The Challenges of Analysing Highly Diverse Picobirnavirus Sequence Data


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
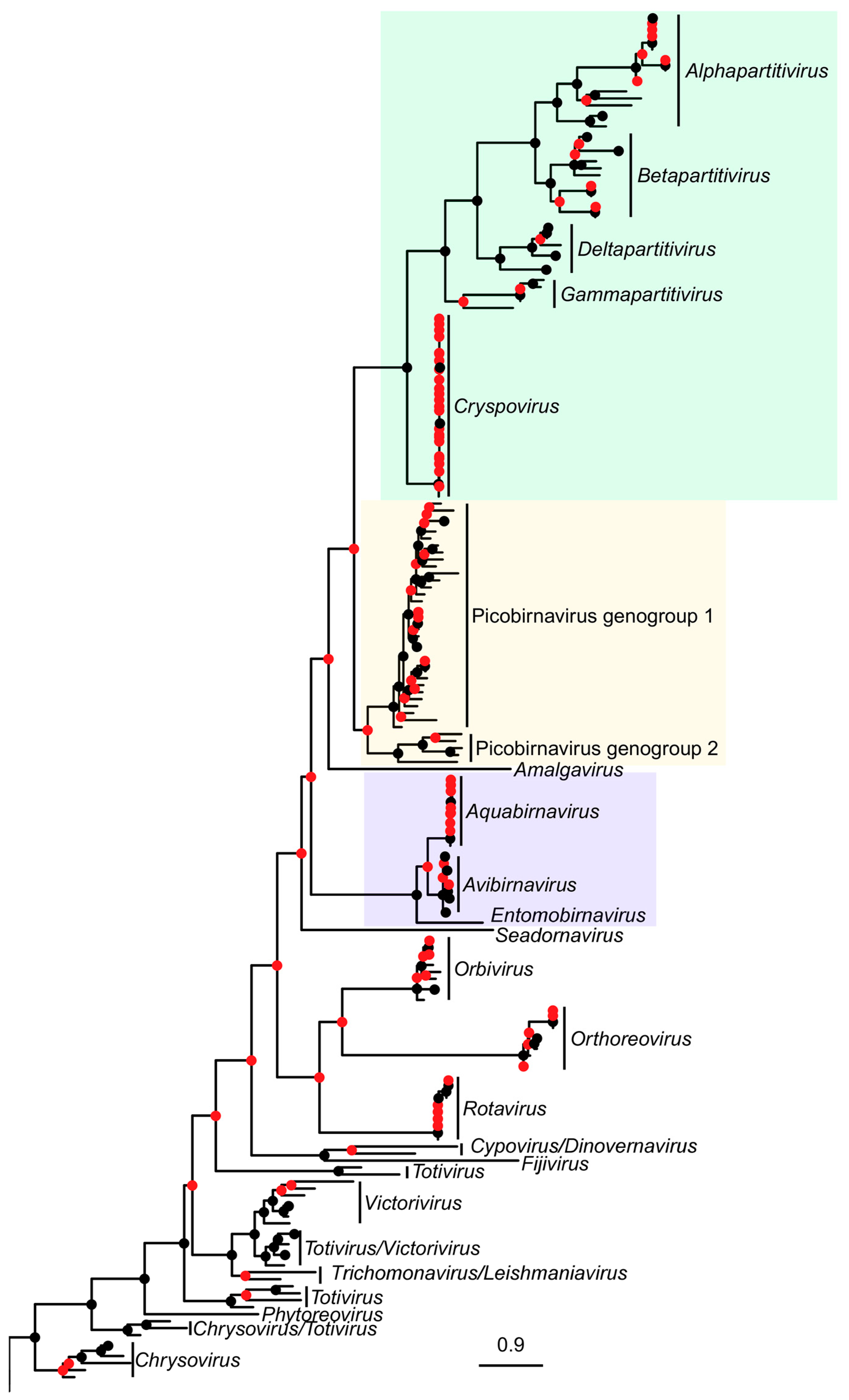
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Doorn, H.R. Emerging infectious diseases. Medicine 2014, 42, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Morse, S.S.; Mazet, J.A.K.; Woolhouse, M.; Parrish, C.R.; Carroll, D.; Karesh, W.B.; Zambrana-Torrelio, C.; Lipkin, W.I.; Daszak, P. Prediction and prevention of the next pandemic zoonosis. Lancet 2012, 380, 1956–1965. [Google Scholar] [CrossRef]
- Taylor, L.H.; Latham, S.M.; Woolhouse, M.E. Risk factors for human disease emergence. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2001, 356, 983–989. [Google Scholar] [CrossRef]
- Delwart, E.L. Viral metagenomics. Rev. Med. Virol. 2007, 17, 115–131. [Google Scholar] [CrossRef]
- Edwards, R.A.; Rohwer, F. Viral metagenomics. Nat. Rev. Microbiol. 2005, 3, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.-D.; Chen, X.; Tian, J.-H.; Chen, L.-J.; Li, K.; Wang, W.; Eden, J.-S.; Shen, J.-J.; Liu, L.; et al. The evolutionary history of vertebrate RNA viruses. Nature 2018, 556, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Rohwer, F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 2005, 13, 278–284. [Google Scholar] [CrossRef]
- Rohwer, F.; Edwards, R. The phage proteomic tree: A genome-based taxonomy for phage. J. Bacteriol. 2002, 184, 4529–4535. [Google Scholar] [CrossRef]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Mokili, J.L.; Rohwer, F.; Dutilh, B.E. Metagenomics and future perspectives in virus discovery. Curr. Opin. Virol. 2012, 2, 63–77. [Google Scholar] [CrossRef]
- Krishnamurthy, S.R.; Wang, D. Origins and challenges of viral dark matter. Virus Res. 2017, 239, 136–142. [Google Scholar] [CrossRef]
- Ng, T.F.; Vega, E.; Kondov, N.O.; Markey, C.; Deng, X.; Gregoricus, N.; Vinje, J.; Delwart, E. Divergent picobirnaviruses in human feces. Genome Announc. 2014, 2, e00415-14. [Google Scholar] [CrossRef]
- Banyai, K.; Jakab, F.; Reuter, G.; Bene, J.; Uj, M.; Melegh, B.; Szucs, G. Sequence heterogeneity among human picobirnaviruses detected in a gastroenteritis outbreak. Arch. Virol. 2003, 148, 2281–2291. [Google Scholar] [CrossRef]
- Ganesh, B.; Nataraju, S.M.; Rajendran, K.; Ramamurthy, T.; Kanungo, S.; Manna, B.; Nagashima, S.; Sur, D.; Kobayashi, N.; Krishnan, T. Detection of closely related picobirnaviruses among diarrhoeic children in Kolkata: Evidence of zoonoses? Infect. Genet. Evol. 2010, 10, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tu, X.; Humphrey, C.; McClure, H.; Jiang, X.; Qin, C.; Glass, R.I.; Jiang, B. Detection of viral agents in fecal specimens of monkeys with diarrhea. J. Med. Primatol. 2007, 36, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, B.; Banyai, K.; Masachessi, G.; Mladenova, Z.; Nagashima, S.; Ghosh, S.; Nataraju, S.M.; Pativada, M.; Kumar, R.; Kobayashi, N. Genogroup I picobirnavirus in diarrhoeic foals: Can the horse serve as a natural reservoir for human infection? Vet. Res. 2011, 42, 52. [Google Scholar] [CrossRef] [PubMed]
- Masachessi, G.; Martinez, L.C.; Giordano, M.O.; Barril, P.A.; Isa, B.M.; Ferreyra, L.; Villareal, D.; Carello, M.; Asis, C.; Nates, S.V. Picobirnavirus (PBV) natural hosts in captivity and virus excretion pattern in infected animals. Arch. Virol. 2007, 152, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.R.; Wang, D. Extensive conservation of prokaryotic ribosomal binding sites in known and novel picobirnaviruses. Virology 2018, 516, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Yinda, C.K.; Ghogomu, S.M.; Conceicao-Neto, N.; Beller, L.; Deboutte, W.; Vanhulle, E.; Maes, P.; Van Ranst, M.; Matthijnssens, J. Cameroonian fruit bats harbor divergent viruses, including rotavirus H, bastroviruses, and picobirnaviruses using an alternative genetic code. Virus Evol. 2018, 4, vey008. [Google Scholar] [CrossRef]
- Pereira, H.G.; Fialho, A.M.; Flewett, T.H.; Teixeira, J.M.S.; Andrade, Z.P. Novel viruses in human feces. Lancet 1988, 2, 103–104. [Google Scholar] [CrossRef]
- Pereira, H.G.; Flewett, T.H.; Candeias, J.A.N.; Barth, O.M. A virus with a bisegmented double-stranded-RNA genome in rat (Oryzomys nigripes) intestines. J. Gen. Virol. 1988, 69, 2749–2754. [Google Scholar] [CrossRef]
- Carstens, E.B.; Ball, L.A. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2008). Arch. Virol. 2009, 154, 1181–1188. [Google Scholar] [CrossRef]
- Rosen, B.I.; Fang, Z.Y.; Glass, R.I.; Monroe, S.S. Cloning of human picobirnavirus genomic segments and development of an RT-PCR detection assay. Virology 2000, 277, 316–329. [Google Scholar] [CrossRef]
- Banyai, K.; Martella, V.; Bogdan, A.; Forgach, P.; Jakab, F.; Meleg, E.; Biro, H.; Melegh, B.; Szucs, G. Genogroup I picobirnaviruses in pigs: Evidence for genetic diversity and relatedness to human strains. J. Gen. Virol. 2008, 89, 534–539. [Google Scholar] [CrossRef]
- Duarte Fregolente, M.C.; de Castro-Dias, E.; Martins, S.S.; Spilki, F.R.; Allegretti, S.M.; Viccari Gatti, M.S. Molecular characterization of picobirnaviruses from new hosts. Virus Res. 2009, 143, 134–136. [Google Scholar] [CrossRef]
- Symonds, E.M.; Griffin, D.W.; Breitbart, M. Eukaryotic viruses in wastewater samples from the United States. Appl. Environ. Microbiol. 2009, 75, 1402–1409. [Google Scholar] [CrossRef]
- Zhang, S.; Bai, R.; Feng, R.; Zhang, H.; Liu, L. Detection and evolutionary analysis of picobirnaviruses in treated wastewater. Microb. Biotechnol. 2015, 8, 474–482. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Rai, R.; Teng, J.L.L.; Lee, P.; Martelli, P.; Hui, S.-W.; Yuen, K.-Y. Complete genome sequence of a novel picobirnavirus, otarine picobirnavirus, discovered in California sea lions. J. Virol. 2012, 86, 6377–6378. [Google Scholar] [CrossRef]
- Verma, H.; Mor, S.K.; Erber, J.; Goyal, S.M. Prevalence and complete genome characterization of turkey picobirnaviruses. Infect. Genet. Evol. 2015, 30, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Kobayashi, N.; Nagashima, S.; Naik, T.N. Molecular characterization of full-length genomic segment 2 of a bovine picobirnavirus (PBV) strain: Evidence for high genetic diversity with genogroup I PBVs. J. Gen. Virol. 2009, 90, 2519–2524. [Google Scholar] [CrossRef]
- Woo, P.C.; Teng, J.L.; Bai, R.; Wong, A.Y.; Martelli, P.; Hui, S.W.; Tsang, A.K.; Lau, C.C.; Ahmed, S.S.; Yip, C.C.; et al. High diversity of genogroup I picobirnaviruses in mammals. Front. Microbiol. 2016, 7, 1886. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, B.; Masachessi, G.; Mladenova, Z. Animal Picobirnavirus. Virus Dis. 2014, 25, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Smits, S.L.; Poon, L.L.; van Leeuwen, M.; Lau, P.N.; Perera, H.K.; Peiris, J.S.; Simon, J.H.; Osterhaus, A.D. Genogroup I and II picobirnaviruses in respiratory tracts of pigs. Emerg. Infect. Dis. 2011, 17, 2328–2330. [Google Scholar] [CrossRef]
- Van Leeuwen, M.; Williams, M.M.W.; Koraka, P.; Simon, J.H.; Smits, S.L.; Osterhaus, A.D. Human picobirnaviruses identified by molecular screening of diarrhea samples. J. Clin. Microbiol. 2010, 48, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, M.C.; Cagua, E.F.; Balbuena, J.A.; Stouffer, D.B.; Poisot, T. paco: Implementing Procrustean Approach to Cophylogeny in R. Methods Ecol. Evol. 2017, 8, 932–940. [Google Scholar] [CrossRef]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Gallo Cassarino, T.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Collier, A.M.; Lyytinen, O.L.; Guo, Y.R.; Toh, Y.; Poranen, M.M.; Tao, Y.J. Initiation of RNA polymerization and polymerase encapsidation by a small dsRNA virus. PLoS Pathog. 2016, 12, e1005523. [Google Scholar] [CrossRef] [PubMed]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Benkert, P.; Kunzli, M.; Schwede, T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009, 37, W510–W514. [Google Scholar] [CrossRef] [PubMed]
- Finkbeiner, S.R.; Allred, A.F.; Tarr, P.I.; Klein, E.J.; Kirkwood, C.D.; Wang, D. Metagenomic analysis of human diarrhea: Viral detection and discovery. PLoS Pathog. 2008, 4, e1000011. [Google Scholar] [CrossRef]
- Simmonds, P.; Adams, M.J.; Benko, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus statement: Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef]
- Smits, S.L.; Schapendonk, C.M.E.; van Beek, J.; Vennema, H.; Schurch, A.C.; Schipper, D.; Bodewes, R.; Haagmans, B.L.; Osterhaus, A.D.; Koopmans, M.P. New viruses in idiopathic human diarrhea cases, the Netherlands. Emerg. Infect. Dis. 2014, 20, 1218–1222. [Google Scholar] [CrossRef]
- Sun, G.; Zang, Q.; Gu, Y.; Niu, G.; Ding, C.; Zhang, P. Viral metagenomics analysis of picobirnavirus-positive feces from children with sporadic diarrhea in China. Arch. Virol. 2016, 161, 971–975. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Teng, J.L.L.; Tsang, A.K.L.; Joseph, M.; Wong, E.Y.M.; Tang, Y.; Sivakumar, S.; Bai, R.; Wernery, R.; et al. Metagenomic analysis of viromes of dromedary camel fecal samples reveals large number and high diversity of circoviruses and picobirnaviruses. Virology 2014, 471, 117–125. [Google Scholar] [CrossRef]
- Conceicao-Neto, N.; Mesquita, J.R.; Zeller, M.; Yinda, C.K.; Alvares, F.; Roque, S.; Petrucci-Fonseca, F.; Godinho, R.; Heylen, E.; Van Ranst, M.; et al. Reassortment among picobirnaviruses found in wolves. Arch. Virol. 2016, 161, 2859–2862. [Google Scholar] [CrossRef] [PubMed]
- Amimo, J.O.; El Zowalaty, M.E.; Githae, D.; Wamalwa, M.; Djikeng, A.; Nasrallah, G.K. Metagenomic analysis demonstrates the diversity of the fecal virome in asymptomatic pigs in East Africa. Arch. Virol. 2016, 161, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Aw, T.G.; Wengert, S.; Rose, J.B. Metagenomic analysis of viruses associated with field-grown and retail lettuce identifies human and animal viruses. Int. J. Food Microbiol. 2016, 223, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Day, J.M.; Zsak, L. Molecular and phylogenetic analysis of a novel turkey-origin picobirnavirus. Avian Dis. 2014, 58, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Hause, B.M.; Duff, J.W.; Scheidt, A.; Anderson, G. Virus detection using metagenomic sequencing of swine nasal and rectal swabs. J. Swine Health Prod. 2016, 24, 304–308. [Google Scholar]
- Kluge, M.; Campos, F.S.; Tavares, M.; de Amorim, D.B.; Valdez, F.P.; Giongo, A.; Roehe, P.M.; Franco, A.C. Metagenomic survey of viral diversity obtained from feces of subantarctic and South American fur seals. PLoS ONE 2016, 11, e0151921. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Giannitti, F.; Low, J.; Keyes, C.; Ullmann, L.S.; Deng, X.; Aleman, M.; Pesavento, P.A.; Pusterla, N.; Delwart, E. Exploring the virome of diseased horses. J. Gen. Virol. 2015, 96, 2721–2733. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knox, M.A.; Gedye, K.R.; Hayman, D.T.S. The Challenges of Analysing Highly Diverse Picobirnavirus Sequence Data. Viruses 2018, 10, 685. https://doi.org/10.3390/v10120685
Knox MA, Gedye KR, Hayman DTS. The Challenges of Analysing Highly Diverse Picobirnavirus Sequence Data. Viruses. 2018; 10(12):685. https://doi.org/10.3390/v10120685
Chicago/Turabian StyleKnox, Matthew A., Kristene R. Gedye, and David T. S. Hayman. 2018. "The Challenges of Analysing Highly Diverse Picobirnavirus Sequence Data" Viruses 10, no. 12: 685. https://doi.org/10.3390/v10120685
APA StyleKnox, M. A., Gedye, K. R., & Hayman, D. T. S. (2018). The Challenges of Analysing Highly Diverse Picobirnavirus Sequence Data. Viruses, 10(12), 685. https://doi.org/10.3390/v10120685