Removal of the C6 Vaccinia Virus Interferon-β Inhibitor in the Hepatitis C Vaccine Candidate MVA-HCV Elicited in Mice High Immunogenicity in Spite of Reduced Host Gene Expression

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Cells and Viruses
2.3. Plasmid Transfer Vector pGem-RG-C6L wm
2.4. Construction of MVA-HCV ΔC6L
2.5. PCR Analysis
2.6. Expression of HCV Proteins
2.7. Analysis of Virus Growth
2.8. RNA Analysis by Quantitative Real-Time PCR
2.9. Microarray Analysis
2.10. Recruitment of Immune Cells in the Peritoneal Cavity of C57BL/6 Mice
2.11. Peptides
2.12. C57BL/6 Mice Immunization Schedule
2.13. ICS Assay
2.14. Enzyme-Linked Immunosorbent Assay (ELISA)
2.15. Statistical Procedures
3. Results
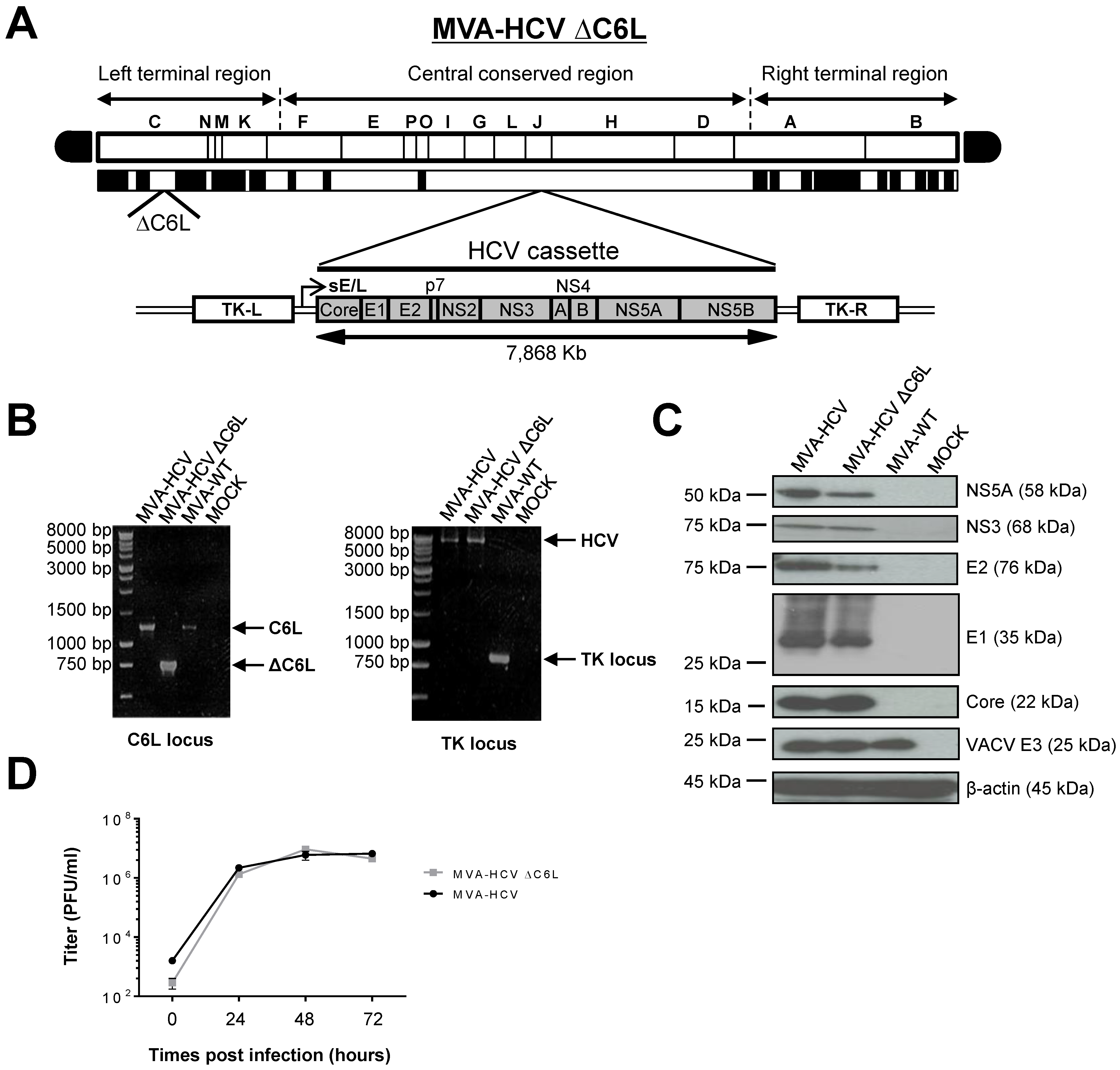
3.1. Generation of MVA-HCV ΔC6L Deletion Mutant
3.2. Expression of HCV Proteins by MVA-HCV ΔC6L
3.3. VACV C6L Gene Is Non-Essential for MVA-HCV Growth
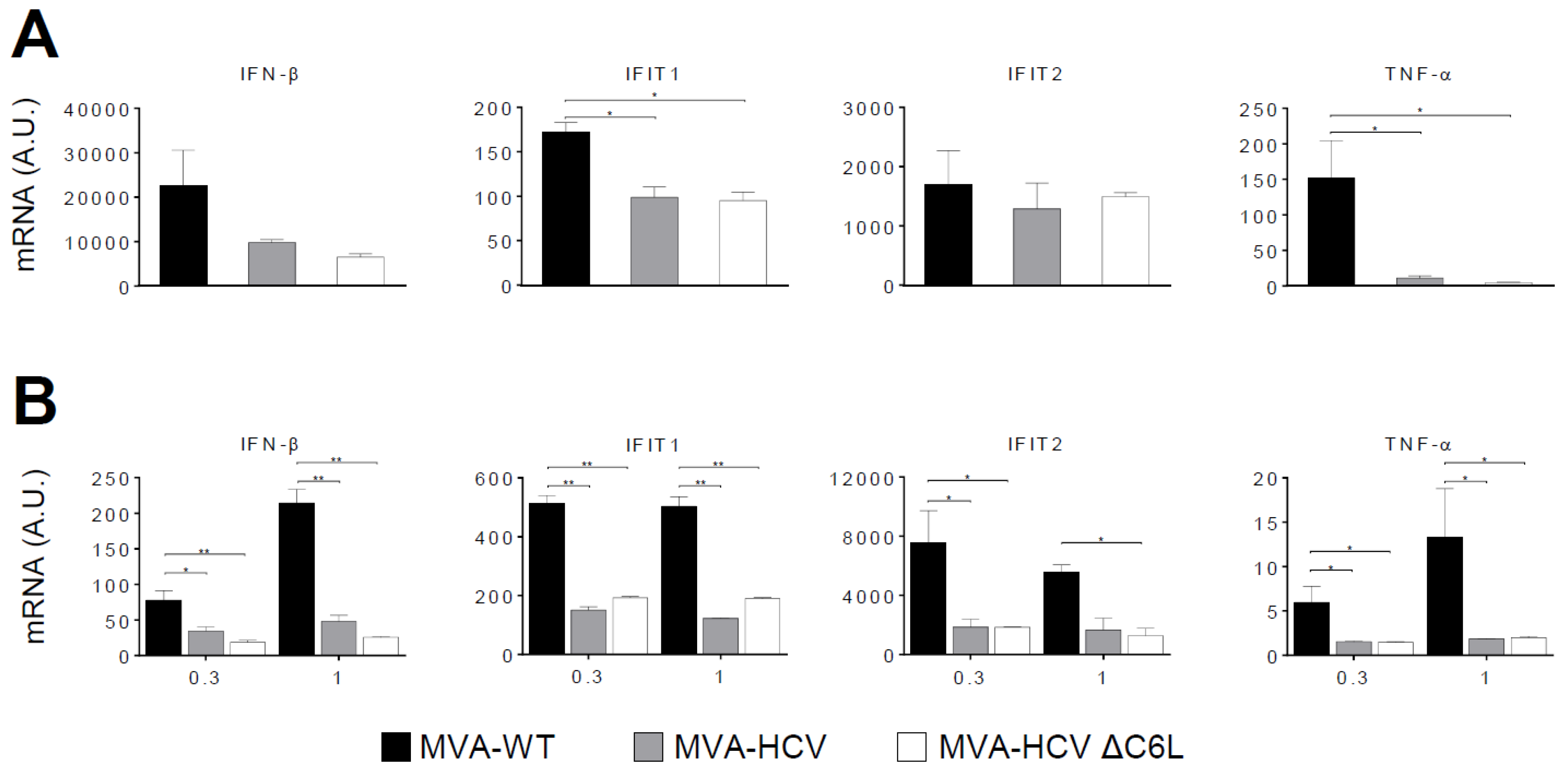
3.4. RT-PCR Showed that MVA-HCV and MVA-HCV ΔC6L Downregulate Expression of Genes Involved in Innate Immunity
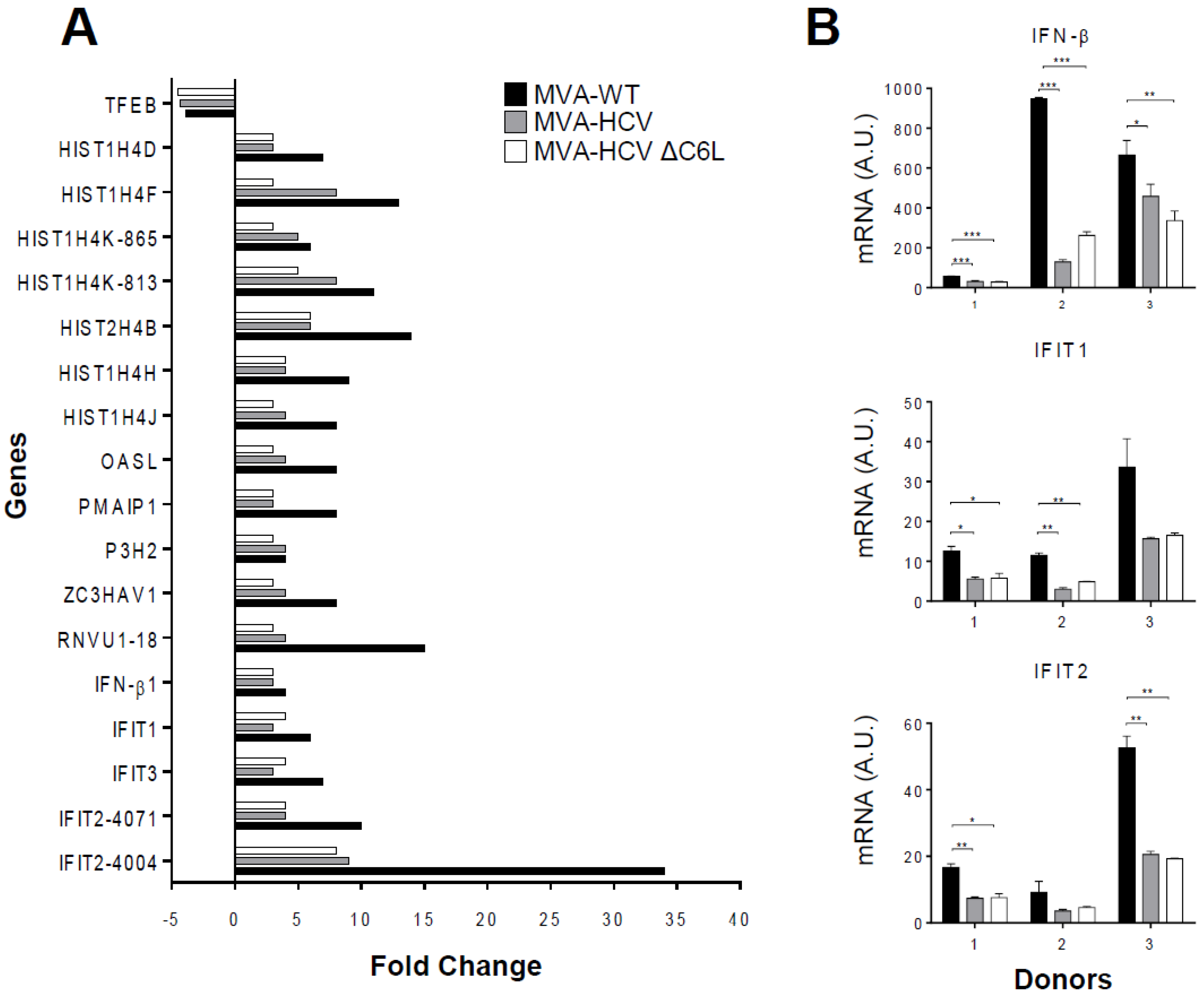
3.5. Microarray Analysis Revealed a Severe Reduction of Host Gene Expression by MVA-HCV and MVA-HCV ΔC6L
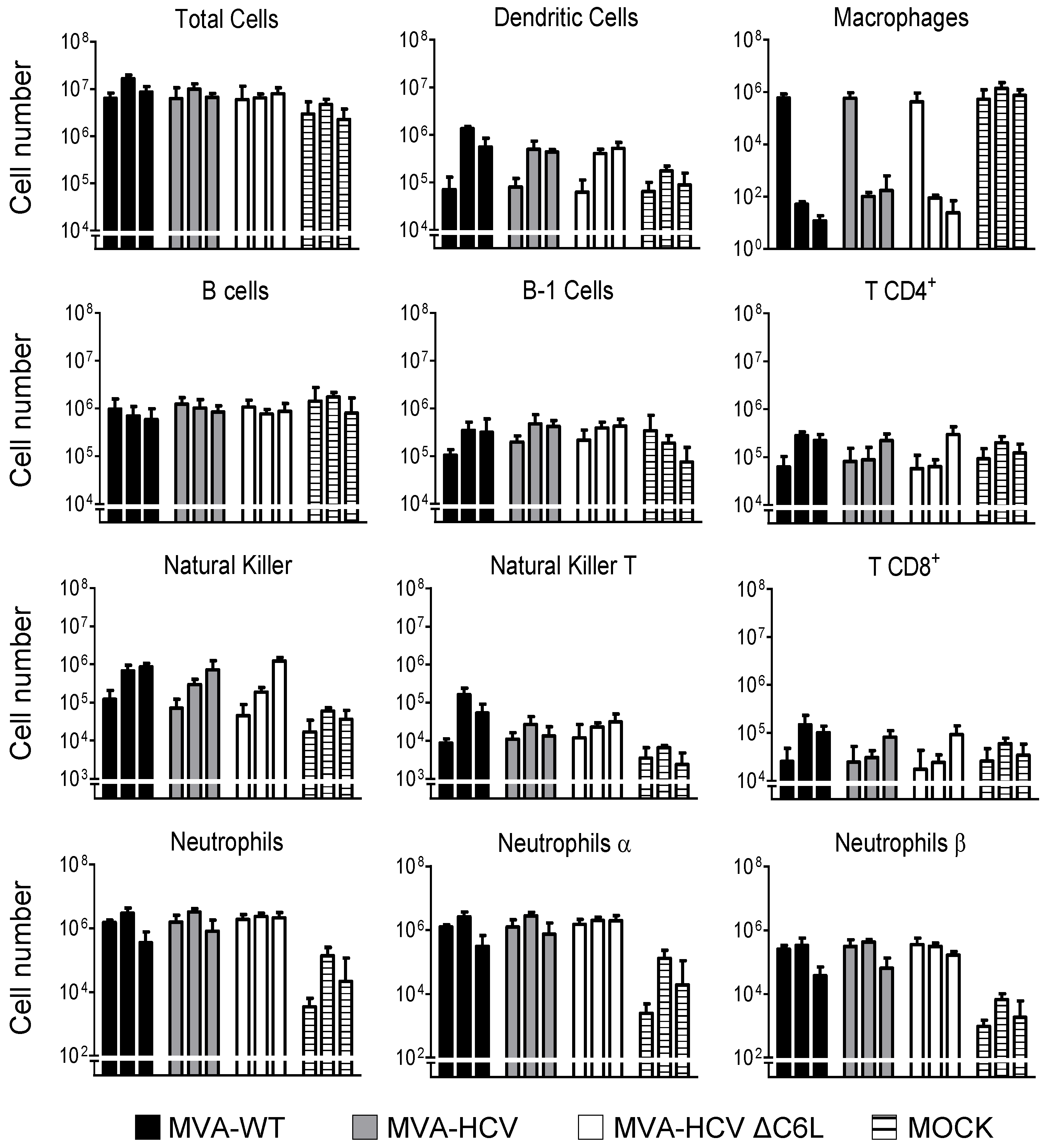
3.6. MVA-HCV and MVA-HCV ΔC6L Triggered Differential Cell Recruitment than MVA-WT in the Peritoneal Cavity of Infected Mice
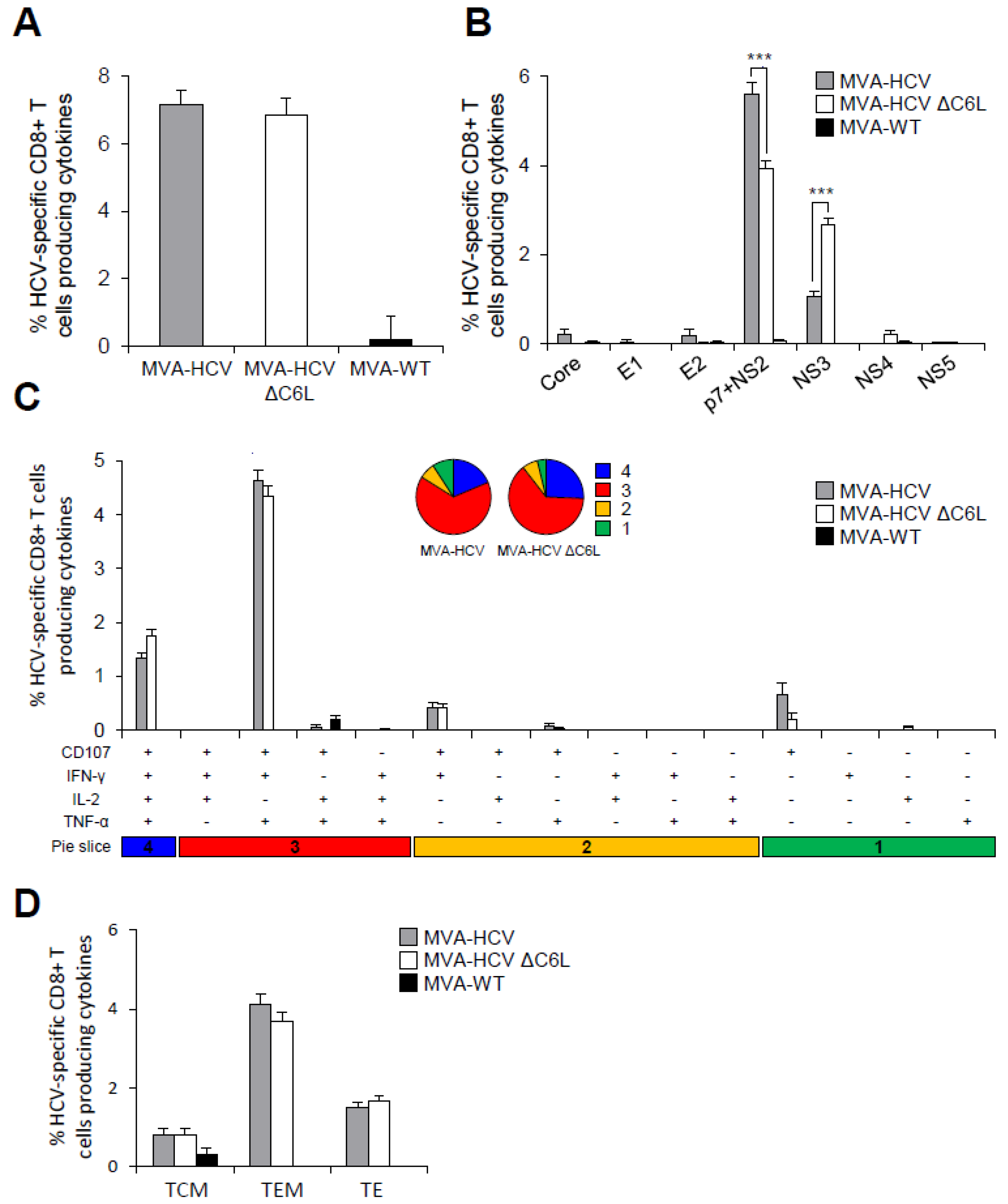
3.7. MVA-HCV and MVA-HCV ΔC6L Induced HCV-Specific T Cell and Humoral Adaptive Immune Responses in Immunized Mice
3.8. MVA-HCV and MVA-HCV ΔC6L Induced HCV-Specific T Cell Memory Immune Responses
3.9. MVA-HCV and MVA-HCV ΔC6L Induced Similar VACV-Specific T Cell and Humoral Immune Responses
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abdelwahab, K.S.; Ahmed Said, Z.N. Status of hepatitis C virus vaccination: Recent update. World J. Gastroenterol. 2016, 22, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Keck, Z.Y.; Foung, S.K. Viral evasion and challenges of hepatitis C virus vaccine development. Curr. Opin. Virol. 2016, 20, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lontok, E.; Harrington, P.; Howe, A.; Kieffer, T.; Lennerstrand, J.; Lenz, O.; McPhee, F.; Mo, H.; Parkin, N.; Pilot-Matias, T.; et al. Hepatitis C virus drug resistance-associated substitutions: State of the art summary. Hepatology 2015, 62, 1623–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarrazin, C.; Isakov, V.; Svarovskaia, E.S.; Hedskog, C.; Martin, R.; Chodavarapu, K.; Brainard, D.M.; Miller, M.D.; Mo, H.; Molina, J.M.; et al. Late relapse versus hepatitis c virus reinfection in patients with sustained virologic response after sofosbuvir-based therapies. Clin. Infect. Dis. 2017, 64, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Torres-Cornejo, A.; Lauer, G.M. Hurdles to the development of effective HBV immunotherapies and HCV vaccines. Pathog. Immun. 2017, 2, 102–125. [Google Scholar] [CrossRef] [PubMed]
- Grebely, J.; Prins, M.; Hellard, M.; Cox, A.L.; Osburn, W.O.; Lauer, G.; Page, K.; Lloyd, A.R.; Dore, G.J. Hepatitis C virus clearance, reinfection, and persistence, with insights from studies of injecting drug users: Towards a vaccine. Lancet Infect. Dis. 2012, 12, 408–414. [Google Scholar] [CrossRef]
- Osburn, W.O.; Fisher, B.E.; Dowd, K.A.; Urban, G.; Liu, L.; Ray, S.C.; Thomas, D.L.; Cox, A.L. Spontaneous control of primary hepatitis C virus infection and immunity against persistent reinfection. Gastroenterology 2010, 138, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Dahari, H.; Feinstone, S.M.; Major, M.E. Meta-analysis of hepatitis C virus vaccine efficacy in chimpanzees indicates an importance for structural proteins. Gastroenterology 2010, 139, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Freeman, Z.T.; Cox, A.L. Lessons from nature: Understanding immunity to HCV to guide vaccine design. PLoS Pathog. 2016, 12, e1005632. [Google Scholar] [CrossRef] [PubMed]
- Holz, L.; Rehermann, B. T cell responses in hepatitis C virus infection: Historical overview and goals for future research. Antivir. Res. 2015, 114, 96–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, C.M. Designing an HCV vaccine: A unique convergence of prevention and therapy? Curr. Opin. Virol. 2017, 23, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Ball, J.K.; Tarr, A.W.; McKeating, J.A. The past, present and future of neutralizing antibodies for hepatitis c virus. Antivir. Res. 2014, 105, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, F.; Rostami, S.; Meshkat, Z. Progress in the development of vaccines for hepatitis C virus infection. World J. Gastroenterol. 2015, 21, 11984–12002. [Google Scholar] [CrossRef] [PubMed]
- Gómez, C.E.; Perdiguero, B.; García-Arriaza, J.; Esteban, M. Clinical applications of attenuated MVA poxvirus strain. Expert Rev. Vaccines 2013, 12, 1395–1416. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sampedro, L.; Perdiguero, B.; Mejías-Pérez, E.; García-Arriaza, J.; Di Pilato, M.; Esteban, M. The evolution of poxvirus vaccines. Viruses 2015, 7, 1726–1803. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.E.; Najera, J.L.; Krupa, M.; Perdiguero, B.; Esteban, M. MVA and NYVAC as vaccines against emergent infectious diseases and cancer. Curr. Gene Ther. 2011, 11, 189–217. [Google Scholar] [CrossRef] [PubMed]
- Volz, A.; Sutter, G. Modified vaccinia virus ankara: History, value in basic research, and current perspectives for vaccine development. Adv. Virus Res. 2017, 97, 187–243. [Google Scholar] [PubMed]
- Heim, M.H. Interferons and hepatitis C virus. Swiss Med. Wkly. 2012, 142, w13586. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Jiang, J.D.; Peng, Z. Recent advances in the anti-HCV mechanisms of interferon. Acta Pharm. Sin. B 2014, 4, 241–247. [Google Scholar] [CrossRef] [PubMed]
- García-Arriaza, J.; Esteban, M. Enhancing poxvirus vectors vaccine immunogenicity. Hum. Vaccines Immunother. 2014, 10, 2235–2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unterholzner, L.; Sumner, R.P.; Baran, M.; Ren, H.; Mansur, D.S.; Bourke, N.M.; Randow, F.; Smith, G.L.; Bowie, A.G. Vaccinia virus protein c6 is a virulence factor that binds tbk-1 adaptor proteins and inhibits activation of irf3 and irf7. PLoS Pathog. 2011, 7, e1002247. [Google Scholar] [CrossRef] [PubMed]
- García-Arriaza, J.; Nájera, J.L.; Gómez, C.E.; Tewabe, N.; Sorzano, C.O.; Calandra, T.; Roger, T.; Esteban, M. A candidate HIV/aids vaccine (MVA-b) lacking vaccinia virus gene c6l enhances memory hiv-1-specific T-cell responses. PLoS ONE 2011, 6, e24244. [Google Scholar] [CrossRef] [PubMed]
- Stuart, J.H.; Sumner, R.P.; Lu, Y.; Snowden, J.S.; Smith, G.L. Vaccinia virus protein c6 inhibits type I IFN signalling in the nucleus and binds to the transactivation domain of stat2. PLoS Pathog. 2016, 12, e1005955. [Google Scholar] [CrossRef] [PubMed]
- García-Arriaza, J.; Arnáez, P.; Gómez, C.E.; Sorzano, C.; Esteban, M. Improving adaptive and memory immune responses of an HIV/aids vaccine candidate MVA-b by deletion of vaccinia virus genes (c6l and k7r) blocking interferon signaling pathways. PLoS ONE 2013, 8, e66894. [Google Scholar] [CrossRef] [PubMed]
- Sumner, R.P.; Ren, H.; Smith, G.L. Deletion of immunomodulator c6 from vaccinia virus strain western reserve enhances virus immunogenicity and vaccine efficacy. J. Gen. Virol. 2013, 94, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Gómez, C.E.; Perdiguero, B.; Cepeda, M.V.; Mingorance, L.; García-Arriaza, J.; Vandermeeren, A.; Sorzano, C.; Esteban, M. High, broad, polyfunctional, and durable T cell immune responses induced in mice by a novel hepatitis C virus (HCV) vaccine candidate (MVA-HCV) based on modified vaccinia virus ankara expressing the nearly full-length HCV genome. J. Virol. 2013, 87, 7282–7300. [Google Scholar] [CrossRef] [PubMed]
- Delaloye, J.; Roger, T.; Steiner-Tardivel, Q.G.; Le Roy, D.; Knaup Reymond, M.; Akira, S.; Petrilli, V.; Gomez, C.E.; Perdiguero, B.; Tschopp, J.; et al. Innate immune sensing of modified vaccinia virus ankara (MVA) is mediated by tlr2-tlr6, mda-5 and the nalp3 inflammasome. PLoS Pathog. 2009, 5, e1000480. [Google Scholar] [CrossRef] [PubMed]
- Gómez, C.E.; Nájera, J.L.; Jiménez, E.P.; Jiménez, V.; Wagner, R.; Graf, M.; Frachette, M.J.; Liljeström, P.; Pantaleo, G.; Esteban, M. Head-to-head comparison on the immunogenicity of two HIV/aids vaccine candidates based on the attenuated poxvirus strains MVA and NYVAC co-expressing in a single locus the HIV-1bx08 gp120 and HIV-1(IIIB) Gag-Pol-Nef proteins of clade B. Vaccine 2007, 25, 2863–2885. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.C.; Gherardi, M.M.; Esteban, M. Biology of attenuated modified vaccinia virus ankara recombinant vector in mice: Virus fate and activation of B- and T-cell immune responses in comparison with the western reserve strain and advantages as a vaccine. J. Virol. 2000, 74, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arriaza, J.; Najera, J.L.; Gomez, C.E.; Sorzano, C.O.; Esteban, M. Immunogenic profiling in mice of a HIV/aids vaccine candidate (MVA-b) expressing four HIV-1 antigens and potentiation by specific gene deletions. PLoS ONE 2010, 5, e12395. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K.; Speed, T. Normalization of cDNA microarray data. Methods 2003, 31, 265–273. [Google Scholar] [CrossRef] [Green Version]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Ihaka, R.; Gentleman, R. R: A language for dataanalysis and graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar]
- Breitling, R.; Armengaud, P.; Amtmann, A.; Herzyk, P. Rank products: A simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett. 2004, 573, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Breitling, R.; McEntee, C.W.; Wittner, B.S.; Nemhauser, J.L.; Chory, J. Rankprod: A bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics 2006, 22, 2825–2827. [Google Scholar] [CrossRef] [PubMed]
- Di Pilato, M.; Mejías-Pérez, E.; Zonca, M.; Perdiguero, B.; Gómez, C.E.; Trakala, M.; Nieto, J.; Nájera, J.L.; Sorzano, C.O.; Combadière, C.; et al. Nfκb activation by modified vaccinia virus as a novel strategy to enhance neutrophil migration and HIV-specific T-cell responses. Proc. Natl. Acad. Sci. USA 2015, 112, E1333–E1342. [Google Scholar] [CrossRef] [PubMed]
- García-Arriaza, J.; Gómez, C.E.; Sorzano, C.; Esteban, M. Deletion of the vaccinia virus N2L gene encoding an inhibitor of IRF3 improves the immunogenicity of modified vaccinia virus ankara expressing hiv-1 antigens. J. Virol. 2014, 88, 3392–3410. [Google Scholar] [CrossRef] [PubMed]
- Najera, J.L.; Gomez, C.E.; Garcia-Arriaza, J.; Sorzano, C.O.; Esteban, M. Insertion of vaccinia virus C7L host range gene into NYVAC-b genome potentiates immune responses against HIV-1 antigens. PLoS ONE 2010, 5, e11406. [Google Scholar] [CrossRef] [PubMed]
- Shoukry, N.H.; Grakoui, A.; Houghton, M.; Chien, D.Y.; Ghrayeb, J.; Reimann, K.A.; Walker, C.M. Memory cd8+ T cells are required for protection from persistent hepatitis C virus infection. J. Exp. Med. 2003, 197, 1645–1655. [Google Scholar] [CrossRef] [PubMed]
- Man John Law, L.; Landi, A.; Magee, W.C.; Lorne Tyrrell, D.; Houghton, M. Progress towards a hepatitis C virus vaccine. Emerg. Microbes Infect. 2013, 2, e79. [Google Scholar] [CrossRef] [PubMed]
- Barnes, E.; Folgori, A.; Capone, S.; Swadling, L.; Aston, S.; Kurioka, A.; Meyer, J.; Huddart, R.; Smith, K.; Townsend, R.; et al. Novel adenovirus-based vaccines induce broad and sustained T cell responses to HCV in man. Sci. Transl. Med. 2012, 4, 115ra1. [Google Scholar] [CrossRef] [PubMed]
- Swadling, L.; Capone, S.; Antrobus, R.D.; Brown, A.; Richardson, R.; Newell, E.W.; Halliday, J.; Kelly, C.; Bowen, D.; Fergusson, J.; et al. A human vaccine strategy based on chimpanzee adenoviral and mva vectors that primes, boosts, and sustains functional HCV-specific T cell memory. Sci. Transl. Med. 2014, 6, 261ra153. [Google Scholar] [CrossRef] [PubMed]
- Habersetzer, F.; Honnet, G.; Bain, C.; Maynard-Muet, M.; Leroy, V.; Zarski, J.P.; Feray, C.; Baumert, T.F.; Bronowicki, J.P.; Doffoel, M.; et al. A poxvirus vaccine is safe, induces T-cell responses, and decreases viral load in patients with chronic hepatitis C. Gastroenterology 2011, 141, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.G.; Zubkova, I.; Kachko, A.; Wells, F.; Adler, H.; Sutter, G.; Major, M.E. Qualitative differences in cellular immunogenicity elicited by hepatitis c virus T-cell vaccines employing prime-boost regimens. PLoS ONE 2017, 12, e0181578. [Google Scholar] [CrossRef] [PubMed]
- Perdiguero, B.; Esteban, M. The interferon system and vaccinia virus evasion mechanisms. J. Interferon Cytokine Res. 2009, 29, 581–598. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.L.; Benfield, C.T.; Maluquer de Motes, C.; Mazzon, M.; Ember, S.W.; Ferguson, B.J.; Sumner, R.P. Vaccinia virus immune evasion: Mechanisms, virulence and immunogenicity. J. Gen. Virol. 2013, 94, 2367–2392. [Google Scholar] [CrossRef] [PubMed]
- Albarnaz, J.D.; Torres, A.A.; Smith, G.L. Modulating vaccinia virus immunomodulators to improve immunological memory. Viruses 2018, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Dolatimehr, F.; Karimi-Sari, H.; Rezaee-Zavareh, M.S.; Alavian, S.M.; Behnava, B.; Gholami-Fesharaki, M.; Sharafi, H. Combination of sofosbuvir, pegylated-interferon and ribavirin for treatment of hepatitis c virus genotype 1 infection: A systematic review and meta-analysis. Daru 2017, 25, 11. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Torres, M.; Lawitz, E.; Kowdley, K.V.; Nelson, D.R.; Dejesus, E.; McHutchison, J.G.; Cornpropst, M.T.; Mader, M.; Albanis, E.; Jiang, D.; et al. Sofosbuvir (gs-7977) plus peginterferon/ribavirin in treatment-naïve patients with HCV genotype 1: A randomized, 28-day, dose-ranging trial. J. Hepatol. 2013, 58, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Lawitz, E.; Poordad, F.; Brainard, D.M.; Hyland, R.H.; An, D.; Dvory-Sobol, H.; Symonds, W.T.; McHutchison, J.G.; Membreno, F.E. Sofosbuvir with peginterferon-ribavirin for 12 weeks in previously treated patients with hepatitis c genotype 2 or 3 and cirrhosis. Hepatology 2015, 61, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Foster, G.R.; Pianko, S.; Brown, A.; Forton, D.; Nahass, R.G.; George, J.; Barnes, E.; Brainard, D.M.; Massetto, B.; Lin, M.; et al. Efficacy of sofosbuvir plus ribavirin with or without peginterferon-alfa in patients with hepatitis C virus genotype 3 infection and treatment-experienced patients with cirrhosis and hepatitis C virus genotype 2 infection. Gastroenterology 2015, 149, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the rig-i antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease ns3/4a cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.E.; Vandermeeren, A.M.; Garcia, M.A.; Domingo-Gil, E.; Esteban, M. Involvement of PKR and RNASE l in translational control and induction of apoptosis after hepatitis c polyprotein expression from a vaccinia virus recombinant. Virol. J. 2005, 2, 81. [Google Scholar] [CrossRef] [PubMed]
- Vandermeeren, A.M.; Gomez, C.E.; Patino, C.; Domingo-Gil, E.; Guerra, S.; Gonzalez, J.M.; Esteban, M. Subcellular forms and biochemical events triggered in human cells by HCV polyprotein expression from a viral vector. Virol. J. 2008, 5, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duong, F.H.; Christen, V.; Lin, S.; Heim, M.H. Hepatitis C virus-induced up-regulation of protein phosphatase 2a inhibits histone modification and DNA damage repair. Hepatology 2010, 51, 741–751. [Google Scholar] [PubMed]
- Chen, S.; Wu, Z.; Wang, M.; Cheng, A. Innate immune evasion mediated by flaviviridae non-structural proteins. Viruses 2017, 9, 291. [Google Scholar] [CrossRef] [PubMed]
- Von dem Bussche, A.; Machida, R.; Li, K.; Loevinsohn, G.; Khander, A.; Wang, J.; Wakita, T.; Wands, J.R.; Li, J. Hepatitis C virus ns2 protein triggers endoplasmic reticulum stress and suppresses its own viral replication. J. Hepatol. 2010, 53, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Foy, E.; Li, K.; Wang, C.; Sumpter, R.; Ikeda, M.; Lemon, S.M.; Gale, M. Regulation of interferon regulatory factor-3 by the hepatitis c virus serine protease. Science 2003, 300, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Yi, G.; Wen, Y.; Shu, C.; Han, Q.; Konan, K.V.; Li, P.; Kao, C.C. Hepatitis C virus ns4b can suppress sting accumulation to evade innate immune responses. J. Virol. 2015, 90, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Kaname, Y.; Hamamoto, I.; Tsuda, Y.; Wen, X.; Taguwa, S.; Moriishi, K.; Takeuchi, O.; Kawai, T.; Kanto, T.; et al. Hepatitis C virus nonstructural protein 5a modulates the toll-like receptor-myd88-dependent signaling pathway in macrophage cell lines. J. Virol. 2007, 81, 8953–8966. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, J.B.; Kim, H.; Ray, R.; Ray, R.B. Hepatitis C virus ns5a protein modulates irf-7-mediated interferon-α signaling. J. Interferon Cytokine Res. 2014, 34, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Kumthip, K.; Chusri, P.; Jilg, N.; Zhao, L.; Fusco, D.N.; Zhao, H.; Goto, K.; Cheng, D.; Schaefer, E.A.; Zhang, L.; et al. Hepatitis c virus ns5a disrupts stat1 phosphorylation and suppresses type i interferon signaling. J. Virol. 2012, 86, 8581–8591. [Google Scholar] [CrossRef] [PubMed]
- Gale, M.J.; Korth, M.J.; Tang, N.M.; Tan, S.L.; Hopkins, D.A.; Dever, T.E.; Polyak, S.J.; Gretch, D.R.; Katze, M.G. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the pkr protein kinase by the nonstructural 5a protein. Virology 1997, 230, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Neumann-Haefelin, C.; Thimme, R. Adaptive immune responses in hepatitis C virus infection. Curr. Top. Microbiol. Immunol. 2013, 369, 243–262. [Google Scholar] [PubMed]
- Tester, I.; Smyk-Pearson, S.; Wang, P.; Wertheimer, A.; Yao, E.; Lewinsohn, D.M.; Tavis, J.E.; Rosen, H.R. Immune evasion versus recovery after acute hepatitis C virus infection from a shared source. J. Exp. Med. 2005, 201, 1725–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, S.; Lauer, G.; Isba, R.; Walker, B.; Klenerman, P. Cellular immune responses against hepatitis C virus: The evidence base 2002. Clin. Exp. Immunol. 2002, 128, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Thimme, R.; Oldach, D.; Chang, K.M.; Steiger, C.; Ray, S.C.; Chisari, F.V. Determinants of viral clearance and persistence during acute hepatitis c virus infection. J. Exp. Med. 2001, 194, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Huang, H.; Xiang, J.; Babiuk, L.A.; van Drunen Littel-van den Hurk, S. Dendritic cells pulsed with hepatitis C virus ns3 protein induce immune responses and protection from infection with recombinant vaccinia virus expressing ns3. J. Gen. Virol. 2006, 87, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Verstrepen, B.E.; Verschoor, E.J.; Fagrouch, Z.C.; Mooij, P.; de Groot, N.G.; Bontrop, R.E.; Bogers, W.M.; Heeney, J.L.; Koopman, G. Strong vaccine-induced cd8 T-cell responses have cytolytic function in a chimpanzee clearing hcv infection. PLoS ONE 2014, 9, e95103. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Hammond, T.; Watson, M.W.; Flexman, J.P.; Cheng, W.; Fernandez, S.; Price, P. Could a loss of memory T cells limit responses to hepatitis C virus (HCV) antigens in blood leucocytes from patients chronically infected with HCV before and during pegylated interferon-alpha and ribavirin therapy? Clin. Exp. Immunol. 2010, 161, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Law, M.; Maruyama, T.; Lewis, J.; Giang, E.; Tarr, A.W.; Stamataki, Z.; Gastaminza, P.; Chisari, F.V.; Jones, I.M.; Fox, R.I.; et al. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 2008, 14, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Farci, P.; Shimoda, A.; Wong, D.; Cabezon, T.; De Gioannis, D.; Strazzera, A.; Shimizu, Y.; Shapiro, M.; Alter, H.J.; Purcell, R.H. Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc. Natl. Acad. Sci. USA 1996, 93, 15394–15399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanwolleghem, T.; Bukh, J.; Meuleman, P.; Desombere, I.; Meunier, J.C.; Alter, H.; Purcell, R.H.; Leroux-Roels, G. Polyclonal immunoglobulins from a chronic hepatitis C virus patient protect human liver-chimeric mice from infection with a homologous hepatitis C virus strain. Hepatology 2008, 47, 1846–1855. [Google Scholar] [CrossRef] [PubMed]
- Drummer, H.E. Editorial on “broadly neutralizing antibodies abrogate established hepatitis C virus infection” published in science translational medicine on 17th September 2014. Ann. Transl. Med. 2015, 3, S6. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name (Probe identity (ID)) a | Accession Number | Fold Change b |
|---|---|---|
| Transmembrane protein 11 (TMEM11) | NM_003876 | +2.05 |
| KIAA0586 (KIAA0586) | NM_014749 | +2.00 |
| CD28 molecule (CD28) | NM_006139 | −2.00 |
| FLJ30313 (lnc-GATA5-2) | lnc-GATA5-2:2 | −2.01 |
| Aquaporin 1 (AQP1) | NM_198098 | −2.03 |
| Gessler Wilms tumor Homo sapiens (SMCR2) | AI821758 | −2.05 |
| Oogenesis homeobox (NOBOX) | NM_001080413 | −2.05 |
| RAS oncogene family (RAB6A) | NM_002869 | −2.08 |
| Ribosomal protein S28 (RPS28) | NM_001031 | −2.08 |
| Major histocompatibility complex, class II, DR beta 5 (HLA-DRB5) | NM_002125 | −2.09 |
| Chimerin 2 (ENST00000409922) | ENST00000409922 | −2.10 |
| Nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent 1 (NFATC1) | NM_172390 | −2.13 |
| RAS guanyl releasing protein 2 (RASGRP2) | ENST00000377494 | −2.14 |
| Glycosyltransferase 3 family (UGT3A1) | NM_152404 | −2.16 |
| Myotubularin related protein 3 (MTMR3) | NM_021090 | −2.17 |
| Keratin associated protein 2-1 (KRTAP2-1) | NM_001123387 | −2.18 |
| keratin associated protein 5-4 (KRTAP5-4) | NM_001012709 | −2.19 |
| G protein-coupled receptor 88 (GPR88) | NM_022049 | −2.19 |
| G protein pathway suppressor 2 pseudogene (ENST00000430745) | ENST00000430745 | −2.19 |
| Secretory trafficking family member B (MON1B) | NM_014940 | −2.21 |
| Solute carrier family 16, member 11 (SLC16A11) | NM_153357 | −2.24 |
| Vacuolar protein sorting 18 homolog (VPS18) | NM_020857 | −2.26 |
| Synapse defective 1, Rho GTPase, homolog 2 (SYDE2) | NM_032184 | −2.27 |
| Neuron-derived neurotrophic factor (NDNF) | NM_024574 | −2.30 |
| Homeobox 1 (EMX1) | BC037242 | −2.32 |
| Actin-like protein 3 (THC2499666) | THC2499666 | −2.32 |
| Interleukin 15 receptor (IL15RA) | ENST00000379971 | −2.39 |
| Chemokine (C-C motif) ligand 18 (CCL18) | NM_002988 | −2.40 |
| cDNA clone NT2RI2025693 (ENST00000587777) | ENST00000587777 | −2.42 |
| Cellular repressor of E1A-stimulated genes 1 (CREG1) | NM_003851 | −2.46 |
| NYN domain and retroviral integrase containing (NYNRIN) | NM_025081 | −2.49 |
| Uncoupling protein 3 (UCP3) | NM_022803 | −2.53 |
| Phospholipase C (PLCH2) | NM_014638 | −2.57 |
| Long intergenic non-protein coding RNA 687 (LINC00687) | NR_110635 | −3.05 |
| Synuclein, beta (SNCB) | NM_001001502 | −3.05 |
| Arylformamidase (AFMID) | NM_001145526 | −3.20 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Q. Marín, M.; Pérez, P.; Gómez, C.E.; S. Sorzano, C.Ó.; Esteban, M.; García-Arriaza, J. Removal of the C6 Vaccinia Virus Interferon-β Inhibitor in the Hepatitis C Vaccine Candidate MVA-HCV Elicited in Mice High Immunogenicity in Spite of Reduced Host Gene Expression. Viruses 2018, 10, 414. https://doi.org/10.3390/v10080414
Q. Marín M, Pérez P, Gómez CE, S. Sorzano CÓ, Esteban M, García-Arriaza J. Removal of the C6 Vaccinia Virus Interferon-β Inhibitor in the Hepatitis C Vaccine Candidate MVA-HCV Elicited in Mice High Immunogenicity in Spite of Reduced Host Gene Expression. Viruses. 2018; 10(8):414. https://doi.org/10.3390/v10080414
Chicago/Turabian StyleQ. Marín, María, Patricia Pérez, Carmen E. Gómez, Carlos Óscar S. Sorzano, Mariano Esteban, and Juan García-Arriaza. 2018. "Removal of the C6 Vaccinia Virus Interferon-β Inhibitor in the Hepatitis C Vaccine Candidate MVA-HCV Elicited in Mice High Immunogenicity in Spite of Reduced Host Gene Expression" Viruses 10, no. 8: 414. https://doi.org/10.3390/v10080414