Genetic Diversity and Phylodynamics of Avian Coronaviruses in Egyptian Wild Birds

 ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling History, Collection, and Virus Isolation
2.2. Polymerase (RdRp) and Spike (S1) Genes Amplification and Sequencing
2.3. Sequence and Phylogenetic Analysis
2.4. Selection Pressure Analysis
3. Results
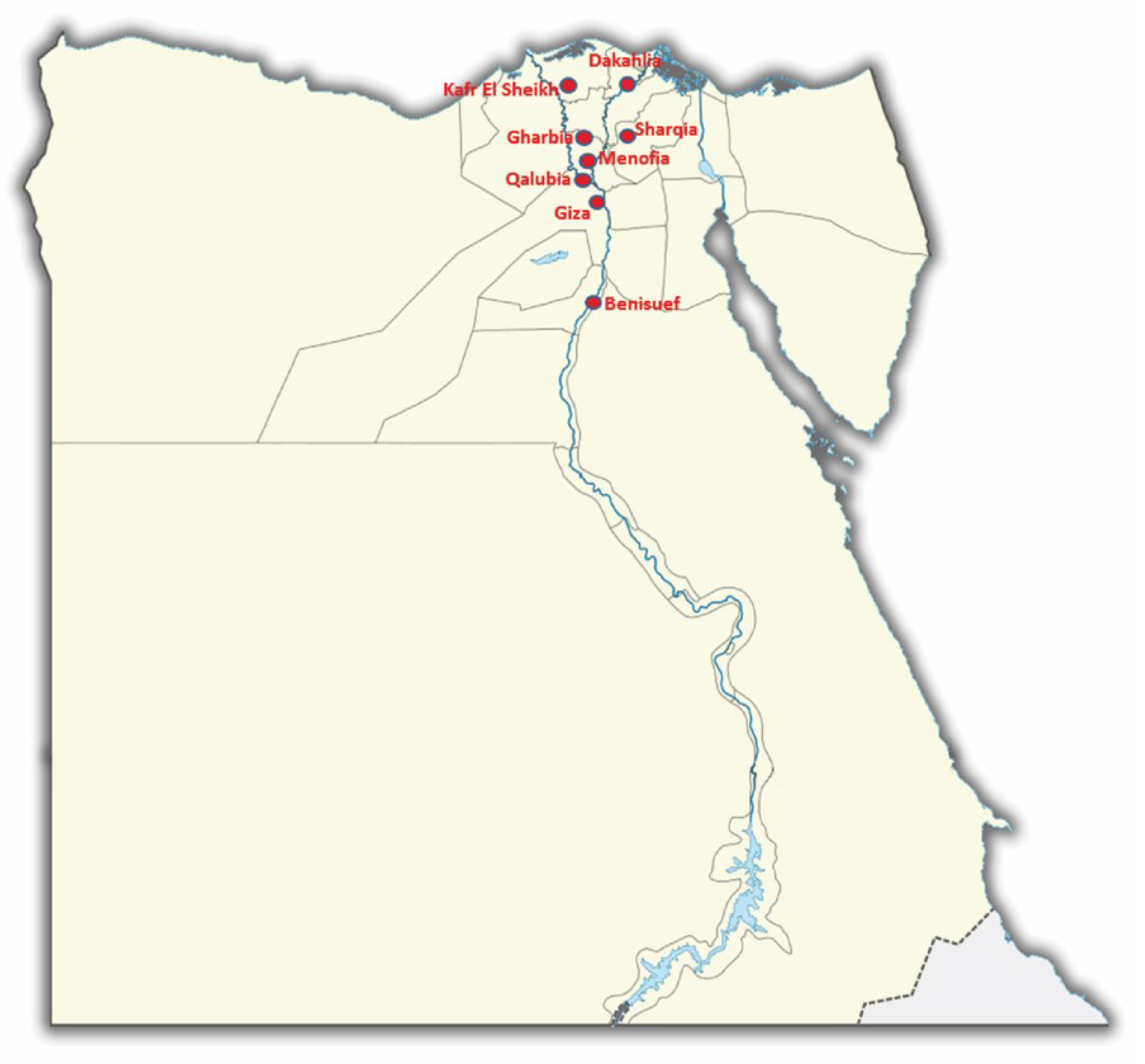
3.1. Occurrence and Distribution of Avian Coronavirus in Wild Birds
3.2. Viral Sequences and Genomics Analysis
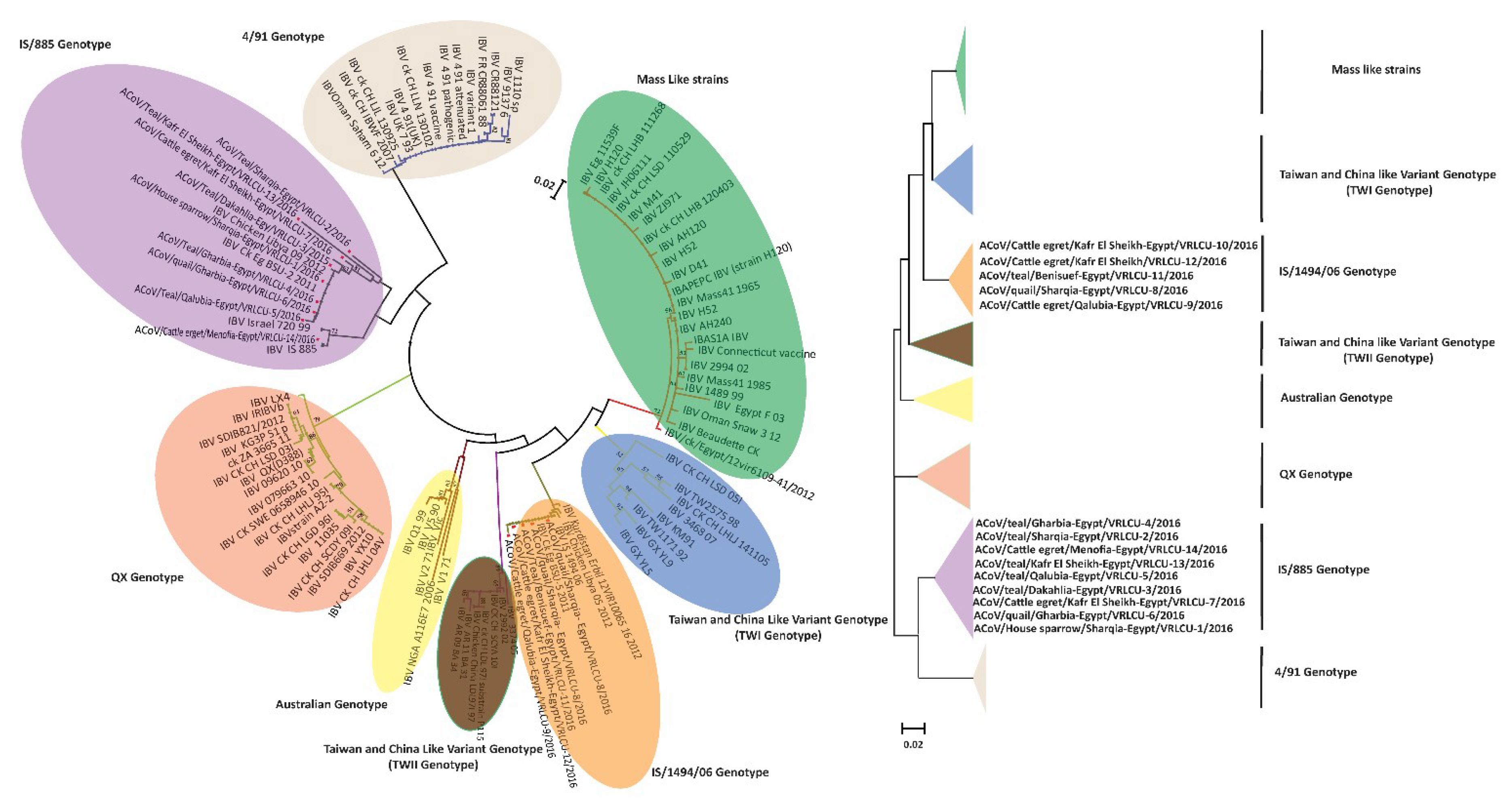
3.3. Phylogenetic Analysis
3.4. Selective Pressure Sites and Recombination Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- ICTV. Virus Taxonomy: 2018. Available online: https://talk.ictvonline.org/taxonomy/ (assessed on 25 December 2018).
- de Groot, R.J.; Baker, S.C.; Baric, R.; Enjuanes, L.; Gorbalenya, A.E.; Holmes, K.V.; Perlman, S.; Poon, L.; Rottier, P.J.; Talbot, P.J.; et al. “Family Coronaviridae”. In Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Lefkowitz, E., Adams, M.J., Carstens, E.B., Eds.; Elsevier: Oxford, UK, 2011; pp. 806–828. [Google Scholar]
- Cavanagh, D. Coronavirus avian infectious bronchitis virus. Vet. Res. 2007, 38, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Wickramasinghe, I.N.A.; de Vries, R.P.; Grone, A.; de Haan, C.A.M.; Verheije, M.H. Binding of Avian Coronavirus Spike Proteins to Host Factors Reflects Virus Tropism and Pathogenicity. J. Virol. 2011, 85, 8903–8912. [Google Scholar] [CrossRef] [Green Version]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef] [PubMed]
- Adzhar, A.; Shaw, K.; Britton, P.; Cavanagh, D. Universal oligonucleotides for the detection of infectious bronchitis virus by the polymerase chain reaction. Avian Pathol. 1996, 25, 817–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kingham, B.F.; Keeler, C.L.; Nix, W.A.; Ladman, B.S.; Gelb, J. Identification of avian infectious bronchitis virus by direct automated cycle sequencing of the S-1 gene. Avian Dis. 2000, 44, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Valastro, V.; Holmes, E.C.; Britton, P.; Fusaro, A.; Jackwood, M.W.; Cattoli, G.; Monne, I. S1 gene-based phylogeny of infectious bronchitis virus: An attempt to harmonize virus classification. Infect. Genet. Evol. 2016, 39, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, M.W. Review of infectious bronchitis virus around the world. Avian Dis. 2012, 56, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Schalk, A.F.; Hawn, M.C. An apparently new respiratory disease of baby chicks. J. Am. Vet. Med. Assoc. 1931, 78, 1931. [Google Scholar]
- Liu, S.; Kong, X. A new genotype of nephropathogenic infectious bronchitis virus circulating in vaccinated and non-vaccinated flocks in China. Avian Pathol. 2004, 33, 321–327. [Google Scholar] [CrossRef] [Green Version]
- McMartin, D.A. Infectious bronchitis. In Virus Infections of Birds; McFerran, J.B., McNulty, M.S., Eds.; Elsevier Science Publishers: Amsterdam, The Netherlands, 1993; pp. 249–274. [Google Scholar]
- Spackman, E. The ecology of avian influenza virus in wild birds: What does this mean for poultry? Poult. Sci. 2009, 88, 847–850. [Google Scholar] [CrossRef] [Green Version]
- Domanska-Blicharz, K.; Smietanka, K.; Minta, Z. Molecular studies on infectious bronchitis virus isolated in Poland. Bull. Vet. Inst. Pulawy 2007, 51, 449–452. [Google Scholar]
- Cavanagh, D.; Gelb, J., Jr. Infectious bronchitis. In Diseases of Poultry, 12th ed.; Saif, Y.M., Fadly, A.M., Glisson, J.R., McDougald, L.R., Nolan, L.K., Swayne, D.E., Eds.; Blackwell Publishing: Hoboken, NJ, USA, 2008; pp. 117–135. [Google Scholar]
- Ahmed, H.N. Incidence and Treatment of Some Infectious Viral Respiratory Diseases of Poultry in Egypt. DVM Thesis, Cairo University, Cairo, Egypt, 1954. [Google Scholar]
- Abdel-Moneim, A.S.; El-Kady, M.F.; Ladman, B.S.; Gelb, J. S1 gene sequence analysis of a nephropathogenic strain of avian infectious bronchitis virus in Egypt. Virol. J. 2006, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Moneim, A.S.; Madbouly, H.M.; Gelb, J.; Ladman, B.S. Isolation and identification of Egypt/Beni-Seuf/01 a novel genotype of infectious bronchitis virus. Vet. Med. J. Giza 2002, 50, 1065–1078. [Google Scholar]
- Hussein, A.H.; Emara, M.; Rohaim, M.; Ganapathy, K.; Arafa, A. Sequence analysis of infectious bronchitis virus IS/1494 like strain isolated from broiler chicken co-infected with Newcastle disease virus in Egypt during 2012. Int. J. Poult. Sci. 2014, 13, 530–536. [Google Scholar] [CrossRef]
- Office International des Epizooties. Manual Diagnostic Tests Vaccines Terrestrial Animals (Mammals, Birds Bees); Office International des Epizooties: Paris, France, 2018. [Google Scholar]
- Muradrasoli, S.; Bálint, Á.; Wahlgren, J.; Waldenström, J.; Belák, S.; Blomberg, J.; Olsen, B. Prevalence and phylogeny of coronaviruses in wild birds from the bering strait area (Beringia). PLoS ONE 2010, 5, e13640. [Google Scholar] [CrossRef] [PubMed]
- Dolz, R.; Pujols, J.; Ordóñez, G.; Porta, R.; Majó, N. Antigenic and molecular characterization of isolates of the Italy 02 infectious bronchitis virus genotype. Avian Pathol. 2006, 35, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Minh, B.Q.; Nguyen, M.A.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Korber, B. HIV Signature and Sequence Variation Analysis. Computational Analysis of HIV Molecular Sequences; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2000; pp. 55–72. [Google Scholar]
- Martin, D.P.; Lemey, P.; Lott, M.; Moulton, V.; Posada, D.; Lefeuvre, P. RDP3: A flexible and fast computer program for analyzing recombination. Bioinformatics 2010, 26, 2462–2463. [Google Scholar] [CrossRef]
- Abdel-Moneim, A.S.; Afifi, M.A.; El-Kady, M.F. Emergence of a novel genotype of avian infectious bronchitis virus in Egypt. Arch. Virol. 2012, 157, 2453–2457. [Google Scholar] [CrossRef]
- Domingo, E.; Sheldon, J.; Perales, C. Viral Quasispecies Evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignjatovic, J.; Sapats, S. Avian infectious bronchitis virus. Rev. Sci. Tech. Off. Int. Epiz. 2000, 19, 493–508. [Google Scholar] [CrossRef]
- Cook, J.K.A. Coronaviridae. In Poultry Diseases, 6th ed.; Pattison, M., Bradbury, A., Eds.; Saunders Elsevier: Amsterdam, The Netherlands, 2008; pp. 340–349. [Google Scholar]
- Cavanagh, D.; Elus, M.M.; Cook, J.K.A. Relationship between sequence variation in the S1 spike protein of infectious bronchitis virus and the extent of cross—Protection in vivo S1 spike protein of infectious bronchitis virus. Avian Pathol. 1997, 26, 63–74. [Google Scholar] [CrossRef]
- Domanska-Blicharz, K.; Jacukowicz, A.; Lisowska, A.; Wyrostek, K.; Minta, Z. Detection and molecular characterization of infectious bronchitis-like viruses in wild bird populations. Avian Pathol. 2014, 43, 406–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Chen, J.; Chen, J.; Kong, X.; Shao, Y.; Han, Z.; Feng, L.; Cai, X.; Gu, S.; Liu, M. Isolation of avian infectious bronchitis coronavirus from domestic peafowl (Pavo cristatus) and teal (Anas). J. Gen. Virol. 2005, 86, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Hughes, L.A.; Savage, C.; Naylor, C.; Bennett, M.; Chantrey, J.; Jones, R. Genetically diverse coronaviruses in wild bird populations of northern England. Emerg. Infect. Dis. 2009, 15, 1091–1094. [Google Scholar] [CrossRef]
- Kahya, S.; Coven, F.; Temelli, S.; Eyigor, A.; Carli, K.T. Presence of IS/1494/06 genotype-related infectious bronchitis virus in breeder and broiler flocks in Turkey. Ankara Üniv. Vet. Fak. Derg. 2013, 60, 27–31. [Google Scholar]
- Ganapathy, K.; Ball, C.; Forrester, A. Genotypes of infectious bronchitis viruses circulating in the Middle East between 2009 and 2014. Virus Res. 2015, 210, 198–204. [Google Scholar] [CrossRef]
- Jackwood, M.W.; Hall, D.; Handel, A. Molecular evolution and emergence of avian gammacoronaviruses. Infect. Genet. Evol. 2012, 12, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Thor, S.W.; Hilt, D.A.; Kissinger, J.C.; Paterson, A.H.; Jackwood, M.W. Recombination in avian gamma-coronavirus infectious bronchitis virus. Viruses 2011, 3, 1777–1799. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, H.N.; Wang, T.; Fan, W.Q.; Zhang, A.Y.; Wei, K.; Tian, G.B.; Yang, X. Complete genome sequence and recombination analysis of infectious bronchitis virus attenuated vaccine strain. HVirus Genes 2010, 41, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Zhang, T.; Xu, Q.; Gao, M.; Chen, Y.; Wang, Q.; Zhao, Y.; Shao, Y.; Li, H.; Kong, X.; et al. Altered pathogenicity of a tl/CH/ LDT3/03 genotype infectious bronchitis coronavirus due to natural recombination in the 5’-17 kb region of the genome. Virus Res. 2016, 213, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, M.W.; Boynton, T.O.; Hilt, D.A.; McKinley, E.T.; Kissinger, J.C.; Paterson, A.H.; Robertson, J.; Lemke, C.; McCall, A.W.; Williams, S.M.; et al. Emergence of a group 3 coronavirus through recombination. Virology 2010, 398, 98–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olvera, A.; Cortey, M.; Segalés, J. Molecular evolution of porcine circovirus type 2 genomes: Phylogeny and clonality. Virology 2007, 357, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shangjin, C.; Cortey, M.; Segalés, J. Phylogeny and evolution of the NS1 and VP1/VP2 gene sequences from porcine parvovirus. Virus Res. 2009, 140, 209–215. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Order | Family | Genus | Species | Positive (n) | Sampled (n) | Rate% |
|---|---|---|---|---|---|---|
| Passeriformes | Passeridae | Passer | P. domesticus | 1 | 140 | 0.7 |
| Galliformes | Phasianidae | Coturnix | C. coturnix | 2 | 136 | 1.5 |
| Anseriformes | Anatidae | Anas | A. crecca | 7 | 150 | 4.7 |
| Pelecaniformes | Ardeidae | Bubulcus | B. ibis | 4 | 131 | 3.1 |
| Total = 4 | 4 | 4 | 4 | 14 | 557 | 2.5 |
| Species | Sharqia | Dakahlia | Kafr El Sheikh | Gharbia | Qalubia | Menofia | Giza | Benisuef | Total |
|---|---|---|---|---|---|---|---|---|---|
| C. coturnix | 23 | 21 | 20 | 22 | 23 | 15 | 7 | 9 | 140 |
| P. domesticus | 22 | 20 | 23 | 18 | 17 | 14 | 12 | 10 | 136 |
| A. crecca | 23 | 22 | 19 | 21 | 20 | 18 | 13 | 14 | 150 |
| B. ibis | 23 | 20 | 18 | 21 | 19 | 11 | 9 | 10 | 131 |
| Total | 91 | 83 | 80 | 82 | 79 | 58 | 41 | 43 | 557 |
| Positive (n) | 3 | 1 | 4 | 2 | 2 | 1 | 0 | 1 | 14 |
| Rate % | 3.3 | 1.2 | 5 | 2.4 | 2.5 | 1.7 | 0 | 2.3 | 2.5 |
| Sequence | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | ID | 100% | 99% | 99% | 99% | 99% | 99% | 92% | 92% | 92% | 92% | 92% | 98% | 98% |
| 2 | 100% | ID | 99% | 99% | 100% | 99% | 99% | 92% | 92% | 92% | 92% | 92% | 98% | 98% |
| 3 | 99% | 99% | ID | 99% | 99% | 99% | 99% | 92% | 92% | 92% | 92% | 92% | 98% | 98% |
| 4 | 99% | 99% | 99% | ID | 99% | 99% | 99% | 92% | 92% | 92% | 92% | 92% | 98% | 98% |
| 5 | 99% | 100% | 99% | 99% | ID | 99% | 99% | 92% | 92% | 93% | 92% | 92% | 98% | 98% |
| 6 | 99% | 99% | 99% | 99% | 99% | ID | 100% | 92% | 92% | 93% | 92% | 92% | 98% | 98% |
| 7 | 99% | 99% | 99% | 99% | 99% | 100% | ID | 92% | 92% | 93% | 92% | 92% | 98% | 98% |
| 8 | 92% | 92% | 92% | 92% | 92% | 92% | 92% | ID | 100% | 99% | 100% | 100% | 92% | 92% |
| 9 | 92% | 92% | 92% | 92% | 92% | 92% | 92% | 100% | ID | 99% | 100% | 100% | 92% | 92% |
| 10 | 92% | 92% | 92% | 92% | 93% | 93% | 93% | 99% | 99% | ID | 99% | 99% | 93% | 92% |
| 11 | 92% | 92% | 92% | 92% | 92% | 92% | 92% | 100% | 100% | 99% | ID | 100% | 92% | 92% |
| 12 | 92% | 92% | 92% | 92% | 92% | 92% | 92% | 100% | 100% | 99% | 100% | ID | 92% | 92% |
| 13 | 98% | 98% | 98% | 98% | 98% | 98% | 98% | 92% | 92% | 93% | 92% | 92% | ID | 98% |
| 14 | 98% | 98% | 98% | 98% | 98% | 98% | 98% | 92% | 92% | 92% | 92% | 92% | 98% | ID |
| Strain | HVR1 (60-88) | HVR2 (115-140) | HVR3 (275-292) |
|---|---|---|---|
| IBV strain H120 | GSSSGCTVGIIHGGRVVNASSIAMTAPSS | YKH--GGCPITGMLQQHSIRVSAMKNGQ | HNETGANPNPSGVQNIQTY |
| IBV strain Ma5 | |||
| house sparrow/Sharqia-Egypt/VRLCU-1 | ..Q.Q..A.A.YWSKNFS.A.V.....QN | ..SSS.S..L...IP..Y..I...R.NS | Y..SN.H..NG..HT.SI. |
| teal/Sharqia -Egypt/VRLCU-2 | ..Q.Q..A.A.YWSKNFS.A.V.....QN | ..SSS.S..L...IP..Y..I...R.NS | Y..SN.H..NG..HT.SI. |
| teal/Dakahlia -Egypt/VRLCU-3 | ..Q.Q..A.A.YWSKNFS.A.V.....QN | ..SSS.S..L...IP..Y..I...R.NS | ...SN.H..NG..HT.SL. |
| teal/Gharbia-Egypt/VRLCU-4 | ..Q.Q..A.A.YWSKNFS.A.V.....QN | ..SSS.S..L...IP.YY..I...R.NS | ...SN.H..NG..HT.SL. |
| teal/Qalubia-Egypt/VRLCU-5 | ..Q.Q..A.A.YWSKNFS.A.V.....QN | ..SSS.S..L...IP.YY..I...R.NS | Y..SN.H..NG..HT.SL. |
| quail/Gharbia-Egypt/VRLCU-6 | ..Q.Q..A.S.YWSKNFS.A.V.....QN | ..SSS.S..L...IP..Y..I...R.NS | Y..SN.H..NG..HT.SL. |
| cattle egret/Kafr El Sheikh-Egypt/VRLCU-7 | ..Q.Q..A.S.YWSKNFS.A.V.....QN | ..SSS.S..L...IP.YY..I...R.NS | Y..SN.H..NG..HT.SL. |
| quail/Sharqia-Egypt/VRLCU-8 | ..G.Q..A.S.YWSKNFT...V.....DT | ..SSS.S..L...IP..Y..I...R.NS | T.VSN.S..TG..NT.NI. |
| cattle egret/Qalubia-Egypt/VRLCU-9 | ..G.Q..A.S.YWSKNFT...V.....DT | ..SSS.S..L...IP..Y..I...R.NS | T.VSN.S..TG..NT.NI. |
| cattle egret/Kafr El Sheikh-Egypt/VRLCU-10 | ..G.Q..A.S.YWSKNFT...V.....DT | ..NGQ.S..L..LIP.NH..I.....SR | T.VSN.S..TG..NT.NI. |
| teal/Benisuef-Egypt/VRLCU-11 | ..G.Q..A.S.YWSKNFT...V.....DT | ..NGQ.S..L..LIP.NH..I.....SR | T.VSN.S..TG..NT.NI. |
| cattle egret/Kafr El Sheikh-Egypt/VRLCU-12 | ..G.Q..A.S.YWSKNFT...V.....DT | ..NGQ.S..L..LIP.NH..I.....SR | T.VSN.S..TG..NT.NI. |
| teal/Kafr El Sheikh-Egypt/VRLCU-13 | ..Q.Q..A.A.YWSKNFS.A.V.....QN | ..NGQ.S..L..LIP.NH..I.....SR | Y..SN.S..SG..NT.NLF |
| cattle egret/Menofia-Egypt/VRLCU-14 | ..E.Q..A.A.YWSKNFS.A.V.....QN | ..NGQ.S..L..LIP.NH..I.....SR | Y..SN.S..SG..NT.NLF |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
A. Rohaim, M.; F. El Naggar, R.; M. Helal, A.; M. Bayoumi, M.; A. El-Saied, M.; A. Ahmed, K.; Z. Shabbir, M.; Munir, M. Genetic Diversity and Phylodynamics of Avian Coronaviruses in Egyptian Wild Birds. Viruses 2019, 11, 57. https://doi.org/10.3390/v11010057
A. Rohaim M, F. El Naggar R, M. Helal A, M. Bayoumi M, A. El-Saied M, A. Ahmed K, Z. Shabbir M, Munir M. Genetic Diversity and Phylodynamics of Avian Coronaviruses in Egyptian Wild Birds. Viruses. 2019; 11(1):57. https://doi.org/10.3390/v11010057
Chicago/Turabian StyleA. Rohaim, Mohammed, Rania F. El Naggar, Ahmed M. Helal, Mahmoud M. Bayoumi, Mohamed A. El-Saied, Kawkab A. Ahmed, Muhammad Z. Shabbir, and Muhammad Munir. 2019. "Genetic Diversity and Phylodynamics of Avian Coronaviruses in Egyptian Wild Birds" Viruses 11, no. 1: 57. https://doi.org/10.3390/v11010057