Deciphering the Human Virome with Single-Virus Genomics and Metagenomics

 and
and 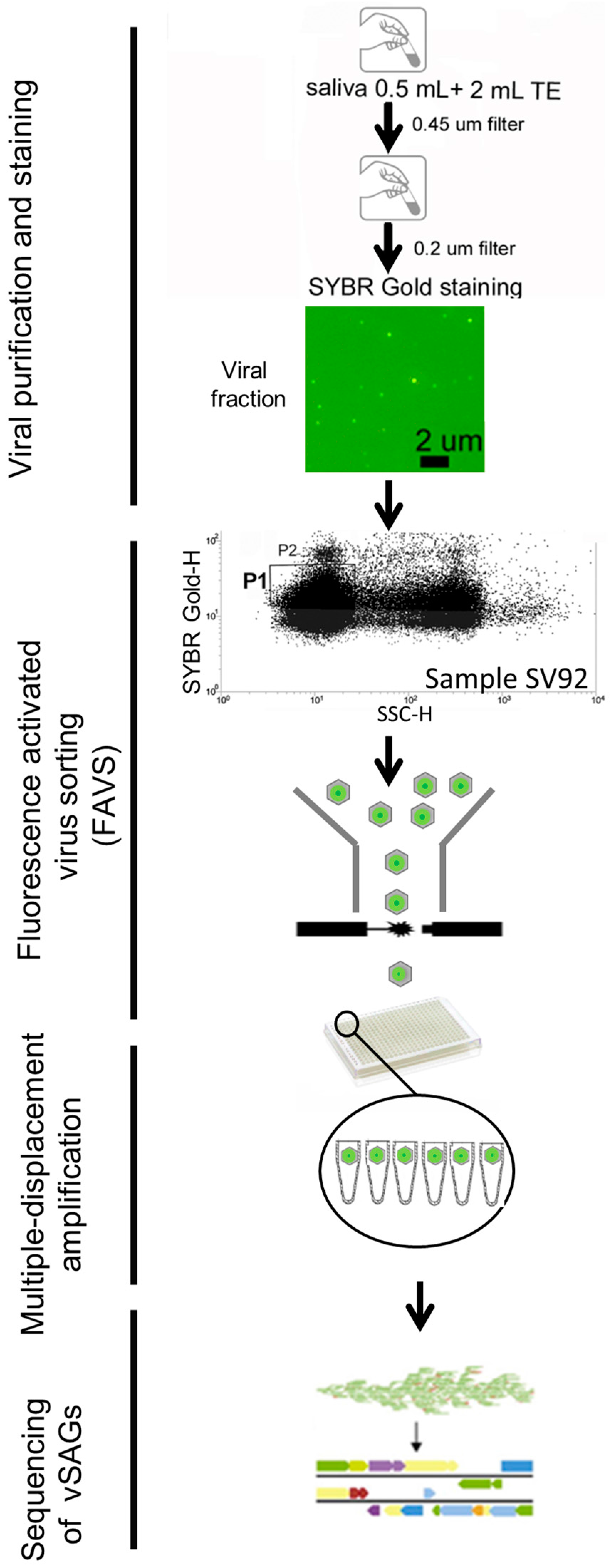{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
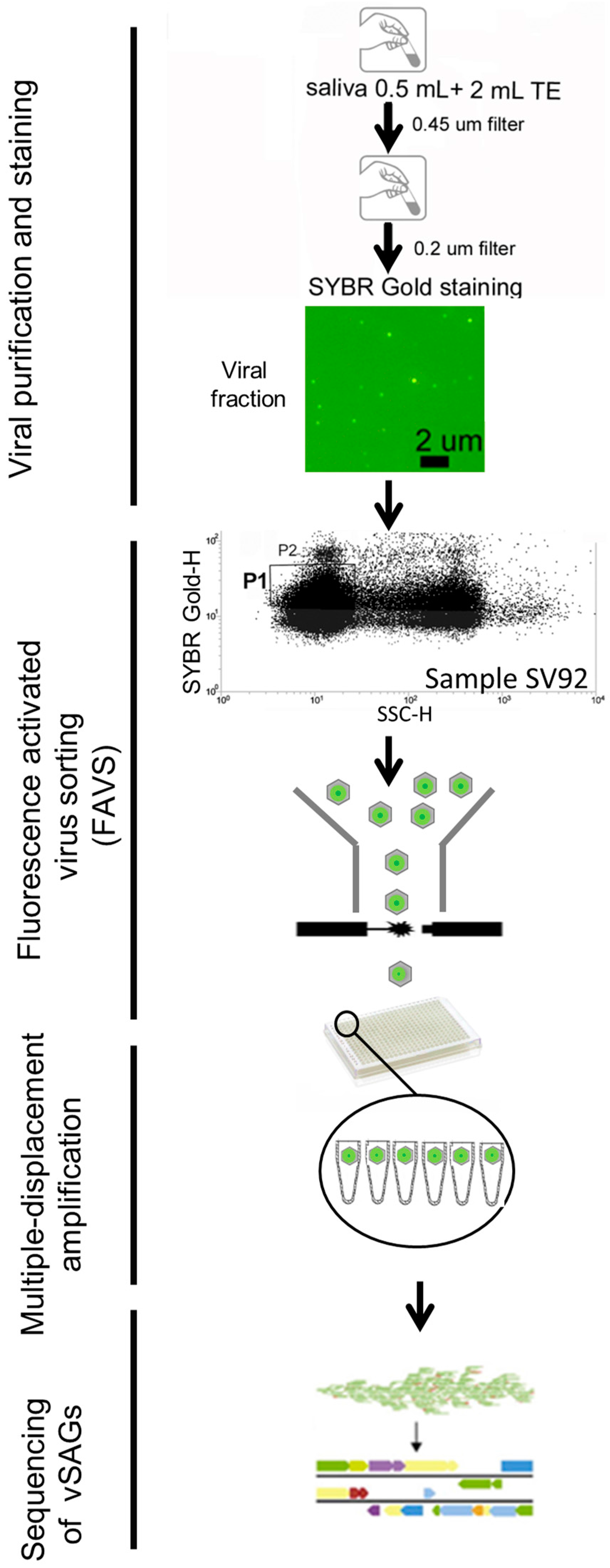
2.1. Human Sample Collection and Processing for Single-Virus Genomics and Viromics
2.2. Sequencing, Assembly, Annotation, and Genome Analyses
2.3. Virome Fragment Recruitment
2.4. Gene Content-Based Network Analysis
3. Results
3.1. Single-Virus Genomics: A New Approach in the Context of the Human Virome
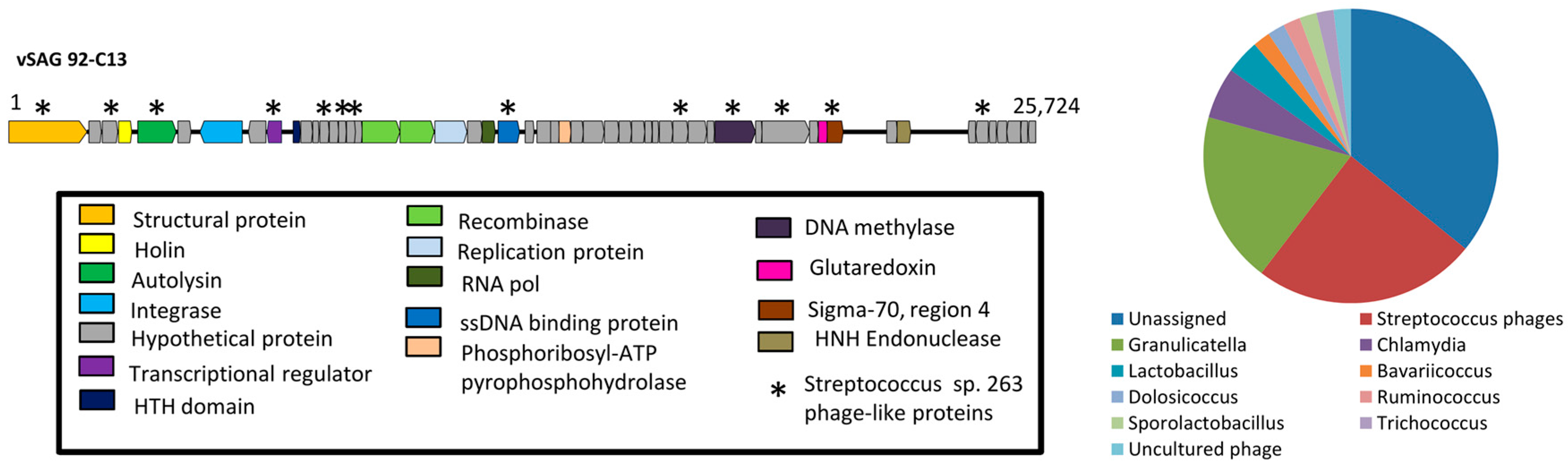
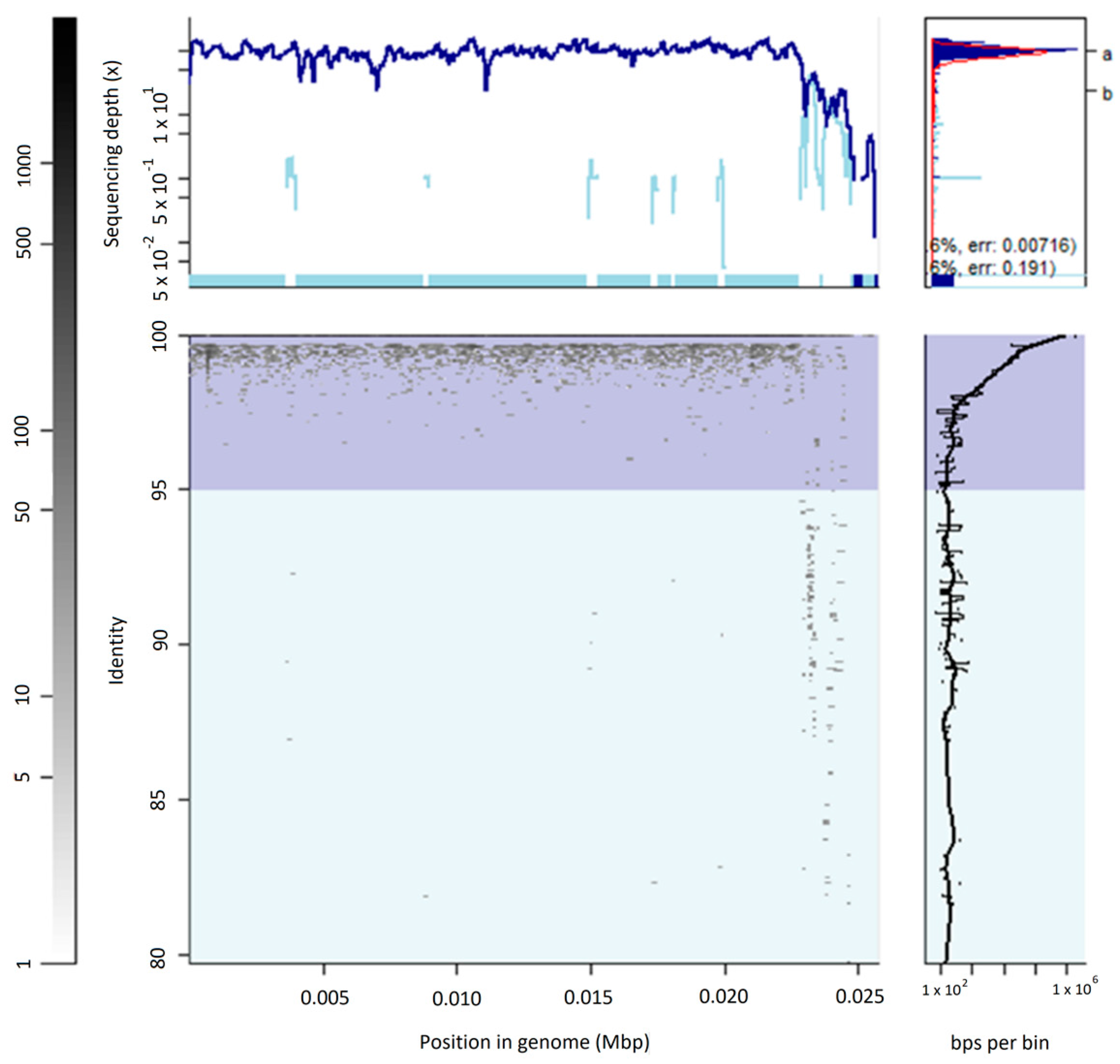
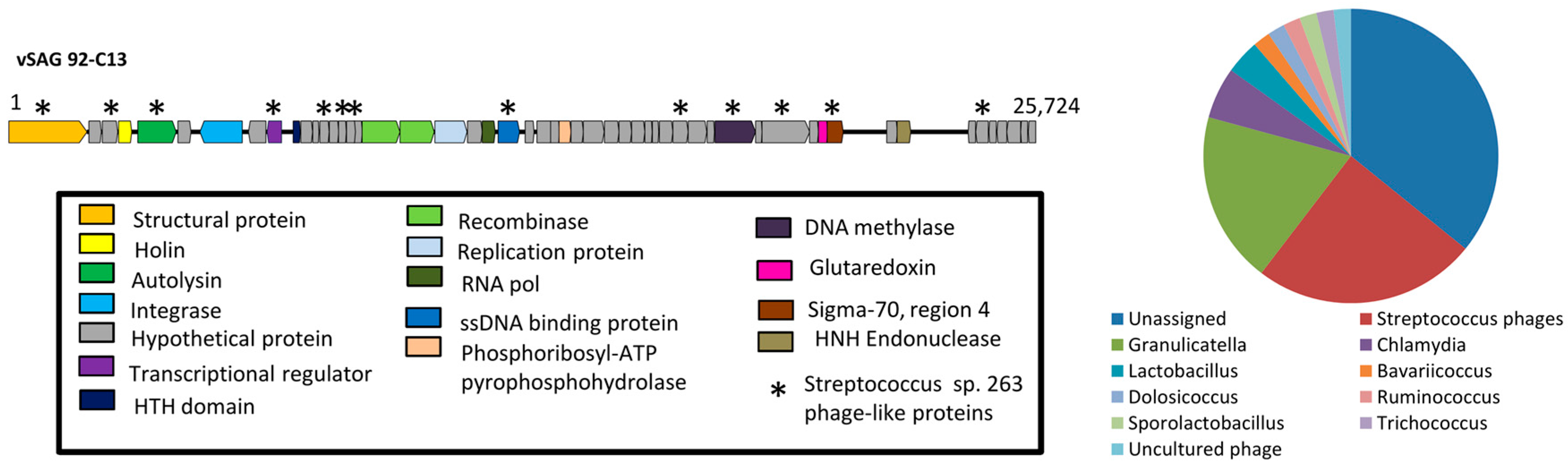
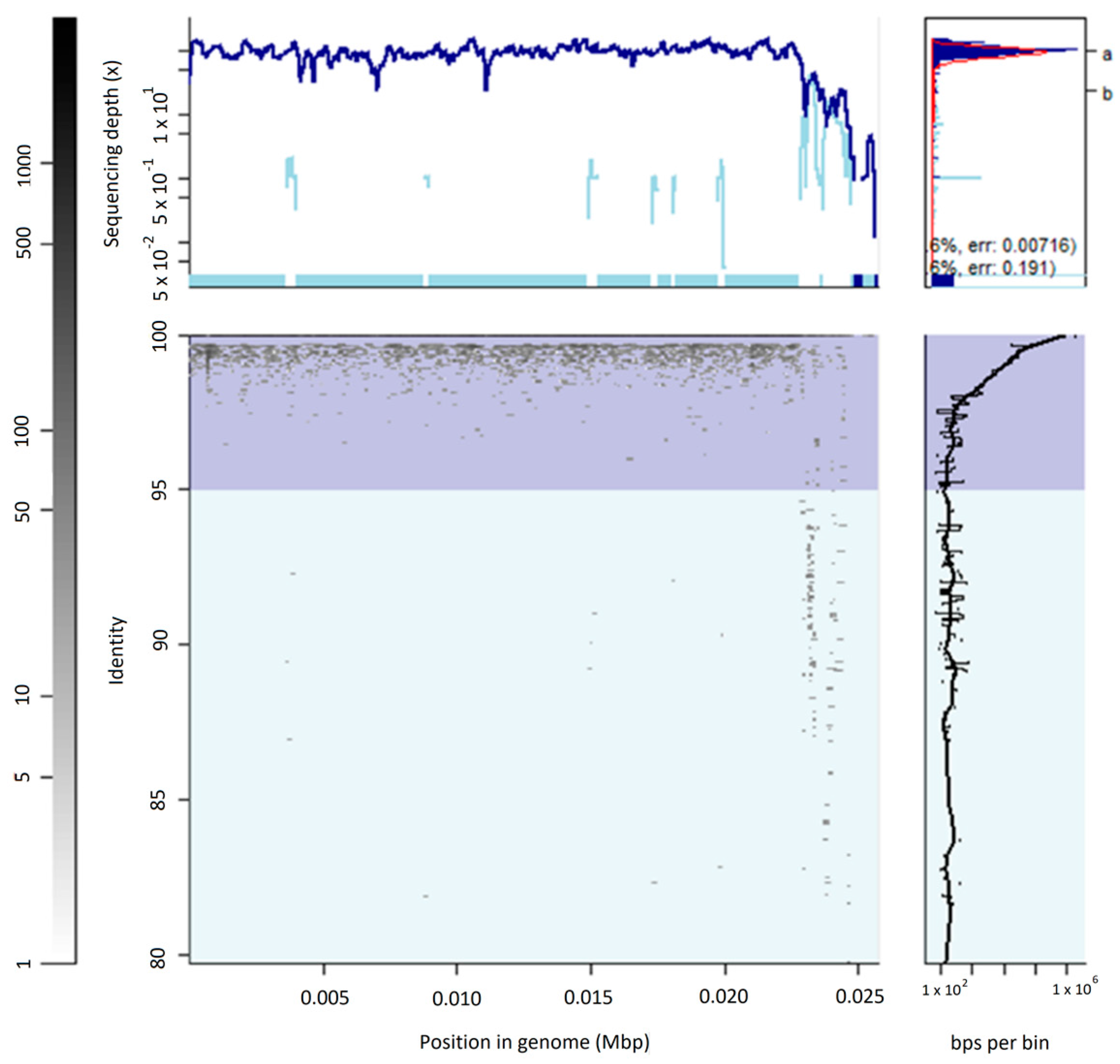
3.2. The Uncultured Abundant Single Virus 92-C13
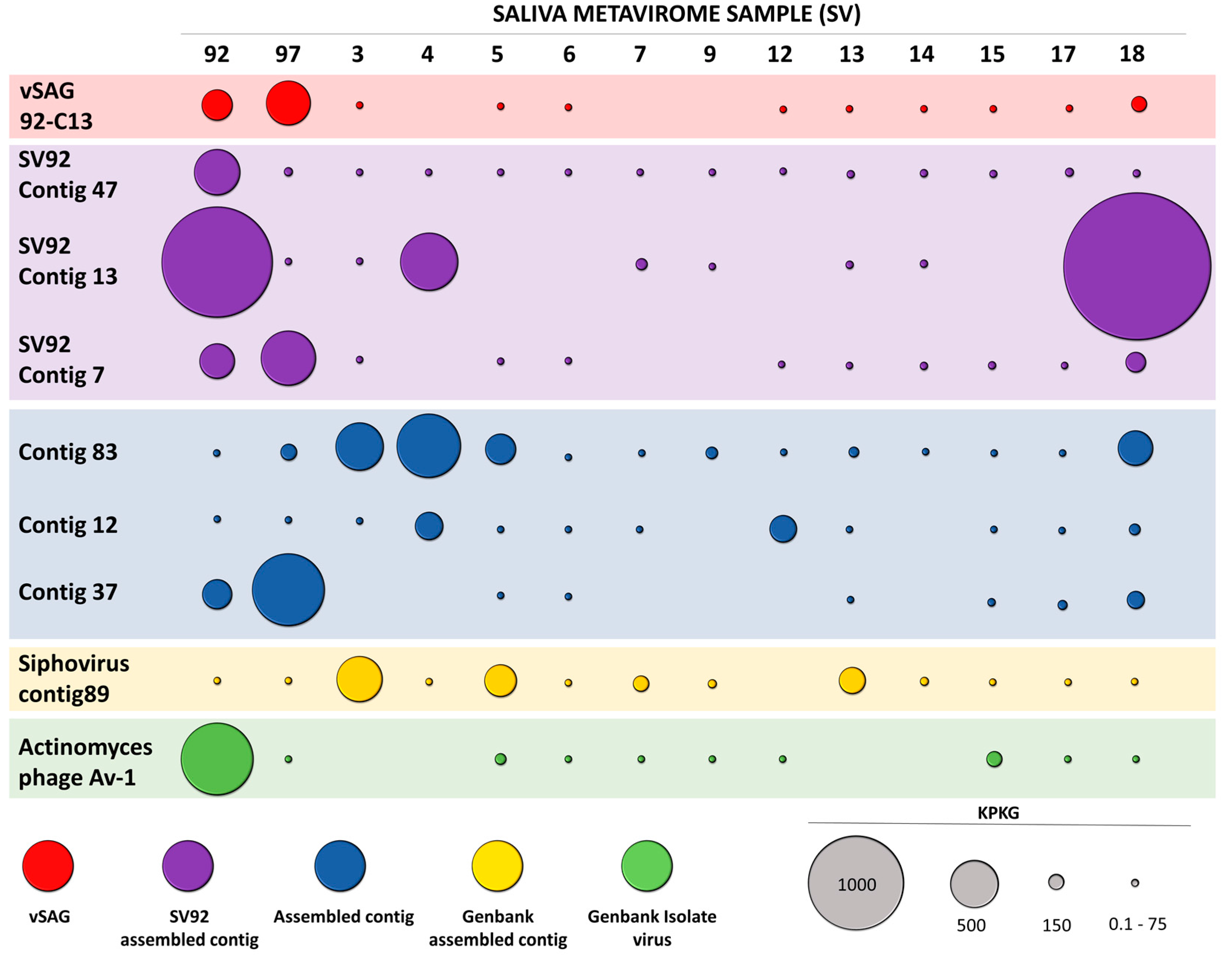
3.3. Abundance and Ubiquity of Viruses in the Oral Virome
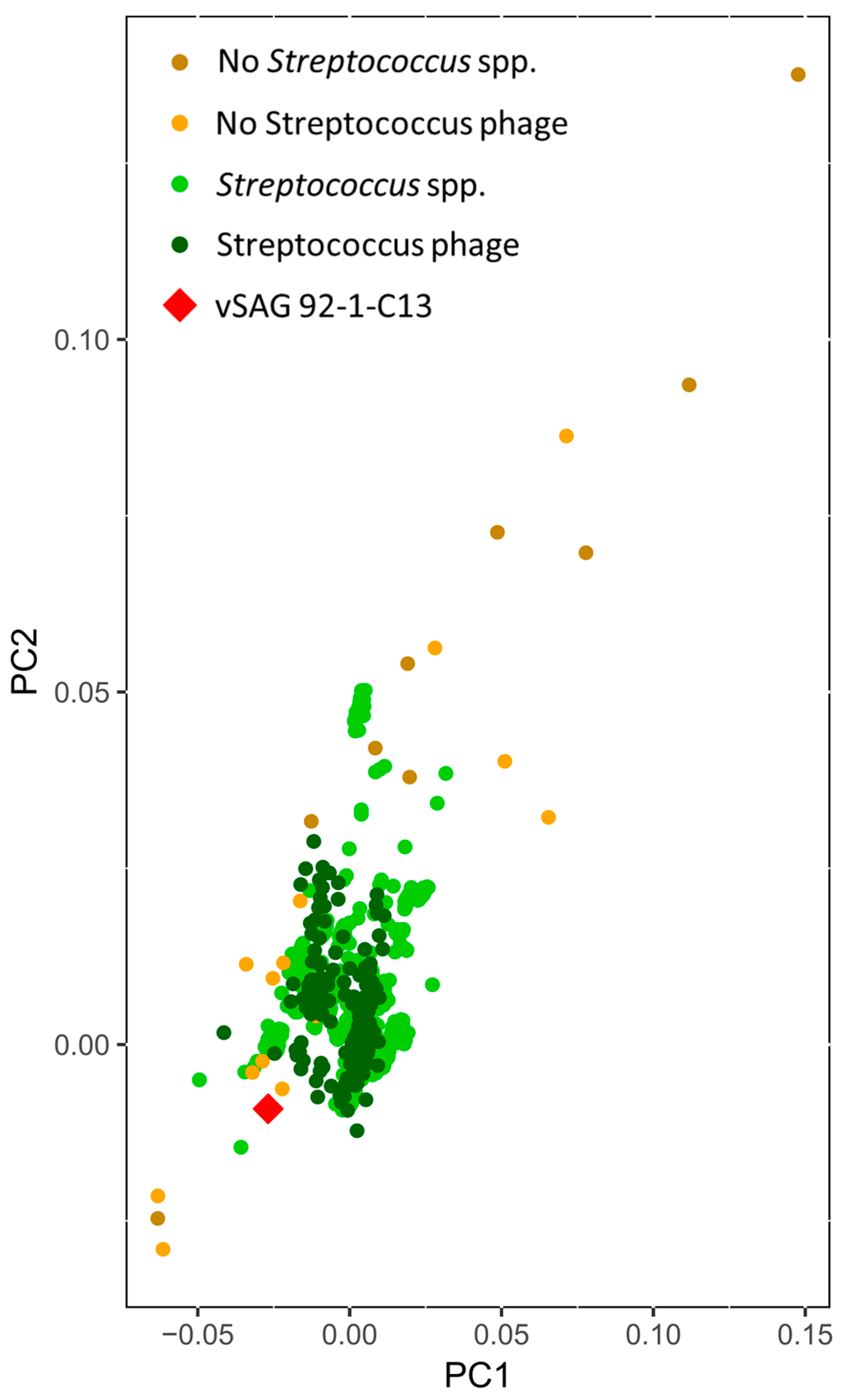
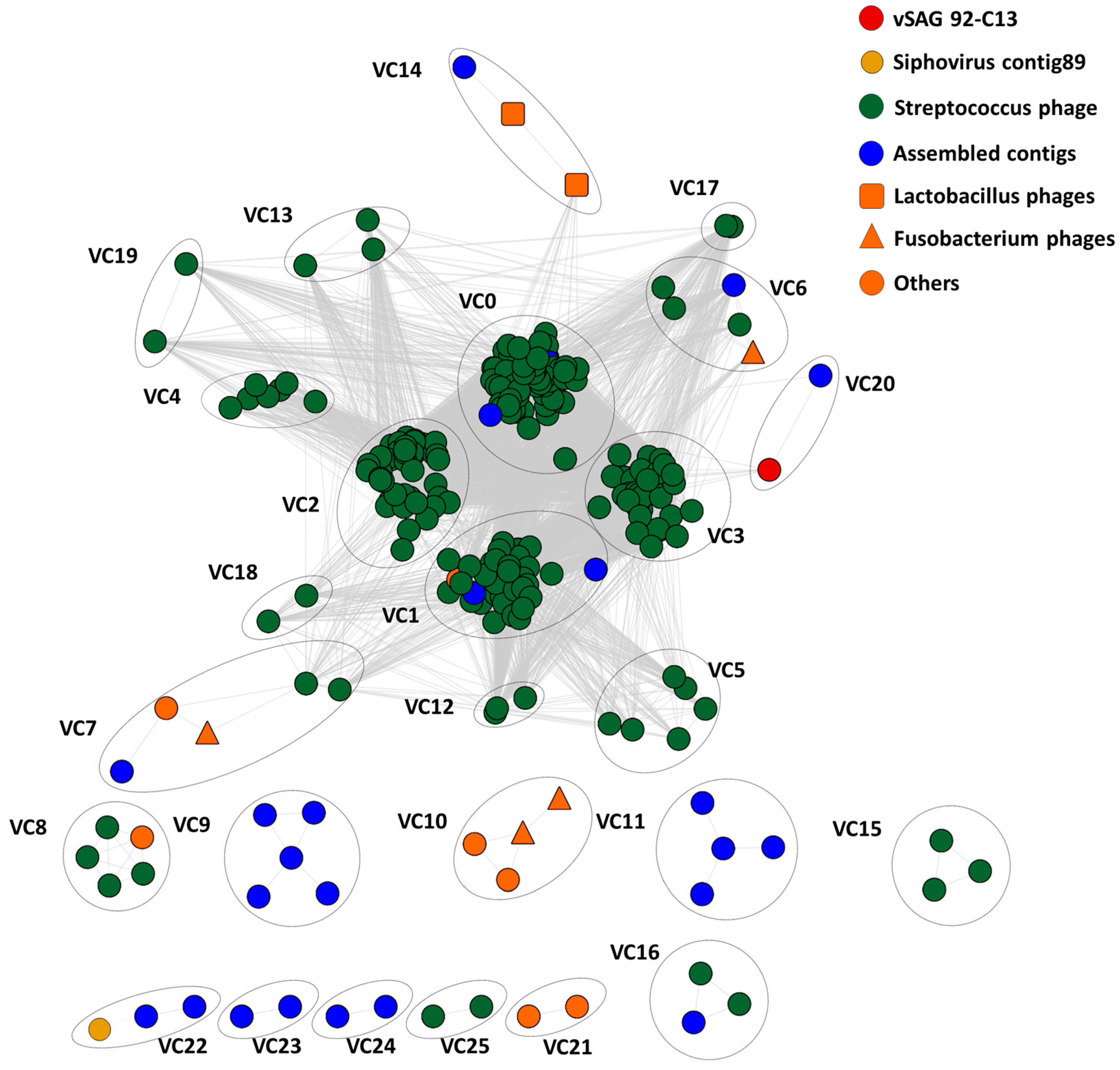
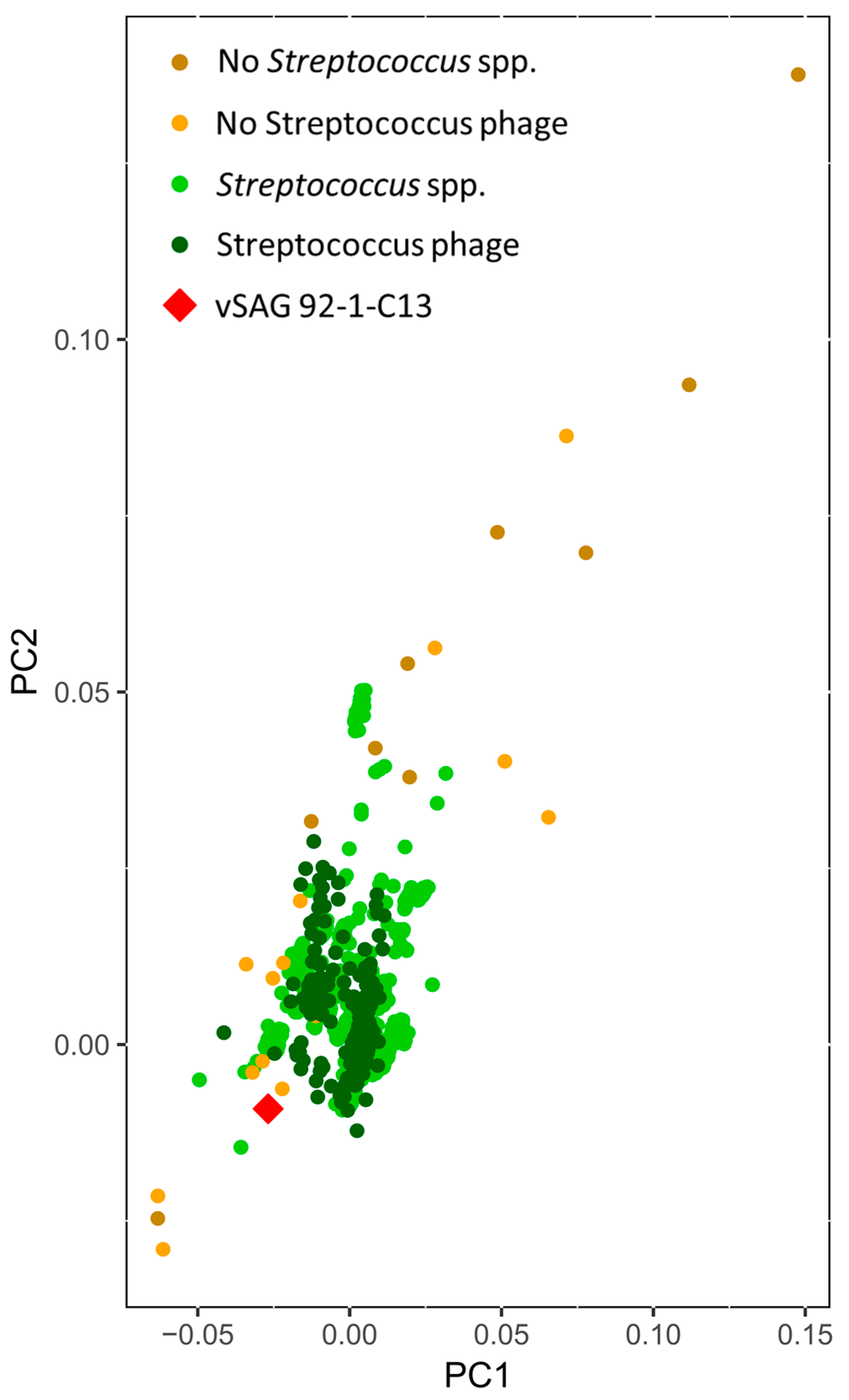
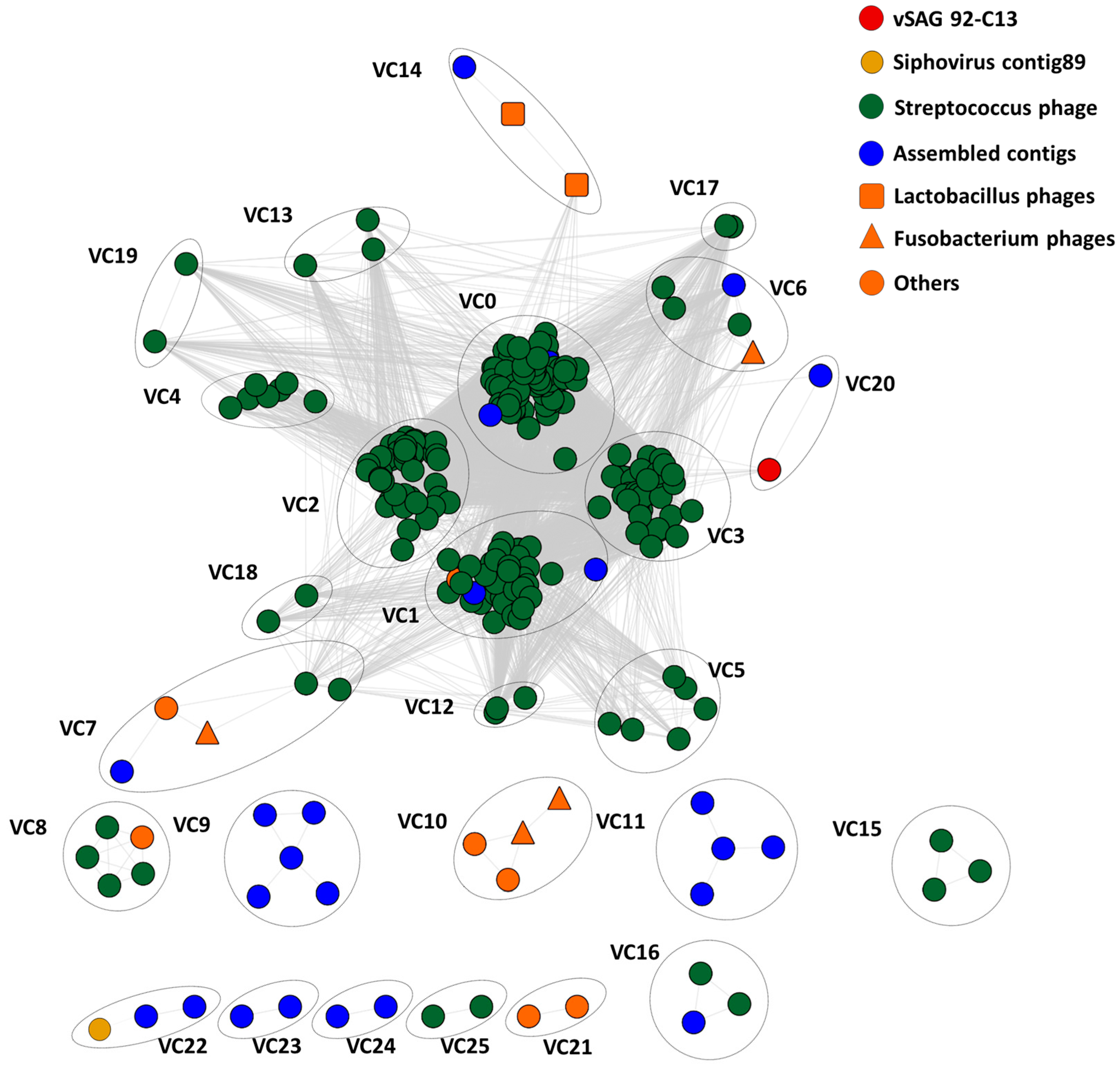
3.4. Tentative Viral Community Structure of the Oral Cavity
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Availability of Data and Materials
References
- Bouziat, R.; Hinterleitner, R.; Brown, J.J.; Stencel-Baerenwald, J.E.; Ikizler, M.; Mayassi, T.; Meisel, M.; Kim, S.M.; Discepolo, V.; Pruijssers, A.J.; et al. Reovirus infection triggers inflammatory responses to dietary antigens and development of celiac disease. Science 2017, 356, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.J.; Auro, R.; Furlan, M.; Whiteson, K.L.; Erb, M.L.; Pogliano, J.; Stotland, A.; Wolkowicz, R.; Cutting, A.S.; Doran, K.S.; et al. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc. Natl. Acad. Sci. USA 2013, 110, 10771–10776. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Haynes, M.; Rohwer, F. The Human Virome. In Metagenomics of the Human Body; Springer: New York, NY, USA, 2011; pp. 63–77. [Google Scholar]
- Willner, D.; Furlan, M.; Haynes, M.; Schmieder, R.; Angly, F.E.; Silva, J.; Tammadoni, S.; Nosrat, B.; Conrad, D.; Rohwer, F. Metagenomic analysis of respiratory tract DNA viral communities in cystic fibrosis and non-cystic fibrosis individuals. PLoS ONE 2009, 4, e7370. [Google Scholar] [CrossRef] [PubMed]
- Wylie, K.M.; Weinstock, G.M.; Storch, G.A. Emerging view of the human virome. Transl. Res. 2012, 160, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Lecuit, M.; Eloit, M. The human virome: New tools and concepts. Trends Microbiol. 2013, 21, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.C.P. The other microbiome. Proc. Natl. Acad. Sci. USA 2013, 110, 2682–2684. [Google Scholar] [CrossRef] [PubMed]
- Wylie, K.M.; Mihindukulasuriya, K.A.; Zhou, Y.; Sodergren, E.; Storch, G.A.; Weinstock, G.M. Metagenomic analysis of double-stranded DNA viruses in healthy adults. BMC Biol. 2014, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus statement: Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Duraisamy, R. Metagenomics and the Human Virome in Asymptomatic Individuals. Annu. Rev. Microbiol. 2016. [Google Scholar] [CrossRef]
- Martinez-Hernandez, F.; Fornas, O.; Lluesma Gomez, M.; Bolduc, B.; de La Cruz Peña, M.J.; Martínez, J.M.; Anton, J.; Gasol, J.M.; Rosselli, R.; Rodriguez-Valera, F.; et al. Single-virus genomics reveals hidden cosmopolitan and abundant viruses. Nat. Commun. 2017, 8, 15892. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Emerson, J.B.; Eloe-Fadrosh, E.A.; Sullivan, M.B. Benchmarking viromics: An in silico evaluation of metagenome-enabled estimates of viral community composition and diversity. PeerJ 2017, 5, e3817. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.Z.; Ishoey, T.; Novotny, M.A.; McLean, J.S.; Lasken, R.S.; Williamson, S.J. Single virus genomics: A new tool for virus discovery. PLoS ONE 2011, 6, e17722. [Google Scholar] [CrossRef] [PubMed]
- Stepanauskas, R.; Fergusson, E.A.; Brown, J.; Poulton, N.J.; Tupper, B.; Labonté, J.M.; Becraft, E.D.; Brown, J.M.; Pachiadaki, M.G.; Povilaitis, T.; et al. Improved genome recovery and integrated cell-size analyses of individual uncultured microbial cells and viral particles. Nat. Commun. 2017, 8, 1–10. [Google Scholar]
- Wilson, W.H.; Gilg, I.C.; Moniruzzaman, M.; Field, E.K.; Koren, S.; LeCleir, G.R.; Martínez Martínez, J.; Poulton, N.J.; Swan, B.K.; Stepanauskas, R.; et al. Genomic exploration of individual giant ocean viruses. ISME J. 2017, 11, 1736–1745. [Google Scholar] [CrossRef] [PubMed]
- Lasken, R.S.; McLean, J.S. Recent advances in genomic DNA sequencing of microbial species from single cells. Nat. Rev. Genet. 2014, 15, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Pride, D.T.; Salzman, J.; Haynes, M.; Rohwer, F.; Davis-Long, C.; White, R.A.; Loomer, P.; Armitage, G.C.; Relman, D.A. Evidence of a robust resident bacteriophage population revealed through analysis of the human salivary virome. ISME J. 2012, 6, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Brussaard, C.P.D. Optimization of Procedures for Counting Viruses by Flow Cytometry. Appl. Environ. Microbiol. 2004, 70, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Laurence, M.; Hatzis, C.; Brash, D.E. Common contaminants in next-generation sequencing that hinder discovery of low-abundance microbes. PLoS ONE 2014, 9, e97876. [Google Scholar] [CrossRef] [PubMed]
- Tennessen, K.; Andersen, E.; Clingenpeel, S.; Rinke, C.; Lundberg, D.S.; Han, J.; Dangl, J.L.; Ivanova, N.; Woyke, T.; Kyrpides, N.; et al. ProDeGe: A computational protocol for fully automated decontamination of genomes. ISME J. 2015, 10, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Tournayre, J.; Mahul, A.; Debroas, D.; Enault, F. Metavir 2: New tools for viral metagenome comparison and assembled virome analysis. BMC Bioinf. 2014, 15, 76. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, V.M.; Chen, I.-M.A.; Chu, K.; Szeto, E.; Palaniappan, K.; Pillay, M.; Ratner, A.; Huang, J.; Pagani, I.; Tringe, S.; et al. IMG/M 4 version of the integrated metagenome comparative analysis system. Nucleic Acids Res. 2014, 42, D568–D573. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinf. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, K.; Parkhill, J.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.-A.; Barrell, B. Artemis: Sequence visualization and annotation. Bioinformatics 2000, 16, 944–945. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Rodriguez-R, L.M.; Konstantinidis, K.T. The enveomics collection: A toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ 2016. [Google Scholar] [CrossRef]
- Roux, S.; Brum, J.R.; Dutilh, B.E.; Sunagawa, S.; Duhaime, M.B.; Loy, A.; Poulos, B.T.; Solonenko, N.; Lara, E.; Poulain, J.; et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 2016, 537, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, H.; Taniguchi, T.; Itoh, T. MetaGeneAnnotator: Detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res. 2008, 15, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, B.; Jang, H.; Doulcier, G.; You, Z.-Q.; Roux, S.; Sullivan, M.B. vConTACT: An iVirus tool to classify double-stranded DNA viruses that infect Archaea and Bacteria. PeerJ 2017, 5, e3243. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Abeles, S.R.; Robles-Sikisaka, R.; Ly, M.; Lum, A.G.; Salzman, J.; Boehm, T.K.; Pride, D.T. Human oral viruses are personal, persistent and gender-consistent. ISME J. 2014, 8, 1753–1767. [Google Scholar] [CrossRef] [PubMed]
- Gawad, C.; Koh, W.; Quake, S.R. Single-cell genome sequencing: Current state of the science. Nat. Rev. Genet. 2016, 17, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Forterre, P.; Soler, N.; Krupovic, M.; Marguet, E.; Ackermann, H.-W. Fake virus particles generated by fluorescence microscopy. Trends Microbiol. 2013, 21, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Biller, S.J.; Schubotz, F.; Roggensack, S.E.; Thompson, A.W.; Summons, R.E.; Chisholm, S.W. Bacterial vesicles in marine ecosystems. Science 2014, 343, 183–186. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De la Cruz Peña, M.J.; Martinez-Hernandez, F.; Garcia-Heredia, I.; Lluesma Gomez, M.; Fornas, Ò.; Martinez-Garcia, M. Deciphering the Human Virome with Single-Virus Genomics and Metagenomics. Viruses 2018, 10, 113. https://doi.org/10.3390/v10030113
De la Cruz Peña MJ, Martinez-Hernandez F, Garcia-Heredia I, Lluesma Gomez M, Fornas Ò, Martinez-Garcia M. Deciphering the Human Virome with Single-Virus Genomics and Metagenomics. Viruses. 2018; 10(3):113. https://doi.org/10.3390/v10030113
Chicago/Turabian StyleDe la Cruz Peña, Maria José, Francisco Martinez-Hernandez, Inmaculada Garcia-Heredia, Mónica Lluesma Gomez, Òscar Fornas, and Manuel Martinez-Garcia. 2018. "Deciphering the Human Virome with Single-Virus Genomics and Metagenomics" Viruses 10, no. 3: 113. https://doi.org/10.3390/v10030113