protease-sensitive sites of PrPSc, their differential sizes imply a shift of these sites. Such a shift can occur via alterations of cross-β spine patterns and β-arches. For example, when conversion to β-sheets and formation of cross-β spines propagate to the whole molecule like a slider and zipper from an amyloid core/interface that is first converted by the template amyloid, it is conceivable that the position of the amyloid core/interface determines the pairing partners of the cross-β spines and that a process starting from another amyloid core results in a distinct pattern of cross-β spines. Indeed, in de novo synthesis of infectious PrPSc prions [8,37,38,39], infectious PrPSc can be induced by either type of in vitro-formed PrP fibril with a protease-resistant core in the N-terminal [40] or C-terminal region [41,42]. The Creutzfeldt-Jakob disease (CJD) with 13-kDa fragments, e.g., sporadic CJD with methionine homozygosity at the codon 129, showed shortened incubation periods in human-mouse chimeric PrP-expressing mice, corroborating the significance of structural variation in strain barriers of PrPSc prions [36,43].
3. Progress in Investigation of αSyn Amyloids
αSyn is a cytosolic protein abundantly expressed in neurons, representing ~1 % of the total cytoplasmic proteins [44]. Since αSyn was cloned as the “non-Aβ component” (NAC) from AD amyloids [45], the first-identified region encompassing residues 61–95 is often referred to as the NAC region. It is localized mainly in presynaptic nerve terminals and contributes to control of neurotransmitter releases [46]. In addition to the diversity of clinicopathological features among synucleinopathies, i.e., PD, DLB, and multiple system atrophy (MSA), familial PD shows clinical variations depending on the causative mutations in the SNCA gene, in age of onset, disease duration, and presence of pyramidal signs [47,48]. Variations in physicochemical properties of αSyn amyloids are also well-documented. For instance, in-vitro formed αSyn fibrils showed at least two types of morphologically-distinguishable strains, “ribbon” and “fibril” types, which are different in dimensions of the fibrils, blotting patterns of protease-resistant fragments, cytotoxicity, and optimal salt conditions for efficient in-vitro propagation [49,50]. Their secondary structures determined by ssNMR revealed strain-specific β-sheet distributions: the ribbon had stable β-sheet structures in the N-terminal region encompassing residues 1–38, whereas the corresponding region of the fibril type was disordered except for a short β-sheet 16–20. On the contrary, residues 44–57 were disordered in the ribbon, while they are in a β-sheet in the fibril type. Although the distal NAC region of the fibril type might be structurally varied [50], positions of β-sheets in the NAC regions were relatively similar between the ribbon- and the fibril-types [49,50,51], similar to Pick’s and AD’s Tau amyloids sharing many β-sheet regions. Besides in vitro-formed fibrils, αSyn amyloids derived from DLB- and MSA-affected brains also differed in the blotting patterns of proteolytic fragments [52]. Like the different optimal propagation conditions between the ribbon and the fibril, MSA-αSyn amyloid is formed in the milieu of oligodendrocytes, whereas DLB-αSyn amyloid is formed in the neurons [52]. Thus, αSyn amyloids can be a good model to investigate strain diversification of pathogenic amyloids because of unequivocal structural differences between strains and structure–phenotype correlations as suggested by proteolytic-fragment patterns.
Another vigorously attempted biochemical approach for structural information of αSyn amyloids is assessment of cross-seeding efficiencies between αSyn molecules with familial-PD-associated mutations, e.g., between A30P and A53T [53]. Evaluation of aggregation formation and cross-reactions of in vitro-translated GFP-fusion mutant αSyn demonstrated that αSyn mutants with A53T, H50Q, or E46K spontaneously form large aggregates and efficiently co-aggregate with each other, whereas they do not co-aggregate with the wild-type or G51D or A30P mutants. The latter group of three αSyn species formed smaller aggregates and co-aggregate with each other [54]. These findings suggested existence of two mutually exclusive aggregation paths of αSyn amyloid formation. Cross-seeding experiments demonstrated contribution of the regions N-terminal- and C-terminal to the Greek-key moiety to efficient propagation and strain-specific properties of αSyn amyloids, as demonstrated by in-vitro fibril formation and in vivo inoculation experiments with various human-mouse chimeric αSyn [55], or with N- and C-terminally truncated mutant αSyn [56]. Not only affecting cross-seeding efficiencies between αSyn molecules, strain-specific structures of αSyn amyloids can also affect interactions with other proteins. For instance, induction of Tau pathologies by αSyn preformed fibrils is a strain-specific phenomenon. Two in vitro-formed strains of αSyn amyloids with distinct proteolytic-fragment patterns exhibited different efficiencies in cross-seeding Tau and induction of phosphorylated Tau in vivo. The results demonstrated that the N-terminal region was important for the cross-seeding of Tau [57]. Whether cross-seeding of other proteins by αSyn amyloids, e.g., Aβ [58] or amylin [59], is also strain-dependent warrants further investigation.
Atomic-level structures of in vitro-formed αSyn amyloids were determined either by ssNMR or cryo-EM [17,23,24]. The conformation determined by ssNMR exhibited a “Greek-key” conformation encompassing residues 35 to 99 with intricate interactions among the constituent β-sheets [17]. Although the majority of αSyn fibrils observed in PD brains were intertwined two-protofibril forms [60], the Greek-key αSyn amyloid was rather stable in MD simulations of 400 ns, even without another protofibril (Figure 2A,B, middle and right panels) [61]. Regardless of relative instability in the region 47–61 of the stack-end molecules, the overall stability of the amyloid stack was sufficient to evaluate the effects of various mutations, e.g., G51D or A53T, on the amyloid structures [61]. The detailed structures of αSyn amyloids determined by cryo-EM revealed co-existence of two apparently distinct types of fibrils formed under the same conditions. Both the polymorphs, “rod” and “twister”, consisted of two intertwined protofibrils and shared the common amyloid “kernel,” but they had different inter-protofibrillar interfaces [24]. The authors therefore postulated that the inter-protofibrillar interface might be an important determinant of αSyn amyloid strains. Although the rod type and the one reported by Guerrero-Ferreira et al. (PDB ID: 6h6b) had Greek-key conformations, they were slightly different from the one identified with ssNMR (PDB ID: 2n0a) in that the side chain of Ala53 pointed outward to constitute the inter-protofibril interface and that the highly-charged region 56–60 formed a β-sheet. Whether the two polymorphs represent two different strains, rather than structural polymorphs of the same strain with the same kernel, would need to be corroborated by strain-specific biological properties of each polymorph.
4. Insights from MD Simulations of αSyn Amyloids
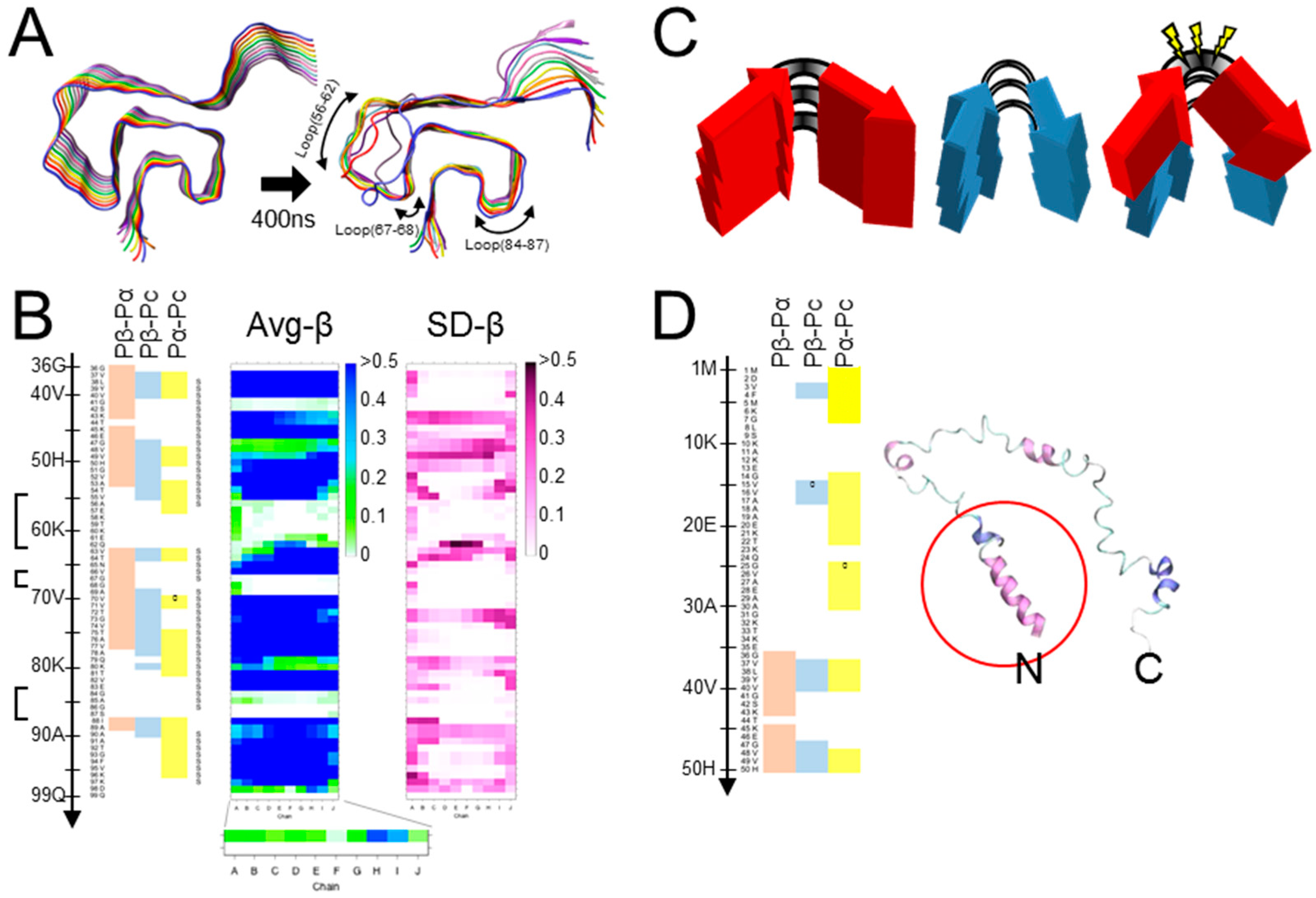
The detailed structures of Tau and αSyn amyloids suggested strain diversification by alterations in positions of β-arches, pairing patterns of cross-β spines, and inter-protofibrillar interfaces, which greatly depend on intricate interactions among the constituent β-sheets. On the other hand, the incompatibility between Pick’s Tau amyloid and 4R-Tau substrate by steric conflicts due to the branched Cβ of Val300 demonstrated that strain barriers can be also posed by such local factors. We hypothesized that conflicts between the local structures of the template amyloid and the corresponding local intrinsic propensity of the substrate peptides can cause a strain barrier, and posited that the intrinsic propensities are predictable by a neural-network secondary structure prediction algorithm (http://cib.cf.ocha.ac.jp/bitool/MIX/) [62,63]. Although the algorithm was originally designed for monomeric proteins, we thought the singular structural feature of in-register parallel β-sheet amyloids would allow for application (Figure 1A). By comparing predicted propensities, i.e., β-sheet propensity (Pβ), α-helix propensity (Pα), and coil propensity (Pc), of PrPs from various species, we previously hypothesized that compatibility of β-arches between PrPSc and PrPC contributes to species barriers (Figure 2C) [63]. Here we test the hypothesis of compatibility between the local conformations and the propensity of substrates with MD simulation, which enable observation of the influences exclusively of the conformations on behaviors of the peptides without any interference from other factors or proteins.
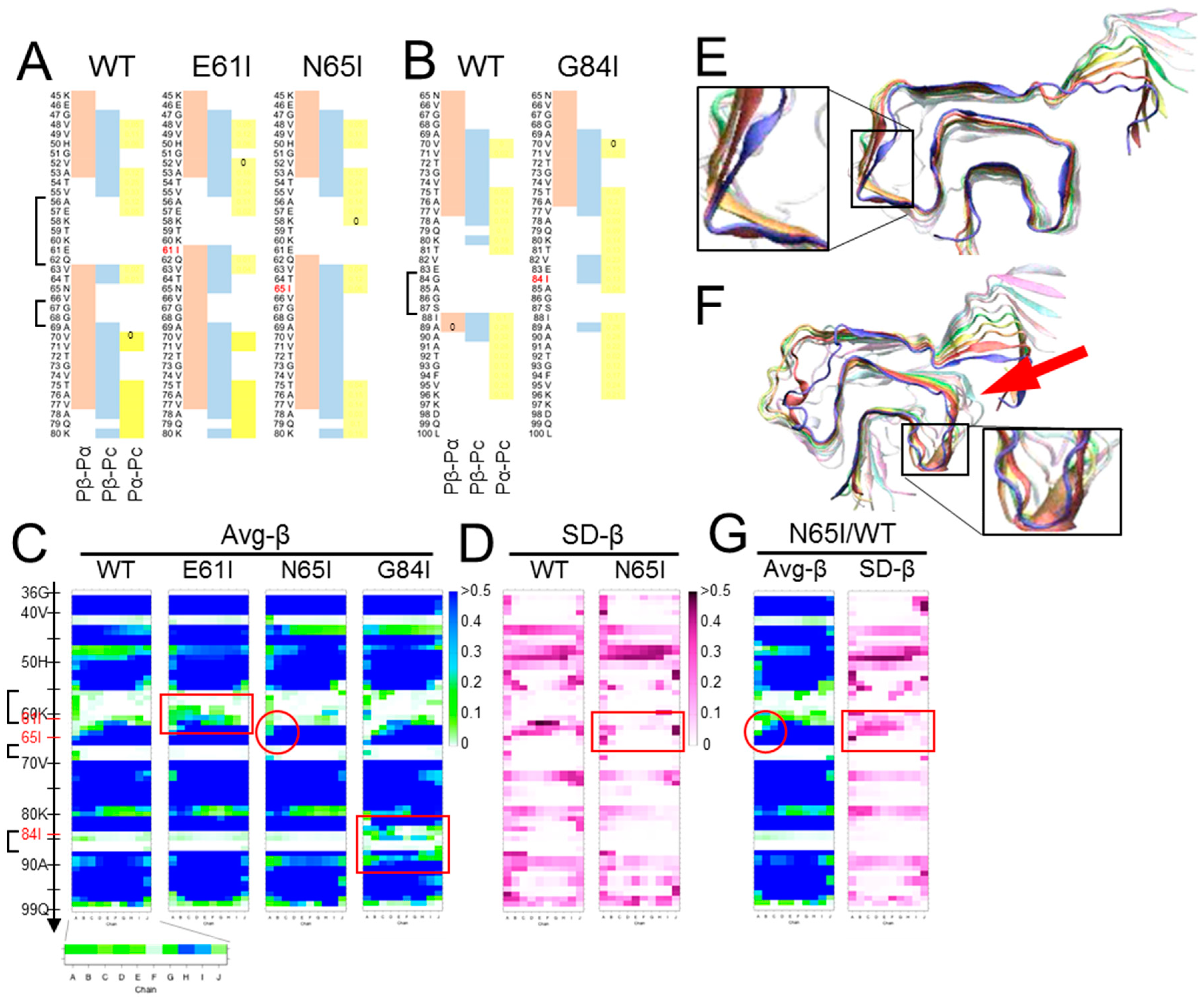
We chose the “Greek-key” αSyn amyloid (PDB ID: 2n0a) [17] as a representative model of an in-register parallel β-sheet amyloid, because of its intricately-interacting β-sheets, absence of proline, and paucity of aromatic amino acids, wherein its π–π interactions are not fully represented by the used force field. First, we compared predicted propensities with the results of MD simulations of the wild-type (WT) αSyn amyloid [61] to assess the relationship of the predicted propensities and the actual structures of αSyn amyloids in silico. Here, we used a set of modified parameters, Pβ-Pα, Pβ-Pc, and Pα-Pc, to clearly visualize magnitude correlations between two of the three conventional parameters, while the algorithm originally assigns a secondary structure of the highest value to a given residue, to assess whether the magnitude correlations between two have any significance on the structures. These predicted propensities exhibited some correlation with the actual average β-sheet propensity from the MD simulations (Figure 2B), suggesting certain influences of intrinsic propensities on structures of the amyloid. Positive (Pα-Pc) values (Figure 2B, left panel) indicated high α-helix propensities in theory, and indeed the N-terminal region with positive (Pα-Pc) values maintained a helix in the MD simulation of a native αSyn (PDB ID: 2kkw [64]), even in the absence of micelles (Figure 2D). The β-sheet in the region 89-95 with positive-(Pα-Pc) values (Figure 2B) yielded a similar contribution of high α-helix propensities to amyloid formation as reported in the literature, which explains why amyloidogenic proteins often have α-helices in native conformations, e.g., Aβ and PrP [65,66,67,68]. Given the correlation of the predicted propensity profiles with the structures of αSyn amyloid in silico, next, we introduced substitution mutations of isoleucine around the three loops encompassing residues 56–62 [loop(56–62)], 67–68 [loop(67–68)], and 84–87 [loop(84–87)] (Figure 2A), specifically Glu61Ile (E61l), Asn65Ile (N65I), and Gly84Ile (G84I), respectively (Figure 3A,B). Then, we analyzed their predicted propensities. G61I locally raised (Pβ-Pα) and (Pβ-Pc) values (Figure 3A, middle). N65I raised (Pβ-Pc) values in the loop (67–68) and the adjacent β-strands (Figure 3A, right). G84I raised (Pα-Pc) values to positivity through the loop and changed the positive-(Pβ-Pc) spot at the residue 80 to the wider one encompassing 82–84 (Figure 3B).
MD simulations of homo-oligomers of mutant αSyn were performed with the same conditions as outlined in [61] and briefly described in the Supplementary Protocol and Figure S1. Influences of N65I were subtle but the β-sheet encompassing 62–66 was more stabilized particularly on the chain-A side (Figure 3C, red circle) with smaller SD-β values than those of WT (Figure 3D). E61I occasionally induced β-strands in the loop (56–62) and tended to stabilize the amyloid stack (Figure 3E, inset). In contrast, G84I substantially destabilized the loop (84–87) and the adjacent structures (Figure 3F, arrow). In accordance with the raised local β-sheet propensity, β-strands were temporarily induced in loop (84–87) (Figure 3F, inset) but could not form a stable β-sheet. Those varied effects of isoleucine substitutions suggested that raised β-sheet propensity either stabilizes or destabilizes the loops depending on their shapes, lengths, flexibility, and orientation of the side chain; the side chain of N65I pointed outward of the β-arch, while those of E61I and G84I inward. A “U-shaped” loop with small turning radius like loop (84–87) might be destabilized because the space inside the β-arch is too small to accommodate the side chain of Ile with Cβ branching inside. This provides a proof-of-concept to the hypothesis that incompatibility between the intrinsic propensities of the substrate with the actual local structures of the template amyloid affects stability of the amyloid [63]. To note, the amyloids contained only mutant αSyn without any mismatch in the primary structures among the peptides in the stack. In seeding reactions of amyloids or PrPSc prions, homology in the primary structures between the seed and the substrate is an important determinant of seeding efficiency; however, the present result corroborated that compatibility of intrinsic propensity of the substrate with local structures of the amyloid is more important than the sequence homology. This helps to interpret the singular inter-species transmissions of PrPSc prions wherein even secondary transmissions are inefficient, despite the homology between the PrPSc in the inoculum and the host PrP [69,70]. Interestingly, the stabilizing effects of E61I or N65I in the homo-oligomers were affected in hetero-oligomers by the combined WT αSyn (Figure 3G). As peptides in an amyloid are incessantly moving in a fine vibratory manner with a twisting tendency, the hetero-oligomers might have discordance in the motions between the heterologous peptides that eventually lead to destabilization.
5. Significance of Stack-End Molecular Behavior
MD simulations of various mutant αSyn amyloids demonstrated that behaviors of the stack-end molecules, i.e., which β-sheets of the stack-ends are stable, were highly varied depending on the primary structures, despite being in the same conformation [61]. Whether the behaviors of the stack-end molecules (Figure 2B, chain A or J) affect incorporation of the substrate to the stack to pose a strain barrier is yet to be investigated; however, if they do, they could modulate levels of barriers without substantial conformational changes depending on environment, e.g., pH or salt strength. Indeed, seeding reactions of amyloids are often affected by reaction conditions [49]. This may also explain why different strains have apparently similar secondary structures [71,72,73].
6. Implications about PrPSc Prion
We previously investigated inter-species transmissions of mammalian prions with a focus on PrPSc as an in-register parallel β-sheet amyloid [63]. Indeed, in-register parallel β-sheet amyloids show many similar properties to PrPSc. For example, the existence of different proteolytic-fragment patterns of Tau and αSyn amyloids [31,49,52] are reminiscent of the 19-kDa and 21-kDa fragments of PrPSc prions [74]. Besides, those polymorphs of Tau and αSyn amyloids are associated with distinct clinicopathological features [14,52]. These proteins are good surrogate models for PrPSc and may elucidate mechanisms of strain diversification and translation of strain-specific structures, particularly because of the aforementioned advantages in experiments.
MD simulations of the mutant αSyn demonstrated that inappropriately high intrinsic β-sheet propensity of loops in certain shapes, e.g., a U-shape, can destabilize the local structures of the amyloid. Inversely, local structures of a given amyloid might be inferable from mutations around putative loop regions; if a mutation that raises local β-sheet propensity inhibits incorporation of the mutant peptide into the amyloid, the local structure around the mutation could be a U-shaped loop which cannot accommodate newly-induced β-sheets or the bulky side chain with Cβ branching. Interestingly, the human PrP with an anti-prion polymorphism Val127 [75] shows positive (Pβ-Pc) values at the residues 126–127 where the PrP with Gly127 has negative values (Figure 4A). As residue 127 is located at the end of the flexible region intervening two high-(Pβ-Pc) regions, AVVGGLGG127YMLGS, like the residue 84 of the αSyn amyloid, it may also affect structures of the loop and destabilize the surrounding structures. Moreover, as other residues associated with strain barriers are often located near presumably flexible regions, e.g., codon 129 of human PrP [74] and codon 136 of ovine PrP [76], the similar mechanism might affect the heterologous transmission efficiencies as well. Notably, in the Groveman’s in-register parallel β-sheet model of PrPSc, i.e., PIRIB model, residue 129 is located in the middle of a long straight stretch [8]. If minimal surface area of a protofibril of amyloid is thermodynamically favorable as in a micelle, curling like a Greek-key conformation is advantageous in this regard. Given the relative instability in the N-terminal region of the PIRIB model [8,42], an in-register parallel β-sheet model of PrPSc with a β-arch around residue 129 might be worth considering. On the other hand, the Silva’s four-rung β-solenoid model of PrPSc has a β-arch centering the Leu125 [77]. Although theoretically its structural stability can be similarly affected by Val127 as in the in-register parallel amyloid model, low sequence homology between layers of β-arch of the β-solenoid model may modify the effects, just as influences of a mutation were masked in heterologous β-arches in the hetero-oligomer αSyn amyloid (Figure 3G). Therefore, investigations specifically into β-solenoids for evaluation of influences of mutations are necessary to test this view.
The region between the first and second α-helices (H1~H2) can be one of the main interfaces between substrate PrPC and template PrPSc, i.e., their first interaction site [78]. Although the predicted β-sheet propensities of this region are not necessarily high because of coil propensity (Figure 4B, left), the conformation of native PrPC possibly restricts the mobility of H1~H2 region to make it more prone to β-sheet formation in effect than the predicted propensity. Remarkably, predicted β-sheet propensities of H1~H2 are highly varied among species (Figure 4B), and the mismatches in the propensities in H1~H2 may underlie species barriers by hampering stable β-sheet formation between the PrPSc and PrPC monomer; if the interface cannot convert to stable β-sheet structures, the conversion reaction would not spread to the rest of the molecule. Other regions with high β-sheet propensities, e.g., second or third α-helix, are stably structured in the native PrPC, and unsuitable for interfaces because they demand the energy to unfold before interaction with the template PrPSc. There can be another interface in more N-terminal regions. Considering that Pro102Leu substitution of Gerstman-Sträussler-Scheinker syndrome (GSS) raises local β-sheet propensities in the N-terminal-side region, the 7-kDa protease-resistant fragment of GSS derived from the N-terminal region [79] may reflect preferential interactions of the region with PrPSc as a consequence of the raised local propensity. Interestingly, Fukuoka-1 mouse-adapted prion, which was originally derived from GSS, has distinct structures in H1~H2 from that of other mouse-adapted strains RML or 22L [80]. If pairing patterns of cross-β spines depend on positions of the initiating amyloid core as hypothesized above, the differences might be explainable: β-sheets of H1~H2 form cross-β spines with the C-terminal region of PrP in the latter strains, while they form cross-β spines with another region in PrPSc of GSS.
7. Implications and Conclusions
After strain-specific structures of amyloids are unveiled, the next question to be addressed is how those structures are translated into strain-specific clinicopathological features. The induction of conformational changes by pathogenic amyloids may also occur to non-amyloidogenic proteins which have optimal intrinsic propensities, via cross-seeding reactions between non-amyloidogenic protein and amyloids. If the same principles as that for between amyloidogenic proteins operate, non-amyloidogenic proteins which preferentially interact with a given amyloid might be predictable from the primary structure.
Herein, we reviewed recent advances in the structures and mechanisms of strain diversity of in-register parallel β-sheet amyloids, particularly Tau and αSyn, and discussed implications regarding PrPSc prions. Strain diversification of in-register parallel β-sheet amyloids seems to have multiple mechanisms due to intricate interactions between the β-sheets and local structures. As in-silico methodology proves to be useful presumably because of the singular structural features, more proactive use of it would foster further advances in our knowledge.
Supplementary Materials
The following are available online at https://www.mdpi.com/1999-4915/11/2/110/s1, Figure S1: Supplementary information about the MD simulation., Table S1: Raw data for the heatmaps, Supplementary Protocols: Protocols for the MD simulation.
Author Contributions
Conceptualization, Y.T.; Methodology, H.O.; Software, H.O.; Validation, Y.T. and N.N.; Formal Analysis, Y.T. and H.O.; Investigation, H.O. and Y.T.; Resources, H.O. and N.N.; Data Curation, H.O. and Y.T.; Writing – Original Draft Preparation, Y.T.; Writing – Review & Editing, Y.T. and N.N.; Visualization, H.O. and Y.T.; Supervision, N.N.; Project Administration, Y.T.; Funding Acquisition, H.O., Y.T. and N.N.
Acknowledgments
We thank Kei Yura, Ochanomizu University, for generously allowing us to use the neural network secondary structure prediction algorithm on his website. The numerical calculations were carried out on the TSUBAME2.5/3.0 supercomputer at the Tokyo Institute of Technology and the Reedbush-U supercomputer at the Information Technology Center, the University of Tokyo. This work was supported by the “TSUBAME Encouragement Program for Young/Female Users” of Global Scientific Information and Computing Center at the Tokyo Institute of Technology, the “Initiative on Promotion of Supercomputing for Young or Women Researchers” from the Information Technology Center, the University of Tokyo, and Takeda Science Foundation (www.takeda-sci.or.jp/).
Conflicts of Interest
The authors declare no conflicts of interest, either financial or non-financial, associated with this manuscript.
References
- Bessen, R.A.; Kocisko, D.A.; Raymond, G.J.; Nandan, S.; Lansbury, P.T.; Caughey, B. Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature 1995, 375, 698–700. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Telling, G.C.; Parchi, P.; DeArmond, S.J.; Cortelli, P.; Montagna, P.; Gabizon, R.; Mastrianni, J.; Lugaresi, E.; Gambetti, P.; Prusiner, S.B. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 1996, 274, 2079–2082. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.A.; Ward, A.; Race, B.; Priola, S.A. Processing of high-titer prions for mass spectrometry inactivates prion infectivity. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Theint, T.; Xia, Y.; Nadaud, P.S.; Mukhopadhyay, D.; Schwieters, C.D.; Surewicz, K.; Surewicz, W.K.; Jaroniec, C.P. Structural Studies of Amyloid Fibrils by Paramagnetic Solid-State Nuclear Magnetic Resonance Spectroscopy. J. Am. Chem. Soc. 2018, 140, 13161–13166. [Google Scholar] [CrossRef] [PubMed]
- Cobb, N.J.; Sönnichsen, F.D.; McHaourab, H.; Surewicz, W.K. Molecular architecture of human prion protein amyloid: A parallel, in-register beta-structure. Proc. Natl. Acad. Sci. USA 2007, 104, 18946–18951. [Google Scholar] [CrossRef] [PubMed]
- Tycko, R.; Savtchenko, R.; Ostapchenko, V.G.; Makarava, N.; Baskakov, I.V. The α-helical C-terminal domain of full-length recombinant PrP converts to an in-register parallel β-sheet structure in PrP fibrils: Evidence from solid state nuclear magnetic resonance. Biochemistry 2010, 49, 9488–9497. [Google Scholar] [CrossRef] [PubMed]
- Groveman, B.R.; Dolan, M.A.; Taubner, L.M.; Kraus, A.; Wickner, R.B.; Caughey, B. Parallel in-register intermolecular β-sheet architectures for prion-seeded prion protein (PrP) amyloids. J. Biol. Chem. 2014, 289, 24129–24142. [Google Scholar] [CrossRef]
- Vázquez-fernández, E.; Vos, M.R.; Afanasyev, P.; Cebey, L.; Sevillano, A.M.; Vidal, E.; Rosa, I.; Renault, L.; Ramos, A.; Peters, P.J.; et al. The Structural Architecture of an Infectious Mammalian Prion Using Electron Cryomicroscopy. PLoS Pathog. 2016, 12, e1005835. [Google Scholar] [CrossRef]
- Mahal, S.P.; Baker, C.A.; Demczyk, C.A.; Smith, E.W.; Julius, C.; Weissmann, C. Prion strain discrimination in cell culture: The cell panel assay. Proc. Natl. Acad. Sci. USA 2007, 104, 20908–20913. [Google Scholar] [CrossRef] [Green Version]
- Marbiah, M.M.; Harvey, A.; West, B.T.; Louzolo, A.; Banerjee, P.; Alden, J.; Grigoriadis, A.; Hummerich, H.; Kan, H.; Cai, Y.; et al. Identification of a gene regulatory network associated with prion replication. EMBO J. 2014, 33, 1527–1547. [Google Scholar] [CrossRef] [PubMed]
- Herbst, A.; Banser, P.; Velasquez, C.D.; Mays, C.E.; Sim, V.L.; Westaway, D.; Aiken, J.M.; McKenzie, D. Infectious prions accumulate to high levels in non proliferative C2C12 myotubes. PLoS Pathog. 2013, 9, e1003755. [Google Scholar] [CrossRef]
- Stöhr, J.; Condello, C.; Watts, J.C.; Bloch, L.; Oehler, A.; Nick, M.; DeArmond, S.J.; Giles, K.; DeGrado, W.F.; Prusiner, S.B. Distinct synthetic Aβ prion strains producing different amyloid deposits in bigenic mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10329–10334. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.; Zhang, W.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Morales, R.; Callegari, K.; Soto, C. Prion-like features of misfolded Aβ and tau aggregates. Virus Res. 2015, 207, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Rohou, A.; Lasker, K.; Yadav, J.K.; Schiene-fischer, C.; Fändrich, M.; Grigorieffb, N. Peptide dimer structure in an Aβ(1–42) fibril visualized with cryo-EM. Proc. Natl. Acad. Sci. USA 2015, 112, 11858–11863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, A.J.; Knowles, T.P.J.; Tartaglia, G.G.; Fitzpatrick, A.W.; Devlin, G.L.; Shammas, S.L.; Waudby, C.A.; Mossuto, M.F.; Meehan, S.; Gras, S.L.; et al. Metastability of native proteins and the phenomenon of amyloid formation. J. Am. Chem. Soc. 2011, 133, 14160–14163. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Ghanati, E.; Schmit, J.D. Theory of Sequence Effects in Amyloid Aggregation. J. Phys. Chem. B 2018, 122, 5567–5578. [Google Scholar] [CrossRef] [PubMed]
- Roterman, I.; Banach, M.; Konieczny, L. Application of the fuzzy oil drop model describes amyloid as a ribbonlike micelle. Entropy 2017, 19, 167. [Google Scholar] [CrossRef]
- Richardson, J.S.; Richardson, D.C. Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc. Natl. Acad. Sci. USA 2002, 99, 2754–2759. [Google Scholar] [CrossRef] [PubMed]
- Polinski, N.K.; Volpicelli-Daley, L.A.; Sortwell, C.E.; Luk, K.C.; Cremades, N.; Gottler, L.M.; Froula, J.; Duffy, M.F.; Lee, V.M.Y.; Martinez, T.N.; et al. Best practices for generating and using alpha-synuclein pre-formed fibrils to model Parkinson’s disease in rodents. J. Parkinsons. Dis. 2018, 8, 303–322. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Ferreira, R.; Taylor, N.M.; Mona, D.; Ringler, P.; Lauer, M.E.; Riek, R.; Britschgi, M.; Stahlberg, H. Cryo-EM structure of alpha-synuclein fibrils. Elife 2018, 7, e36402. [Google Scholar] [CrossRef]
- Li, B.; Ge, P.; Murray, K.A.; Sheth, P.; Zhang, M.; Nair, G.; Sawaya, M.R.; Shin, W.S.; Boyer, D.R.; Ye, S.; et al. Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat. Commun. 2018, 9, 3609. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M. Tau filaments in neurodegenerative diseases. FEBS Lett. 2018, 592, 2383–2391. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M. Molecular mechanisms in the pathogenesis of alzheimer’s disease and Tauopathies-Prion-Like seeded aggregation and phosphorylation. Biomolecules 2016, 6, 24. [Google Scholar] [CrossRef]
- Sergeant, N.; Bretteville, A.; Hamdane, M.; Caillet-Boudin, M.-L.; Grognet, P.; Bombois, S.; Blum, D.; Delacourte, A.; Pasquier, F.; Vanmechelen, E.; et al. Biochemistry of Tau in Alzheimer’s disease and related neurological disorders. Expert Rev. Proteom. 2008, 5, 207–224. [Google Scholar] [CrossRef]
- Kajava, A.V.; Baxa, U.; Steven, A.C. Beta arcades: Recurring motifs in naturally occurring and disease-related amyloid fibrils. FASEB J. 2010, 24, 1311–1319. [Google Scholar] [CrossRef]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.Ø.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross-β spine of amyloid-like fibrils. Nature 2005, 435, 773–778. [Google Scholar] [CrossRef]
- Taniguchi-Watanabe, S.; Arai, T.; Kametani, F.; Nonaka, T.; Masuda-Suzukake, M.; Tarutani, A.; Murayama, S.; Saito, Y.; Arima, K.; Yoshida, M.; et al. Biochemical classification of tauopathies by immunoblot, protein sequence and mass spectrometric analyses of sarkosyl-insoluble and trypsin-resistant tau. Acta Neuropathol. 2016, 131, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Saijo, E.; Ghetti, B.; Zanusso, G.; Oblak, A.; Furman, J.L.; Diamond, M.I.; Kraus, A.; Caughey, B. Ultrasensitive and selective detection of 3-repeat tau seeding activity in Pick disease brain and cerebrospinal fluid. Acta Neuropathol. 2017, 133, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Kraus, A.; Saijo, E.; Metrick, M.A.; Newell, K.; Sigurdson, C.J.; Zanusso, G.; Ghetti, B.; Caughey, B. Seeding selectivity and ultrasensitive detection of tau aggregate conformers of Alzheimer disease. Acta Neuropathol. 2018. [Google Scholar] [CrossRef]
- Notari, S.; Strammiello, R.; Capellari, S.; Giese, A.; Cescatti, M.; Grassi, J.; Ghetti, B.; Langeveld, J.P.M.; Zou, W.-Q.; Gambetti, P.; et al. Characterization of truncated forms of abnormal prion protein in Creutzfeldt-Jakob disease. J. Biol. Chem. 2008, 283, 30557–30565. [Google Scholar] [CrossRef] [PubMed]
- Zanusso, G.; Farinazzo, A.; Prelli, F.; Fiorini, M.; Gelati, M.; Ferrari, S.; Righetti, P.G.; Rizzuto, N.; Frangione, B.; Monaco, S. Identification of distinct N-terminal truncated forms of prion protein in different Creutzfeldt-Jakob disease subtypes. J. Biol. Chem. 2004, 279, 38936–38942. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Muramoto, T.; Tomoyuki, T.; Noritoshi, K.; Ironside, J.W.; Kazuo, N.; Masahito, Y.; Takeshi, S.; Mohri, S.; Tetsuyuki, K. Association of an 11–12 kDa protease-resistant prion protein fragment with subtypes of dura graft-associated Creutzfeldt-Jakob disease and other prion diseases. J. Gen. Virol. 2003, 84, 2885–2893. [Google Scholar] [CrossRef] [PubMed]
- Legname, G.; Baskakov, I.V.; Nguyen, H.-O.B.; Riesner, D.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Synthetic Mammalian Prions. Science 2004, 305, 673–676. [Google Scholar] [CrossRef]
- Wang, F.; Wang, X.; Yuan, C.-G.; Ma, J. Generating a Prion with Bacterially Expressed Recombinant Prion Protein. Science 2010, 327, 1132–1135. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-I.; Cali, I.; Surewicz, K.; Kong, Q.; Raymond, G.J.; Atarashi, R.; Race, B.; Qing, L.; Gambetti, P.; Caughey, B.; et al. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J. Biol. Chem. 2010, 285, 14083–14087. [Google Scholar] [CrossRef]
- Choi, J.; Cali, I.; Surewicz, K.; Kong, Q.; Gambetti, P.; Surewicz, W.K. Amyloid fibrils from the N-terminal prion protein fragment are infectious. Proc. Natl. Acad. Sci. USA 2016, 113, 13851–13856. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Makarava, N.; Savtchenko, R.; Baskakov, I.V. Atypical and classical forms of the disease-associated state of the prion protein exhibit distinct neuronal tropism, deposition patterns, and lesion profiles. Am. J. Pathol. 2013, 183, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Groveman, B.R.; Raymond, G.J.; Campbell, K.J.; Race, B.; Raymond, L.D.; Hughson, A.G.; Orrú, C.D.; Kraus, A.; Phillips, K.; Caughey, B. Role of the central lysine cluster and scrapie templating in the transmissibility of synthetic prion protein aggregates. PLoS Pathog. 2017, 13, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, Y.; Mohri, S.; Ironside, J.W.; Muramoto, T.; Kitamoto, T. Humanized knock-in mice expressing chimeric prion protein showed varied susceptibility to different human prions. Am. J. Pathol. 2003, 163, 2585–2593. [Google Scholar] [CrossRef]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; Rohan de Silva, H.A.; Kittel, A.; Saitoh, T. The precursor protein of non-Aβ component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef]
- Ueda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.A.C.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286. [Google Scholar] [CrossRef]
- Lautenschläger, J.; Kaminski, C.F.; Kaminski Schierle, G.S. α-Synuclein—Regulator of Exocytosis, Endocytosis, or Both? Trends Cell Biol. 2017, 27, 468–479. [Google Scholar] [CrossRef]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Pieri, L.; Madiona, K.; Alexandra, D.; Melki, R.; Verny, C.; Brice, A. G51D α-Synuclein Mutation Causes a Novel Parkinsonian—Pyramidal Syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef]
- Petrucci, S.; Ginevrino, M.; Valente, E.M. Parkinsonism and Related Disorders Phenotypic spectrum of alpha-synuclein mutations: New insights from patients and cellular models. Park. Relat. Disord. 2016, 22 (Suppl. 1), S16–S20. [Google Scholar] [CrossRef]
- Bousset, L.; Pieri, L.; Ruiz-Arlandis, G.; Gath, J.; Jensen, P.H.; Habenstein, B.; Madiona, K.; Olieric, V.; Böckmann, A.; Meier, B.H.; et al. Structural and functional characterization of two alpha-synuclein strains. Nat. Commun. 2013, 4, 2575. [Google Scholar] [CrossRef]
- Gath, J.; Bousset, L.; Habenstein, B.; Melki, R.; Böckmann, A.; Meier, B.H. Unlike twins: An NMR comparison of two α-synuclein polymorphs featuring different toxicity. PLoS ONE 2014, 9, e90659. [Google Scholar] [CrossRef]
- Verasdonck, J.; Bousset, L.; Gath, J.; Melki, R.; Böckmann, A.; Meier, B.H. Further exploration of the conformational space of α-synuclein fibrils: solid-state NMR assignment of a high-pH polymorph. Biomol. NMR Assign. 2016, 10, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Gathagan, R.J.; Covell, D.J.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.C.; et al. Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature 2018, 557, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, A.; Segers-Nolten, I.; Subramaniam, V. Conformational Compatibility Is Essential for Heterologous Aggregation of α- Synuclein. ACS Chem. Neurosci. 2016, 7, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Sierecki, E.; Giles, N.; Bowden, Q.; Polinkovsky, M.E.; Steinbeck, J.; Arrioti, N.; Rahman, D.; Bhumkar, A.; Nicovich, P.R.; Ross, I.; et al. Nanomolar oligomerization and selective co-aggregation of α-synuclein pathogenic mutants revealed by single-molecule fluorescence. Sci. Rep. 2016, 6, 37630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luk, K.C.; Covell, D.J.; Kehm, V.M.; Zhang, B.; Song, I.Y.; Byrne, M.D.; Pitkin, R.M.; Decker, S.C.; Trojanowski, J.Q.; Lee, V.M.Y. Molecular and Biological Compatibility with Host Alpha-Synuclein Influences Fibril Pathogenicity. Cell Rep. 2016, 16, 3373–3387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terada, M.; Suzuki, G.; Nonaka, T.; Kametani, F.; Tamaoka, A.; Hasegawa, M. The effect of truncation on prion-like properties of α-synuclein. J. Biol. Chem. 2018, 293, 13910–13920. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Covell, D.J.; Daniels, J.P.; Iba, M.; Stieber, A.; Zhang, B.; Riddle, D.M.; Kwong, L.K.; Xu, Y.; Trojanowski, J.Q.; et al. Distinct α-Synuclein Strains Differentially Promote Tau Inclusions in Neurons. Cell 2013, 154, 103–117. [Google Scholar] [CrossRef] [Green Version]
- Ono, K.; Takahashi, R.; Ikeda, T.; Yamada, M. Cross-seeding effcts of amyloid β-protein and α-synuclein. J. Neurochem. 2012, 112, 883–890. [Google Scholar] [CrossRef]
- Horvath, I.; Wittung-stafshede, P. Cross-talk between amyloidogenic proteins in type-2 diabetes and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2016, 113, 12473–12477. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef]
- Otaki, H.; Taguchi, Y.; Nishida, N. Molecular dynamics simulation reveals that switchable combinations of β-sheets underlie the prion-like properties of α-synuclein amyloids. bioRxiv 2018. [Google Scholar] [CrossRef]
- Qian, N.; Sejnowski, T.J. Predicting the secondary structure of globular proteins using neural network models. J. Mol. Biol. 1988, 202, 865–884. [Google Scholar] [CrossRef]
- Taguchi, Y.; Nishida, N. Secondary-structure prediction revisited: Theoretical β -sheet propensity and coil propensity represent structures of amyloids and aid in elucidating phenomena involved in interspecies transmission of prions. PLoS ONE 2017, 12, e0171974. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.N.; Jao, C.C.; Hegde, B.G.; Langen, R.; Ulmer, T.S. A combinatorial NMR and EPR approach for evaluating the structural ensemble of partially folded proteins. J. Am. Chem. Soc. 2010, 132, 8657–8668. [Google Scholar] [CrossRef] [PubMed]
- Abedini, A.; Raleigh, D.P. A critical assessment of the role of helical intermediates in amyloid formation by natively unfolded proteins and polypeptides. Protein Eng. Des. Sel. 2009, 22, 453–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pannuzzo, M.; Raudino, A.; Milardi, D.; La Rosa, C.; Karttunen, M. α-Helical structures drive early stages of self-assembly of amyloidogenic amyloid polypeptide aggregate formation in membranes. Sci. Rep. 2013, 3, 2781. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Do, T.D.; Hayden, E.Y.; Teplow, D.B.; Bowers, M.T.; Shea, J.E. Aggregation of chameleon peptides: Implications of α-helicity in fibril formation. J. Phys. Chem. B 2016, 120, 5874–5883. [Google Scholar] [CrossRef]
- Haimov, B.; Srebnik, S. The Relation between α-Helical Conformation and Amyloidogenicity. Biophys. J. 2018, 114, 1869–1877. [Google Scholar] [CrossRef] [Green Version]
- Meade-White, K.D.; Barbian, K.D.; Race, B.; Favara, C.; Gardner, D.; Taubner, L.; Porcella, S.; Race, R. Characteristics of 263K scrapie agent in multiple hamster species. Emerg. Infect. Dis. 2009, 15, 207–215. [Google Scholar] [CrossRef]
- Bian, J.; Khaychuk, V.; Angers, R.C.; Fernández-borges, N.; Vidal, E.; Meyerett-reid, C.; Kima, S.; Calvia, C.L.; Bartz, J.C.; Hoovera, E.A.; et al. Prion replication without host adaptation during interspecies transmissions. Proc. Natl. Acad. Sci. USA 2016, 114, 1141–1146. [Google Scholar] [CrossRef]
- Lemkau, L.R.; Comellas, G.; Kloepper, K.D.; Woods, W.S.; George, J.M.; Rienstra, C.M. Mutant protein A30P α-synuclein adopts wild-type fibril structure, despite slower fibrillation kinetics. J. Biol. Chem. 2012, 287, 11526–11532. [Google Scholar] [CrossRef] [PubMed]
- Lemkau, L.R.; Comellas, G.; Lee, S.W.; Rikardsen, L.K.; Woods, W.S.; George, J.M.; Rienstra, C.M. Site-Specific Perturbations of Alpha-Synuclein Fibril Structure by the Parkinson’s Disease Associated Mutations A53T and E46K. PLoS ONE 2013, 8, e49750. [Google Scholar] [CrossRef] [PubMed]
- Lv, G.; Kumar, A.; Giller, K.; Orcellet, M.L.; Riedel, D.; Fernández, C.O.; Becker, S.; Lange, A. Structural Comparison of Mouse and Human α -Synuclein Amyloid Fibrils by Solid-State NMR. J. Mol. Biol. 2012, 420, 99–111. [Google Scholar] [CrossRef]
- Parchi, P.; Giese, A.; Capellari, S.; Brown, P.; Schulz-Schaeffer, W.; Windl, O.; Zerr, I.; Budka, H.; Kopp, N.; Piccardo, P.; et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann. Neurol. 1999, 46, 224–233. [Google Scholar] [CrossRef]
- Asante, E.A.; Smidak, M.; Grimshaw, A.; Houghton, R.; Tomlinson, A.; Jeelani, A.; Jakubcova, T.; Hamdan, S.; Richard-Londt, A.; Linehan, J.M.; et al. A naturally occurring variant of the human prion protein completely prevents prion disease. Nature 2015, 522, 478–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saijo, E.; Kang, H.-E.; Bian, J.; Bowling, K.G.; Browning, S.; Kim, S.; Hunter, N.; Telling, G.C. Epigenetic dominance of prion conformers. PLoS Pathog. 2013, 9, e1003692. [Google Scholar] [CrossRef]
- Silva, C.J.; Vázquez-Fernández, E.; Onisko, B.; Requena, J.R. Proteinase K and the structure of PrPSc: The good, the bad and the ugly. Virus Res. 2015, 207, 120–126. [Google Scholar] [CrossRef]
- Taguchi, Y.; Mistica, A.M.; Kitamoto, T.; Schätzl, H.M. Critical significance of the region between Helix 1 and 2 for efficient dominant-negative inhibition by conversion-incompetent prion protein. PLoS Pathog. 2013, 9, e1003466. [Google Scholar] [CrossRef]
- Parchi, P. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann-Straussler-Scheinker disease. Proc. Natl. Acad. Sci. USA 1998, 95, 8322–8327. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, Y.; Lu, L.; Marrero-Winkens, C.; Otaki, H.; Nishida, N.; Schatzl, H.M. Disulfide-crosslink scanning reveals prion–induced conformational changes and prion strain–specific structures of the pathological prion protein PrPSc. J. Biol. Chem. 2018, 293, 14925. [Google Scholar] [CrossRef]
Figure 1.
Uniqueness of in-register parallel β-sheet structures. (A). Schematics illustrating the primary structure of a peptide (top), a layer of the peptide in β-sheet structure (lower left), a stack of the in-register parallel β-sheets of the peptide (middle). Note that the identical groups of amino acids align along the fibril axis of the amyloid with ~4.8 Å intervals, like columns of the same groups of amino acids (right). (B). An example of a substitution mutation of the peptide, Leu for Glu (top). The substitution replaces a whole column (bracket) from the negatively-charged to the hydrophobic, creating a large hydrophobic patch encompassing the entire length of the fibril.
Figure 1.
Uniqueness of in-register parallel β-sheet structures. (A). Schematics illustrating the primary structure of a peptide (top), a layer of the peptide in β-sheet structure (lower left), a stack of the in-register parallel β-sheets of the peptide (middle). Note that the identical groups of amino acids align along the fibril axis of the amyloid with ~4.8 Å intervals, like columns of the same groups of amino acids (right). (B). An example of a substitution mutation of the peptide, Leu for Glu (top). The substitution replaces a whole column (bracket) from the negatively-charged to the hydrophobic, creating a large hydrophobic patch encompassing the entire length of the fibril.

Figure 2.
Correlation between results of MD simulation and secondary-structure prediction. (A). The “Greek-key” conformation of αSyn amyloid (PDB ID: 2n0a) (left). The status of the stack of αSyn amyloid after 400 ns of MD simulation (right). The arrows indicate the three loops by or in which Ile-substitution mutations are introduced. Protocols for the MD simulations are described in [61] and in the Supplementary Protocols. The ten αSyn molecules in the stack are designated as chain A (blue, the nearest) to J (gray) (Figure S1A). (B). (Left) Propensity profiles predicted from the primary structure of αSyn by a neural network secondary structure prediction [62]. The heatmap exhibits the magnitude correlations between the conventional β-sheet propensity (Pβ), α-helix propensity (Pα) and coil propensity (Pc) by the new set of parameters, (Pβ-Pα), (Pβ-Pc) and (Pα-Pc), where zero- (“0”) and positive-value residues of each are indicated in red, blue and yellow, respectively. The three loops in Figure 2A are indicated with brackets. (Middle) A heatmap of the average β-sheet propensity (Avg-β) of αSyn amyloid observed in five independent runs of MD simulation. The color codes indicates the proportion of time when a given residue is in β-sheet structures during the runs. For all the heatmaps of Avg-β and SD-β, the vertical and horizontal axes represent the residues 36-99 and the ten chains A-J of the amyloid stack, respectively. (Right) A heatmap of the standard deviation values of β-sheet propensities (SD-β) of the αSyn amyloid observed in the MD simulation of five independent runs. (C). A schematic illustrating a hypothetical local incompatibility between the template amyloid and a heterologous substrate in a cross-seeding reaction. The red peptide has a non-flexible loop, whereas the blue peptide has a more flexible loop. When the red peptide is cross-seeded by the blue amyloid, the substrate red peptide might undergo strain from the incompatibility with the actual structure. (D). (Left) A predicted propensity profiles of the N-terminal region of αSyn encompassing the residues 1-50. (Right) A final snap shot of native-form αSyn (PDB ID: 2kkw) after 5 ns of MD simulation without any micelle: Only the N-terminal side encompassing 1-100 was used. Note that regions with high (Pα-Pc) values relatively maintain α-helix structure (red circle). N and C, N- and C-terminus, respectively.
Figure 2.
Correlation between results of MD simulation and secondary-structure prediction. (A). The “Greek-key” conformation of αSyn amyloid (PDB ID: 2n0a) (left). The status of the stack of αSyn amyloid after 400 ns of MD simulation (right). The arrows indicate the three loops by or in which Ile-substitution mutations are introduced. Protocols for the MD simulations are described in [61] and in the Supplementary Protocols. The ten αSyn molecules in the stack are designated as chain A (blue, the nearest) to J (gray) (Figure S1A). (B). (Left) Propensity profiles predicted from the primary structure of αSyn by a neural network secondary structure prediction [62]. The heatmap exhibits the magnitude correlations between the conventional β-sheet propensity (Pβ), α-helix propensity (Pα) and coil propensity (Pc) by the new set of parameters, (Pβ-Pα), (Pβ-Pc) and (Pα-Pc), where zero- (“0”) and positive-value residues of each are indicated in red, blue and yellow, respectively. The three loops in Figure 2A are indicated with brackets. (Middle) A heatmap of the average β-sheet propensity (Avg-β) of αSyn amyloid observed in five independent runs of MD simulation. The color codes indicates the proportion of time when a given residue is in β-sheet structures during the runs. For all the heatmaps of Avg-β and SD-β, the vertical and horizontal axes represent the residues 36-99 and the ten chains A-J of the amyloid stack, respectively. (Right) A heatmap of the standard deviation values of β-sheet propensities (SD-β) of the αSyn amyloid observed in the MD simulation of five independent runs. (C). A schematic illustrating a hypothetical local incompatibility between the template amyloid and a heterologous substrate in a cross-seeding reaction. The red peptide has a non-flexible loop, whereas the blue peptide has a more flexible loop. When the red peptide is cross-seeded by the blue amyloid, the substrate red peptide might undergo strain from the incompatibility with the actual structure. (D). (Left) A predicted propensity profiles of the N-terminal region of αSyn encompassing the residues 1-50. (Right) A final snap shot of native-form αSyn (PDB ID: 2kkw) after 5 ns of MD simulation without any micelle: Only the N-terminal side encompassing 1-100 was used. Note that regions with high (Pα-Pc) values relatively maintain α-helix structure (red circle). N and C, N- and C-terminus, respectively.

Figure 3.
Inappropriately high β-sheet propensity in a loop region can destabilize amyloid structures. (A) and (B). Comparison of predicted propensity profiles between WT-, E61I-, N65I- and G84I-αSyn. The mutation sites are indicated in red letters. Isoleucine substitutions were introduced near the loop regions based on the assumption that loops with higher β-sheet propensities are less flexible. The brackets indicate the three loops in Figure 2A. (C). Comparison of heatmaps of Avg-β between WT-, E61I-, N65I- and G84I-αSyn. They represent five (for WT) or three (for the others) independent simulations. Note that all the mutants have higher Avg-β values near the mutations (more green/blue cells in the red boxes for E61I and G84I, and in the red circle for N65I). (D). Comparison of the heatmaps of SD-β between WT- and N65I-αSyn. They represent five and three independent runs, respectively. Note that the region comprising N65I mutation shows more stable β-sheet with smaller SD than WT-αSyn (red box). (E). A final snap shot of E61I-αSyn after 400 ns of simulation. The mutation tended to stabilize the global structures of the amyloid and occasionally induced new β-sheets in the loop (56–62) (inset). (F). A final snap shot of G84I-αSyn after 400 ns of simulation. The mutation destabilized structures around the mutation and the adjacent regions (arrow). Occasionally β-strands were temporarily induced in the loop (inset). The differential effects of the isoleucine substitutions on the amyloid structures might be attributable to the shapes of the loops. The loop (56–62) is long and makes obtuse angles with the flanking β-strands. The loop (67–68) is short but rather flexible with two glycine residues in a row. The loop (84–87) is a U-shaped loop with a relatively-small turning radius, which could not accommodate Cβ-branching side chain of Ile. (G). Heatmaps of a hetero-oligomer amyloid containing N65I-αSyn in chain A-E and WT-αSyn in chain F-J (N65I/WT). Despite N65I mutation, Avg-β and SD-β values of chain A-F are similar to those of WT (red circle and red box).
Figure 3.
Inappropriately high β-sheet propensity in a loop region can destabilize amyloid structures. (A) and (B). Comparison of predicted propensity profiles between WT-, E61I-, N65I- and G84I-αSyn. The mutation sites are indicated in red letters. Isoleucine substitutions were introduced near the loop regions based on the assumption that loops with higher β-sheet propensities are less flexible. The brackets indicate the three loops in Figure 2A. (C). Comparison of heatmaps of Avg-β between WT-, E61I-, N65I- and G84I-αSyn. They represent five (for WT) or three (for the others) independent simulations. Note that all the mutants have higher Avg-β values near the mutations (more green/blue cells in the red boxes for E61I and G84I, and in the red circle for N65I). (D). Comparison of the heatmaps of SD-β between WT- and N65I-αSyn. They represent five and three independent runs, respectively. Note that the region comprising N65I mutation shows more stable β-sheet with smaller SD than WT-αSyn (red box). (E). A final snap shot of E61I-αSyn after 400 ns of simulation. The mutation tended to stabilize the global structures of the amyloid and occasionally induced new β-sheets in the loop (56–62) (inset). (F). A final snap shot of G84I-αSyn after 400 ns of simulation. The mutation destabilized structures around the mutation and the adjacent regions (arrow). Occasionally β-strands were temporarily induced in the loop (inset). The differential effects of the isoleucine substitutions on the amyloid structures might be attributable to the shapes of the loops. The loop (56–62) is long and makes obtuse angles with the flanking β-strands. The loop (67–68) is short but rather flexible with two glycine residues in a row. The loop (84–87) is a U-shaped loop with a relatively-small turning radius, which could not accommodate Cβ-branching side chain of Ile. (G). Heatmaps of a hetero-oligomer amyloid containing N65I-αSyn in chain A-E and WT-αSyn in chain F-J (N65I/WT). Despite N65I mutation, Avg-β and SD-β values of chain A-F are similar to those of WT (red circle and red box).

Figure 4.
Influences of primary structures on predicted propensity profiles of PrP. (A). Comparison of predicted propensity profiles between two codon-127 polymorphs of human PrP, Gly127 (left) and Val127 (right). (B). Comparison of predicted propensity profiles of H1~H2 regions between human, elk and rabbit PrPs. Hu #, residue numbers in human numbering. Species-specific residues are denoted in bold letters.
Figure 4.
Influences of primary structures on predicted propensity profiles of PrP. (A). Comparison of predicted propensity profiles between two codon-127 polymorphs of human PrP, Gly127 (left) and Val127 (right). (B). Comparison of predicted propensity profiles of H1~H2 regions between human, elk and rabbit PrPs. Hu #, residue numbers in human numbering. Species-specific residues are denoted in bold letters.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Taguchi, Y.; Otaki, H.; Nishida, N. Mechanisms of Strain Diversity of Disease-Associated in-Register Parallel β-Sheet Amyloids and Implications About Prion Strains. Viruses 2019, 11, 110. https://doi.org/10.3390/v11020110
AMA Style
Taguchi Y, Otaki H, Nishida N. Mechanisms of Strain Diversity of Disease-Associated in-Register Parallel β-Sheet Amyloids and Implications About Prion Strains. Viruses. 2019; 11(2):110. https://doi.org/10.3390/v11020110
Chicago/Turabian StyleTaguchi, Yuzuru, Hiroki Otaki, and Noriyuki Nishida. 2019. "Mechanisms of Strain Diversity of Disease-Associated in-Register Parallel β-Sheet Amyloids and Implications About Prion Strains" Viruses 11, no. 2: 110. https://doi.org/10.3390/v11020110
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.