Inter- and Intra-Host Nucleotide Variations in Hepatitis A Virus in Culture and Clinical Samples Detected by Next-Generation Sequencing

Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses, Cell Culture, and Clinical Samples
2.2. Sample Preparation, RNA Isolation, and Viral RNA Quantification by RT-PCR (RT-qPCR)
2.3. Library Generation and Sequencing
2.4. De Novo Assembly and Reference-Based Mapping
2.5. Single Nucleotide Variation Calling from NGS Reads and Confirmation by Pyrosequencing
2.6. GenBank Accession Number
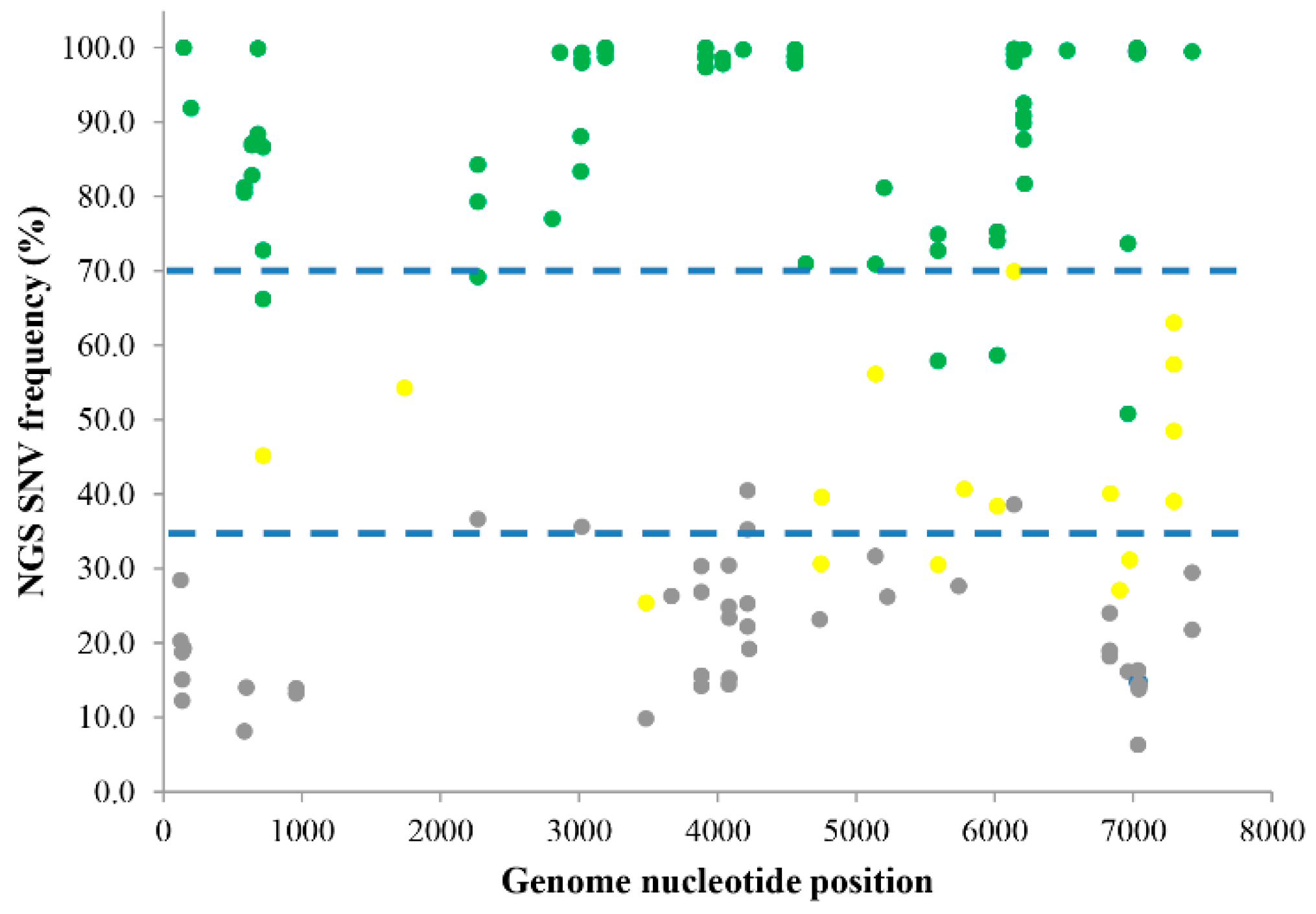
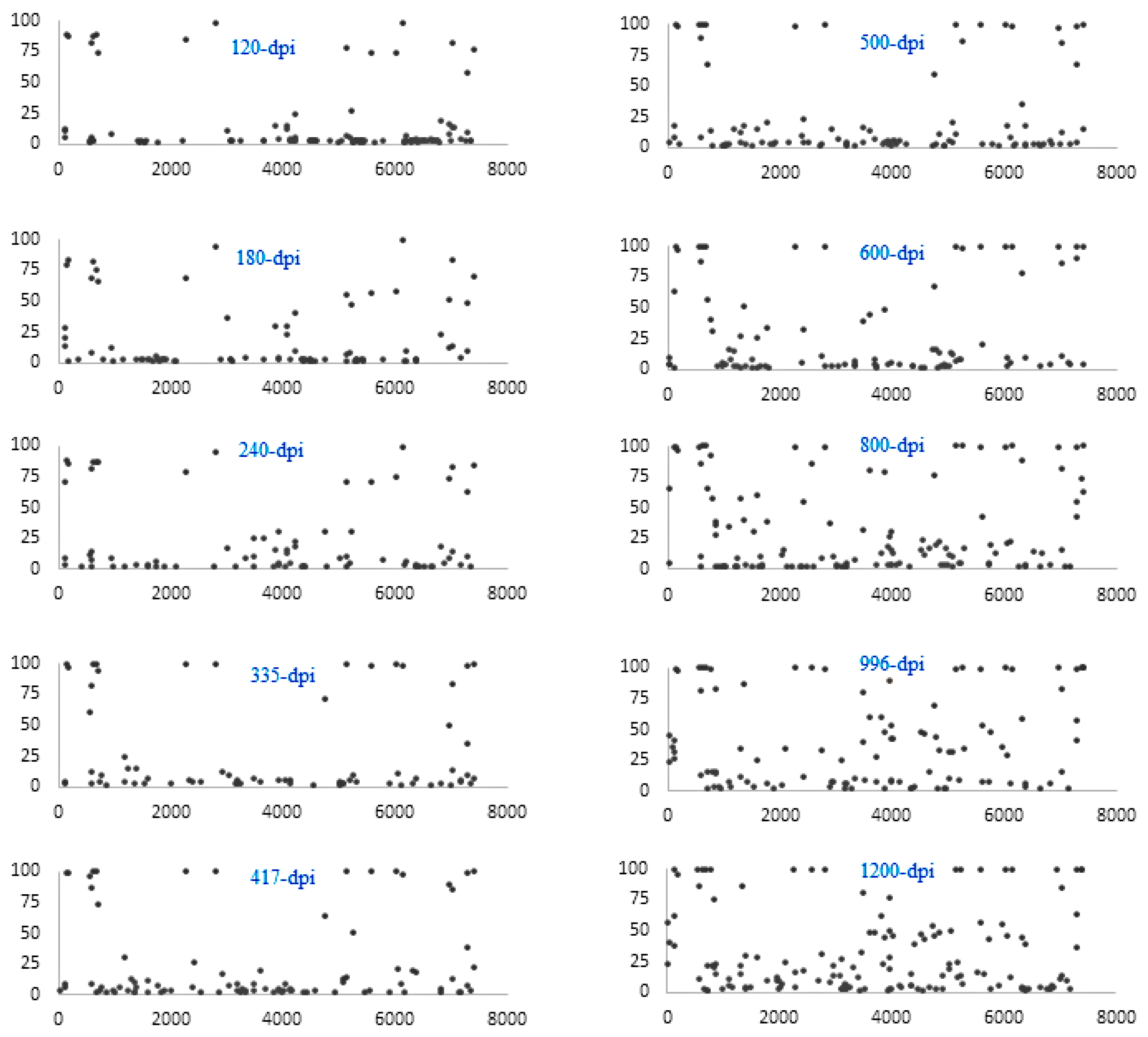
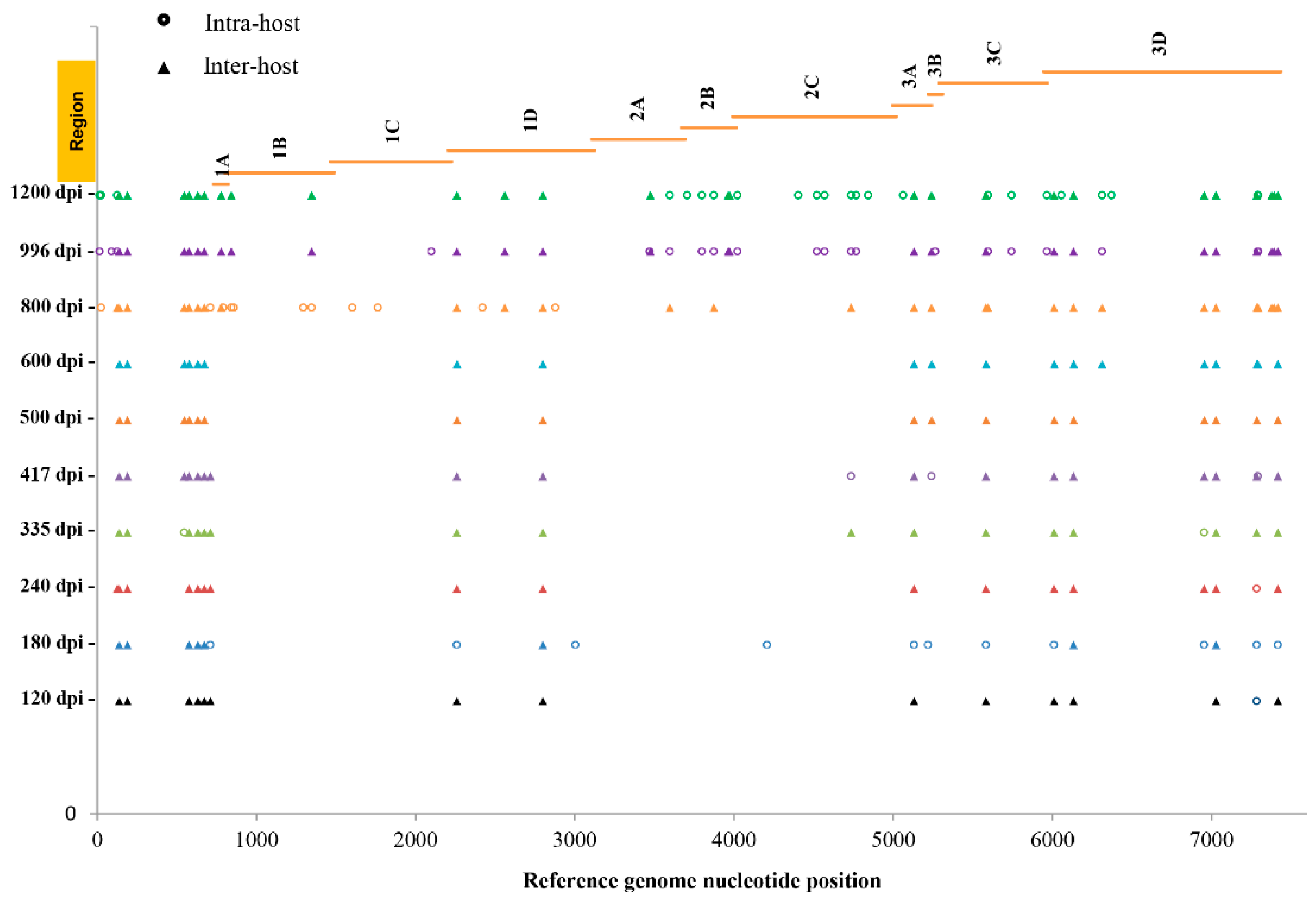
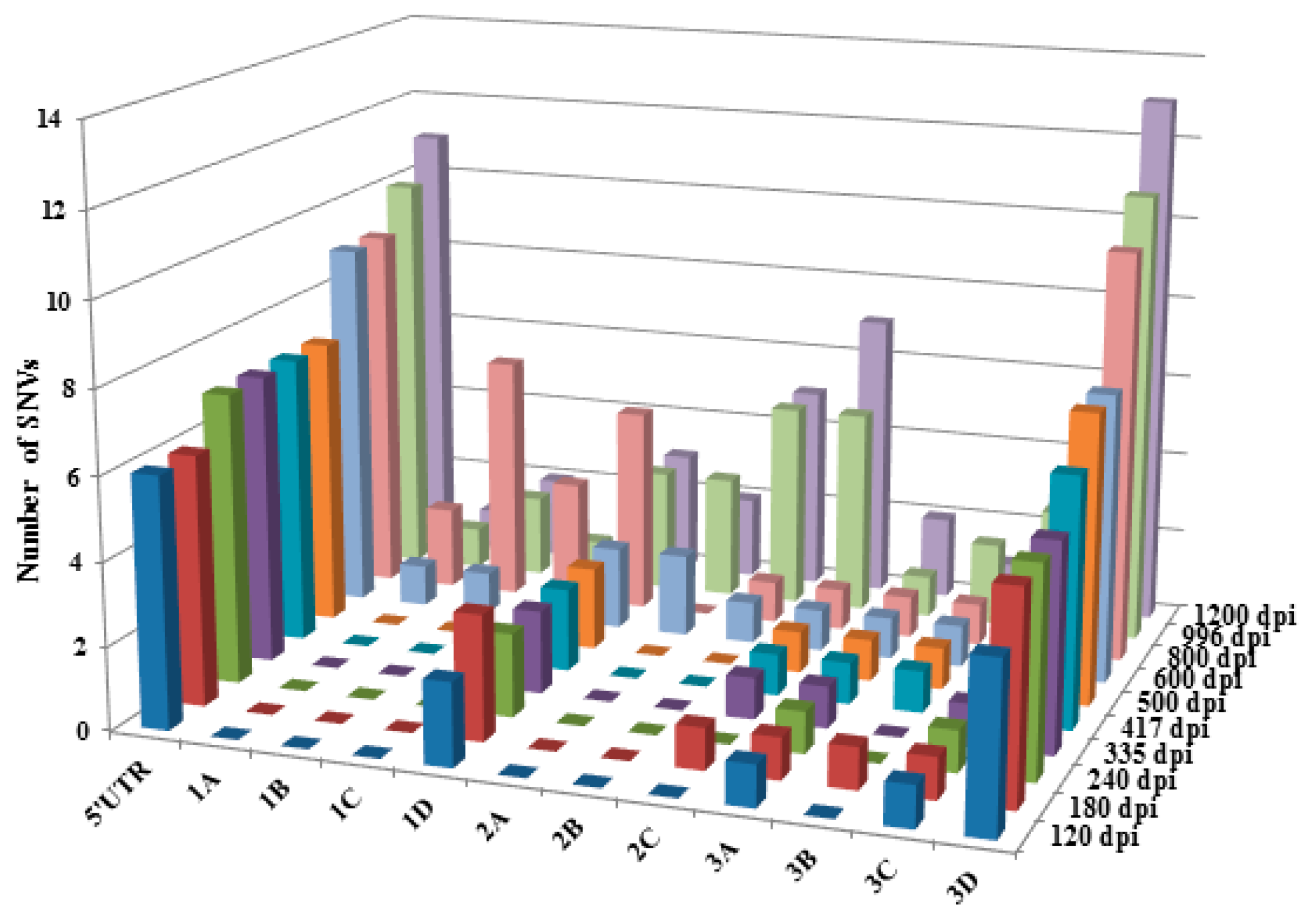
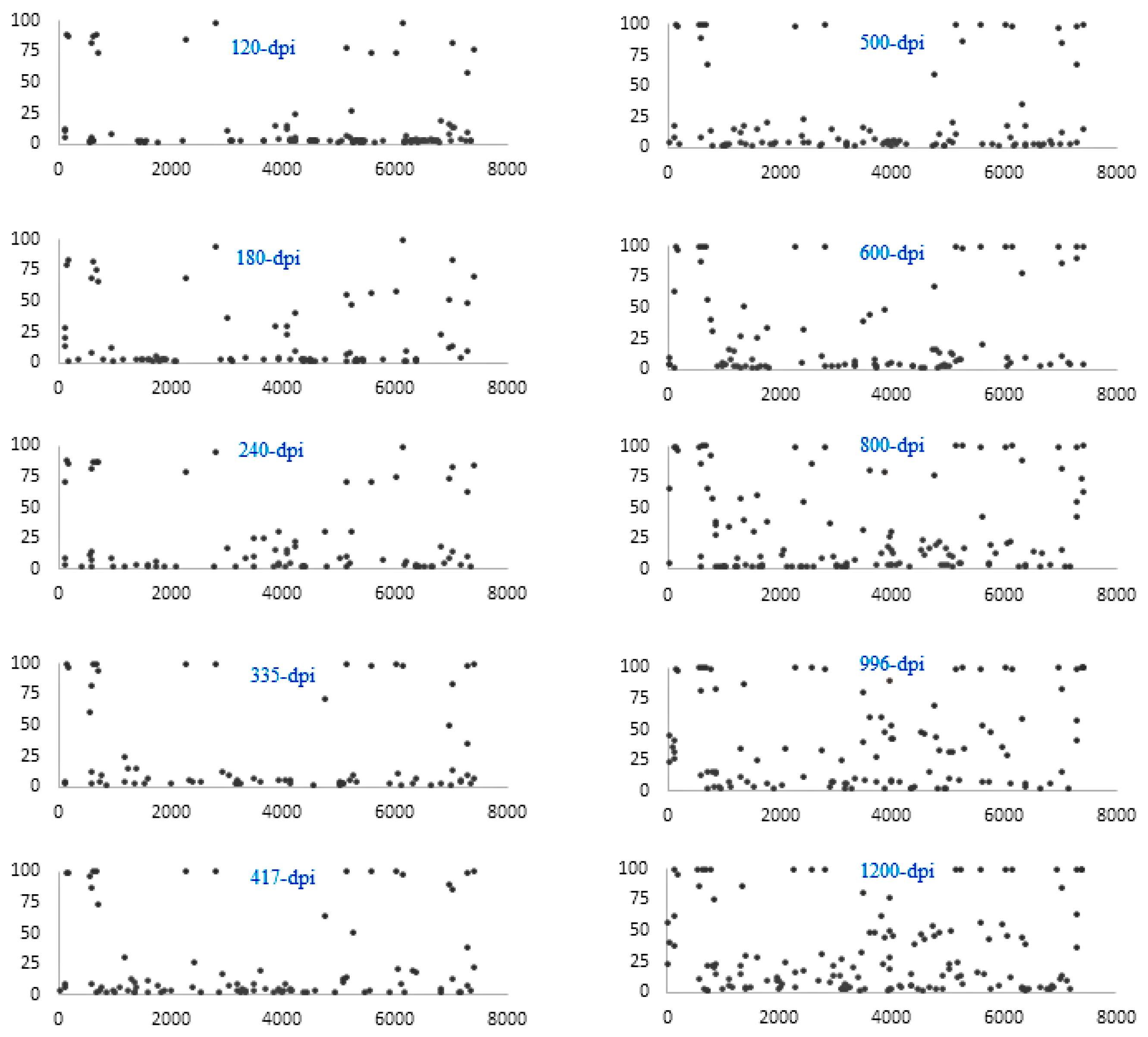
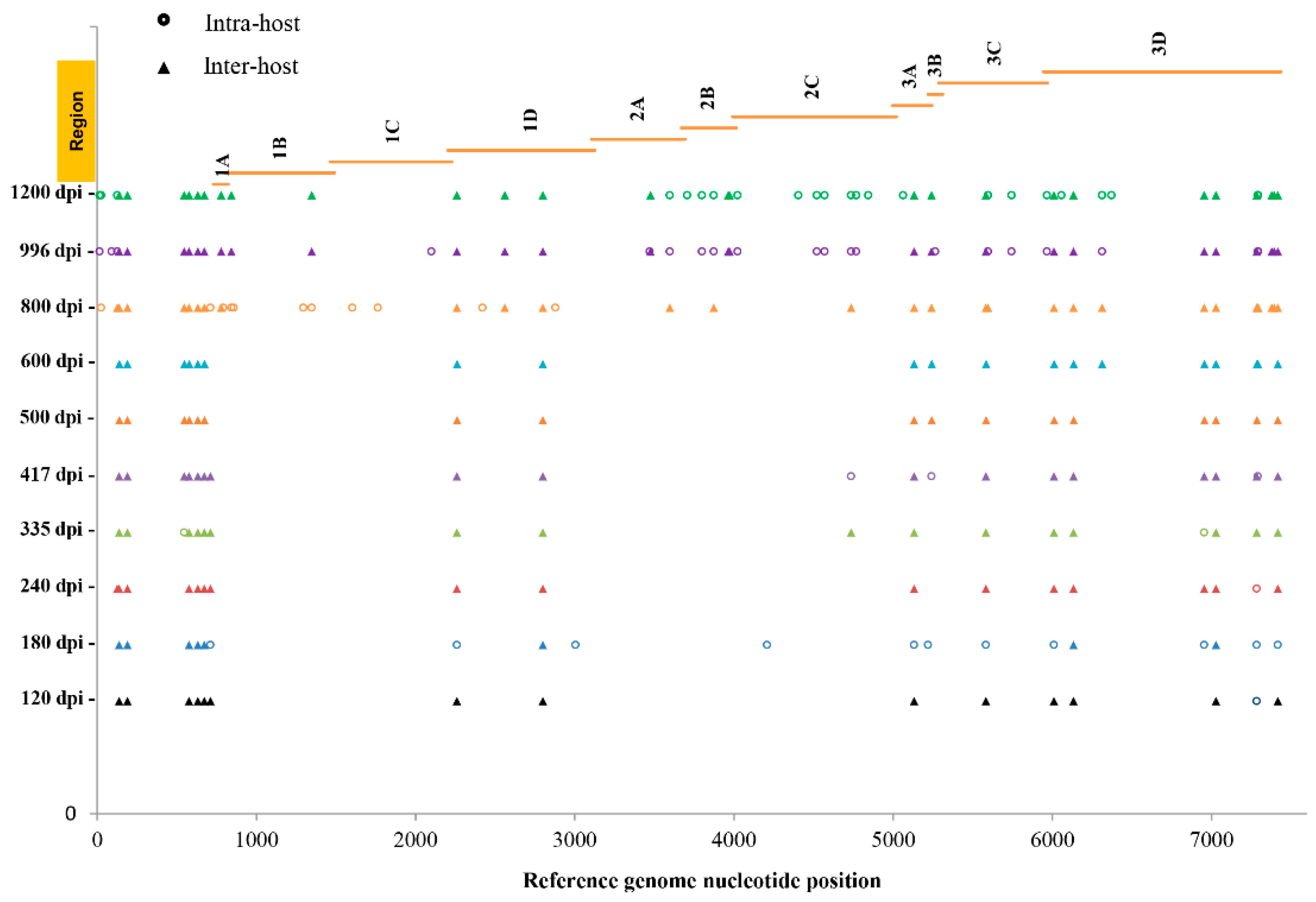
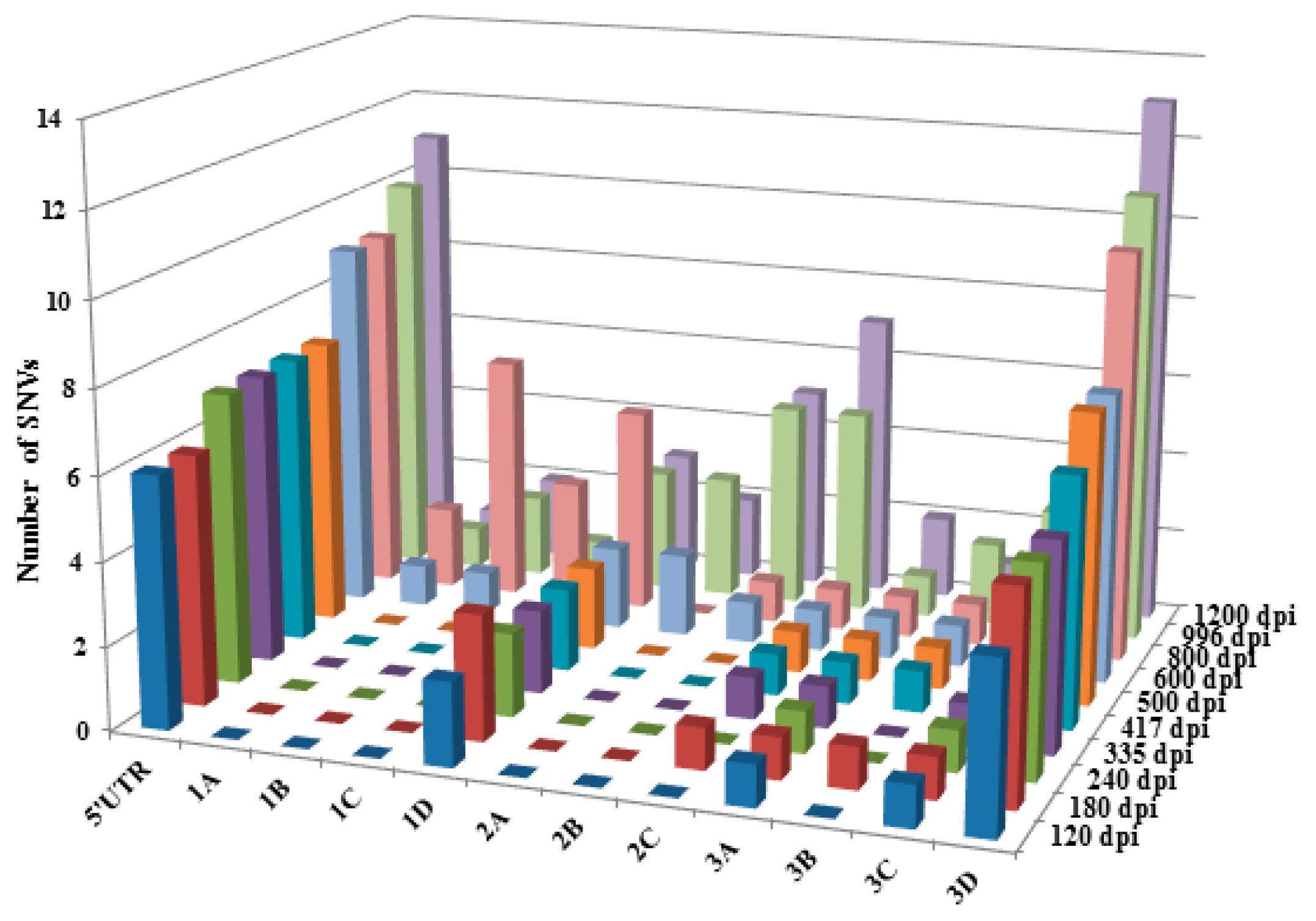
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, Z.; Mammel, M.; Papafragkou, E.; Hida, K.; Elkins, C.A.; Kulka, M. Application of next generation sequencing toward sensitive detection of enteric viruses isolated from celery samples as an example of produce. Int. J. Food Microbiol. 2017, 261, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, T.A. Exonucleolytic proofreading. Cell 1988, 53, 837–840. [Google Scholar] [CrossRef]
- Vaughan, G.; Goncalves Rossi, L.M.; Forbi, J.C.; de Paula, V.S.; Purdy, M.A.; Xia, G.; Khudyakov, Y.E. Hepatitis A virus: Host interactions, molecular epidemiology and evolution. Infect. Genet. Evol. 2014, 21, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.A. What is a quasispecies? Trends Ecol. Evol. 1992, 7, 118–121. [Google Scholar] [CrossRef]
- Barik, S.; Das, S.; Vikalo, H. QSdpR: Viral quasispecies reconstruction via correlation clustering. Genomics 2017. [Google Scholar] [CrossRef] [PubMed]
- Scallan, E.; Hoekstra, R.M.; Angulo, F.J.; Tauxe, R.V.; Widdowson, M.A.; Roy, S.L.; Jones, J.L.; Griffin, P.M. Foodborne illness acquired in the United States—Major pathogens. Emerg. Infect. Diseas. 2011, 17, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Werzberger, A.; Mensch, B.; Kuter, B.; Brown, L.; Lewis, J.; Sitrin, R.; Miller, W.; Shouval, D.; Wiens, B.; Calandra, G.; et al. A controlled trial of a formalin-inactivated hepatitis A vaccine in healthy children. N. Engl. J. Med. 1992, 327, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Innis, B.L.; Snitbhan, R.; Kunasol, P.; Laorakpongse, T.; Poopatanakool, W.; Kozik, C.A.; Suntayakorn, S.; Suknuntapong, T.; Safary, A.; Tang, D.B.; et al. Protection against hepatitis A by an inactivated vaccine. JAMA 1994, 271, 1328–1334. [Google Scholar] [CrossRef] [PubMed]
- Lanini, S.; Minosse, C.; Vairo, F.; Garbuglia, A.; di Bari, V.; Agresta, A.; Rezza, G.; Puro, V.; Pendenza, A.; Loffredo, M.R.; et al. A large ongoing outbreak of hepatitis A predominantly affecting young males in Lazio, Italy; August 2016–March 2017. PLoS ONE 2017, 12, e0185428. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.; Kim, J.S.; Oh, K.H.; Oh, S.S.; Kwon, M.; Kim, S.; Park, J.; Kwak, H.S.; Chung, G.T.; Kim, C.J.; et al. A waterborne outbreak involving hepatitis A virus genotype IA at a residential facility in the Republic of Korea in 2015. J. Clin. Virol. 2017, 94, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Bruni, R.; Taffon, S.; Equestre, M.; Cella, E.; Lo Presti, A.; Costantino, A.; Chionne, P.; Madonna, E.; Golkocheva-Markova, E.; Bankova, D.; et al. Hepatitis a virus genotypes and strains from an endemic area of Europe, Bulgaria 2012–2014. BMC Infect. Dis. 2017, 17, 497. [Google Scholar] [CrossRef] [PubMed]
- Collier, M.G.; Khudyakov, Y.E.; Selvage, D.; Adams-Cameron, M.; Epson, E.; Cronquist, A.; Jervis, R.H.; Lamba, K.; Kimura, A.C.; Sowadsky, R.; et al. Outbreak of hepatitis A in the USA associated with frozen pomegranate arils imported from Turkey: An epidemiological case study. Lancet Infect. Dis. 2014, 14, 976–981. [Google Scholar] [CrossRef]
- Costa, A.M.; Amado, L.A.; Paula, V.S.D. Detection of replication-defective hepatitis A virus based on the correlation between real-time polymerase chain reaction and ELISA in situ results. Mem. Inst. Oswaldo Cruz 2013, 108, 36–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa-Mattioli, M.; Cristina, J.; Romero, H.; Perez-Bercof, R.; Casane, D.; Colina, R.; Garcia, L.; Vega, I.; Glikman, G.; Romanowsky, V.; et al. Molecular Evolution of Hepatitis A Virus: A New Classification Based on the Complete VP1 Protein. J. Virol. 2002, 76, 9516–9525. [Google Scholar] [CrossRef] [PubMed]
- Feinstone, S.M.; Kapikian, A.Z.; Purceli, R.H. Hepatitis A: Detection by immune electron microscopy of a viruslike antigen associated with acute illness. Science 1973, 182, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Robertson, B.H.; Jansen, R.W.; Khanna, B.; Totsuka, A.; Nainan, O.V.; Siegl, G.; Widell, A.; Margolis, H.S.; Isomura, S.; Ito, K.; et al. Genetic relatedness of hepatitis A virus strains recovered from different geographical regions. J. Gen. Virol. 1992, 73, 1365–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, E.A.; Jansen, R.W.; Lemon, S.M. Characterization of a simian hepatitis A virus (HAV): Antigenic and genetic comparison with human HAV. J. Virol. 1989, 63, 4932–4937. [Google Scholar] [PubMed]
- Forbi, J.C.; Esona, M.D.; Agwale, S.M. Molecular characterization of hepatitis A virus isolates from Nigeria. Intervirology 2013, 56, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mattioli, M.; Domingo, E.; Cristina, J. Analysis of sequential hepatitis A virus strains reveals coexistence of distinct viral subpopulations. J. Gen. Virol. 2006, 87, 115–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulbaran, Y.; Gutierrez, C.R.; Marquez, B.; Rojas, D.; Sanchez, D.; Navas, J.; Rovallo, E.; Pujol, F.H. Hepatitis A virus genetic diversity in Venezuela: Exclusive circulation of subgenotype IA and evidence of quasispecies distribution in the isolates. J. Med. Virol. 2010, 82, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, G.; Bosch, A.; Gómez-Mariano, G.; Domingo, E.; Pintó, R. Evidence for quasispecies distributions in the human Hepatitis A virus genome. Virology 2003, 315, 34–42. [Google Scholar] [CrossRef]
- Vaughan, G.; Xia, G.; Forbi, J.C.; Purdy, M.A.; Rossi, L.M.; Spradling, P.R.; Khudyakov, Y.E. Genetic relatedness among hepatitis A virus strains associated with food-borne outbreaks. PLoS ONE 2013, 8, e74546. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Leonard, S.R.; Mammel, M.K.; Elkins, C.A.; Kulka, M. Towards next-generation sequencing analytics for foodborne RNA viruses: Examining the effect of RNA input quantity and viral RNA purity. J. Virol. Methods 2016, 236, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Goswami, B.B.; Kulka, M.; Ngo, D.; Cebula, T.A. Apoptosis induced by a cytopathic hepatitis A virus is dependent on caspase activation following ribosomal RNA degradation but occurs in the absence of 2’–5’ oligoadenylate synthetase. Antivir. Res. 2004, 63, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Kulka, M.; Calvo, M.S.; Ngo, D.T.; Wales, S.Q.; Goswami, B.B. Activation of the 2-5OAS/RNase L pathway in CVB1 or HAV/18f infected FRhK-4 cells does not require induction of OAS1 or OAS2 expression. Virology 2009, 388, 169–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wales, S.Q.; Ngo, D.; Hida, K.; Kulka, M. Temperature and density dependent induction of a cytopathic effect following infection with non-cytopathic HAV strains. Virology 2012, 430, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Bravo, H.C.; Irizarry, R.A. Model-based quality assessment and base-calling for second-generation sequencing data. Biometrics 2010, 66, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Beerenwinkel, N.; Gunthard, H.F.; Roth, V.; Metzner, K.J. Challenges and opportunities in estimating viral genetic diversity from next-generation sequencing data. Front. Microbiol. 2012, 3, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayford, A.E.; Mammel, M.K.; Lacher, D.W.; Brown, E.W. Single nucleotide polymorphism (SNP)-based differentiation of Shigella isolates by pyrosequencing. Infect. Genet. Evol. 2011, 11, 1761–1768. [Google Scholar] [CrossRef] [PubMed]
- Janecek, E.; Streichan, S.; Strube, C. SNP-based real-time pyrosequencing as a sensitive and specific tool for identification and differentiation of Rickettsia species in Ixodes ricinus ticks. BMC Infect. Dis. 2012, 12, 1471–2334. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.H.; Ruzin, A.; Tuckman, M.; Visalli, M.A.; Petersen, P.J.; Bradford, P.A. Pyrosequencing using the single-nucleotide polymorphism protocol for rapid determination of TEM- and SHV-type extended-spectrum β-lactamases in clinical isolates and identification of the novel beta-lactamase genes blaSHV-48, blaSHV-105, and blaTEM-155. Antimicrob. Agents Chemother. 2009, 53, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Satkoski, J.A.; Malhi, R.; Kanthaswamy, S.; Tito, R.; Malladi, V.; Smith, D. Pyrosequencing as a method for SNP identification in the rhesus macaque (Macaca mulatta). BMC Genomics 2008, 9, 256. [Google Scholar] [CrossRef] [PubMed]
- Pu, D.; Pan, R.; Liu, W.; Xiao, P. Quantitative analysis of single-nucleotide polymorphisms by pyrosequencing with di-base addition. Electrophoresis 2017, 38, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Ajay, S.S.; Parker, S.C.; Abaan, H.O.; Fajardo, K.V.; Margulies, E.H. Accurate and comprehensive sequencing of personal genomes. Genome Res. 2011, 21, 1498–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerenwinkel, N.; Zagordi, O. Ultra-deep sequencing for the analysis of viral populations. Curr. Opin. Virol. 2011, 1, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Zagordi, O.; Klein, R.; Daumer, M.; Beerenwinkel, N. Error correction of next-generation sequencing data and reliable estimation of HIV quasispecies. Nucleic Acids Res. 2010, 38, 7400–7409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bull, R.A.; Luciani, F.; McElroy, K.; Gaudieri, S.; Pham, S.T.; Chopra, A.; Cameron, B.; Maher, L.; Dore, G.J.; White, P.A.; et al. Sequential bottlenecks drive viral evolution in early acute hepatitis C. virus infection. PLoS Pathog. 2011, 7, e1002243. [Google Scholar] [CrossRef] [PubMed]
- Belalov, I.S.; Isaeva, O.V.; Lukashev, A.N. Recombination in hepatitis A virus: Evidence for reproductive isolation of genotypes. J. Gen. Virol. 2011, 92, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Desbois, D.; Couturier, E.; Mackiewicz, V.; Graube, A.; Letort, M.J.; Dussaix, E.; Roque-Afonso, A.M. Epidemiology and genetic characterization of hepatitis A. virus genotype IIA. J. Clin. Microbiol. 2010, 48, 3306–3315. [Google Scholar] [CrossRef] [PubMed]
- Robertson, B.H.; Khanna, B.; Nainan, O.V.; Margolis, H.S. Epidemiologic patterns of wild-type hepatitis A virus determined by genetic variation. J. Infect. Dis. 1991, 163, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Jeong, H.; Yun, H.; Kim, K.; Kim, J.H.; Yang, J.M.; Cheon, D.S. Genetic analysis of hepatitis A virus strains that induced epidemics in Korea during 2007–2009. J. Clin. Microbiol. 2012, 50, 1252–1257. [Google Scholar] [CrossRef] [PubMed]
- Sarawaneeyaruk, S.; Iwakawa, H.-O.; Mizumoto, H.; Murakami, H.; Kaido, M.; Mise, K.; Okuno, T. Host-dependent roles of the viral 5′ untranslated region (UTR) in RNA stabilization and cap-independent translational enhancement mediated by the 3′ UTR of Red clover necrotic mosaic virus RNA1. Virology 2009, 391, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Svitkin, Y.V.; Imataka, H.; Khaleghpour, K.; Kahvejian, A.; Liebig, H.D.; Sonenberg, N. Poly(A)-binding protein interaction with elF4G stimulates picornavirus IRES-dependent translation. RNA 2001, 7, 1743–1752. [Google Scholar] [PubMed]
- Pestova, T.V.; Kolupaeva, V.G. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev. 2002, 16, 2906–2922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozak, M. The scanning model for translation: An update. J. Cell. Biol. 1989, 108, 229–241. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference Position a | Coding Region | Reference b | Change c | Coverage d | Frequency e | SNV Called by NGS f | Amino Acid Change g | SNV Identified by PYROSEQUENCING |
|---|---|---|---|---|---|---|---|---|
| 1742 | 1C | G | A | 22,779 | 54.2 | mixed | mixed | |
| 2864 | 1D | T | A | 24,773 | 99.3 | single | single | |
| 4185 | 2C | G | A | 69,820 | 99.7 | single | AAA45465.1:p.[Glu1151Lys] | single |
| 5204 | 3A | G | A | 138,614 | 81.1 | mixed | single | |
| 6216 | 3D | T | C | 166,496 | 81.7 | mixed | single | |
| 6522 | 3D | T | A | 297,276 | 99.6 | single | AAA45465.1:p.[Ser1930Thr] | single |
| 6836 | 3D | C | T | 210,792 | 40.1 | mixed | mixed | |
| 7042 | 3D | - | A | 260,563 | 13.8 | mixed | AAA45465.1:p.[Gln2103fs] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Z.; Mammel, M.; Whitehouse, C.A.; Ngo, D.; Kulka, M. Inter- and Intra-Host Nucleotide Variations in Hepatitis A Virus in Culture and Clinical Samples Detected by Next-Generation Sequencing. Viruses 2018, 10, 619. https://doi.org/10.3390/v10110619
Yang Z, Mammel M, Whitehouse CA, Ngo D, Kulka M. Inter- and Intra-Host Nucleotide Variations in Hepatitis A Virus in Culture and Clinical Samples Detected by Next-Generation Sequencing. Viruses. 2018; 10(11):619. https://doi.org/10.3390/v10110619
Chicago/Turabian StyleYang, Zhihui, Mark Mammel, Chris A. Whitehouse, Diana Ngo, and Michael Kulka. 2018. "Inter- and Intra-Host Nucleotide Variations in Hepatitis A Virus in Culture and Clinical Samples Detected by Next-Generation Sequencing" Viruses 10, no. 11: 619. https://doi.org/10.3390/v10110619