HIV-1 Subtypes and 5’LTR-Leader Sequence Variants Correlate with Seroconversion Status in Pumwani Sex Worker Cohort

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Genomic DNA Isolation and Nested PCR Amplification of Partial 5′LTR of HIV-1
2.3. Cloning and Sequencing of Amplified Partial 5′LTR Sequences
2.4. Sequence and Phylogenetic Analyses
2.5. Sequence Variant Classification by Recursive Partitioning Analysis
3. Results
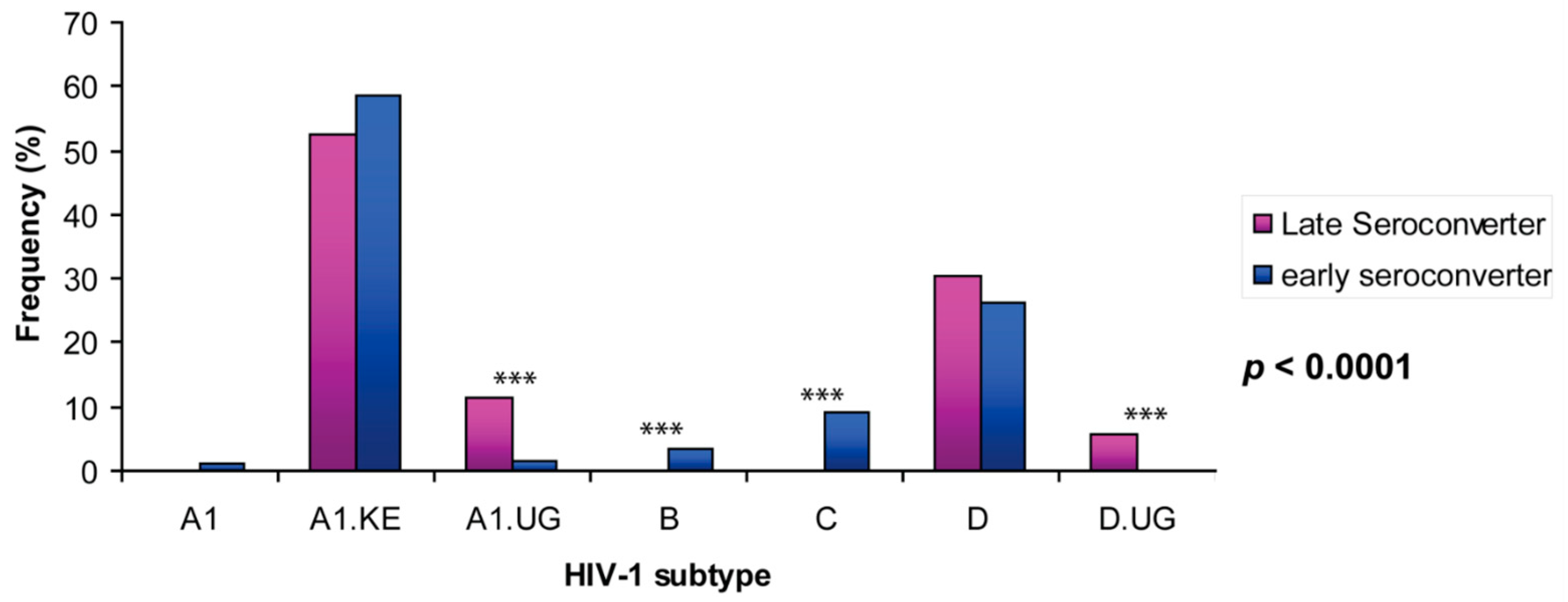
3.1. Uganda A1 and D Subtype 5′LTR-Leader Sequences Were Significantly Enriched in HIV Viral Population from Late Seroconverters
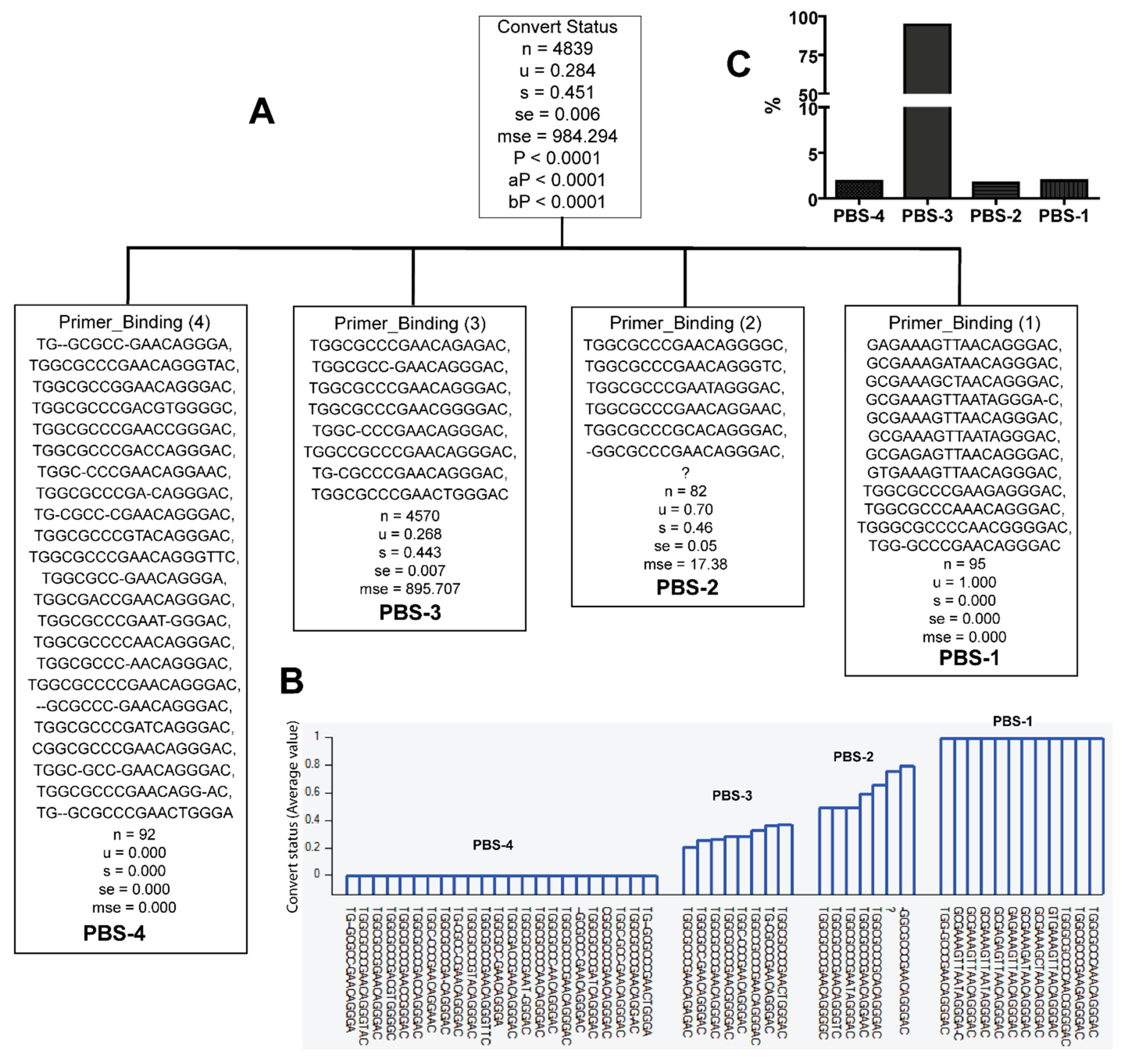
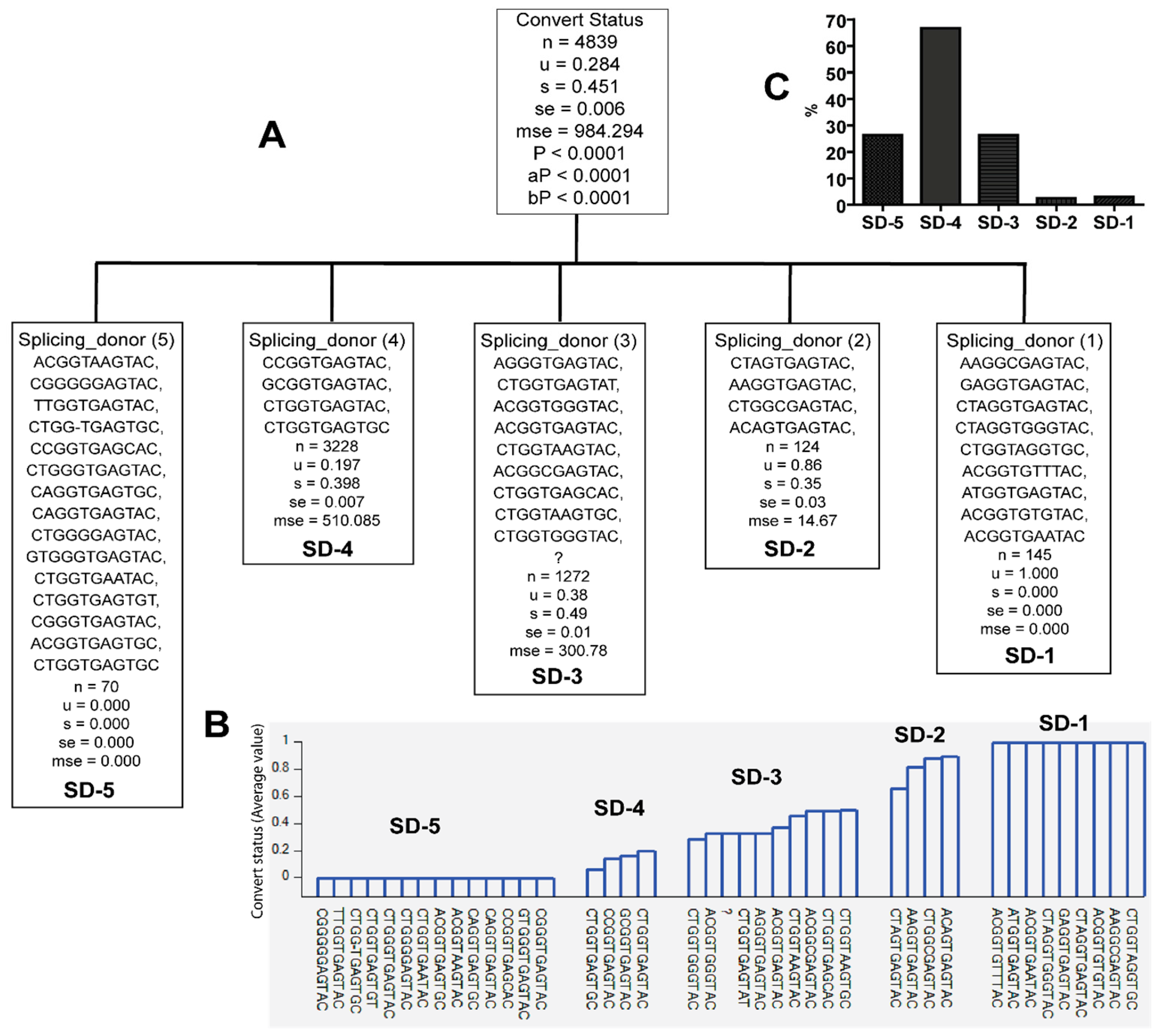
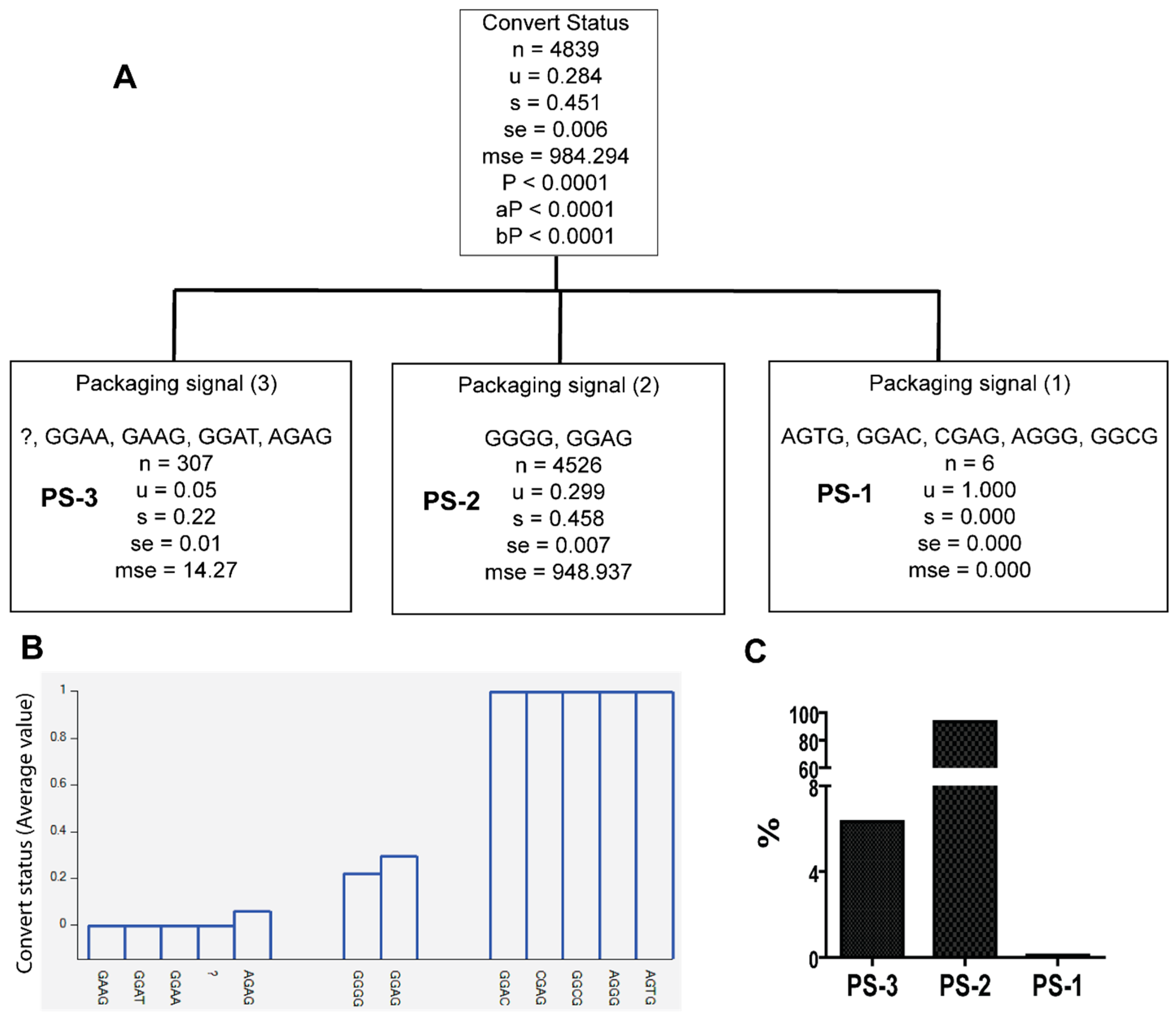
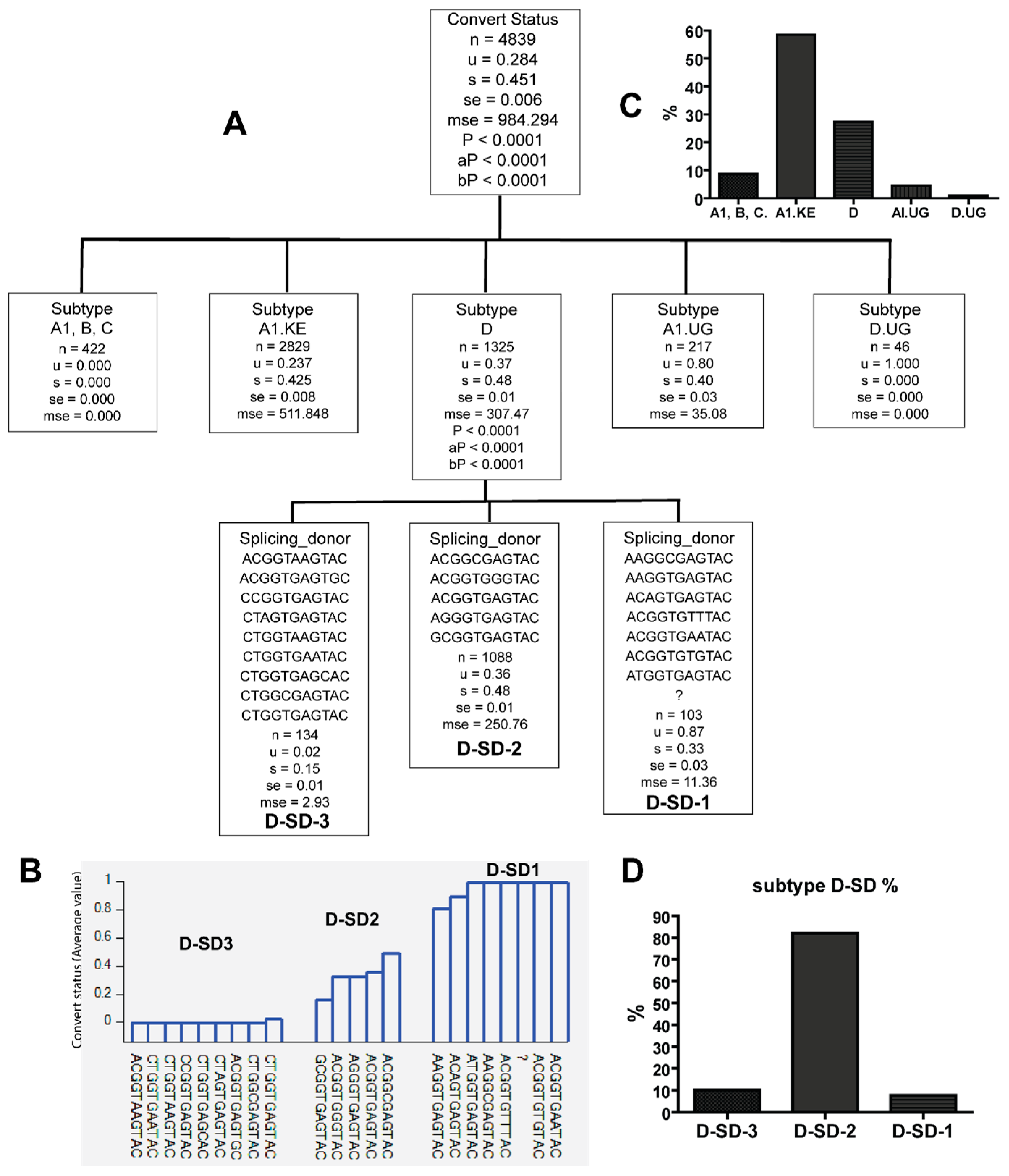
3.2. Unique Sequences and Combinations of PBS, SD, and Ps Sequences in Late Seroconverters
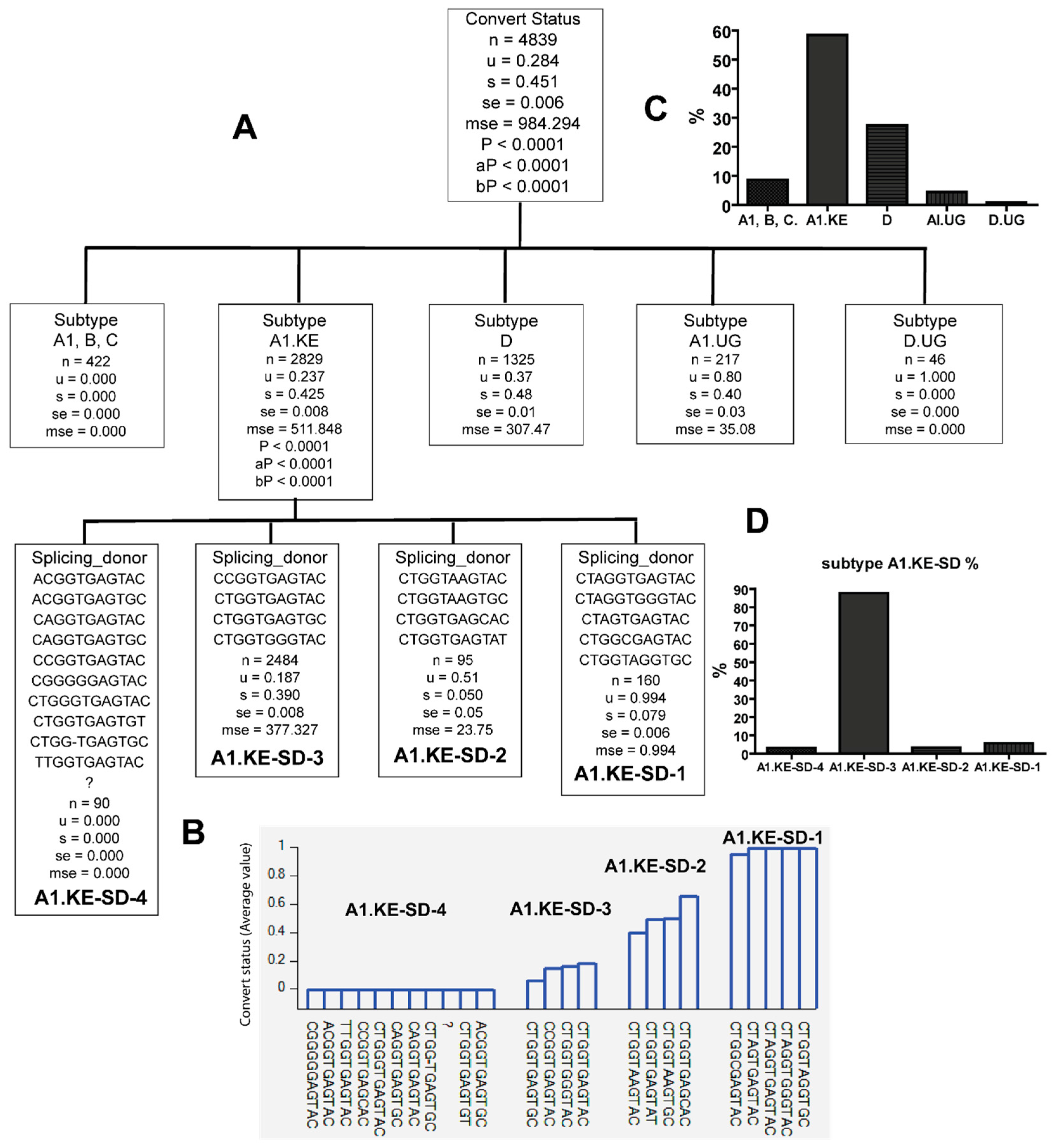
3.3. Combinations of Subtype A1.KE or D with Unique PBS and SD Sequence Variants in Late Seroconverters
3.4. Potential Functional Differences among PBS Variants in Late Seroconverters
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- UNAIDS Data 2017. Available online: http://www.unaids.org/sites/default/files/media_asset/20170720_Data_book_2017_en.pdf (accessed on 5 October 2017).
- Cohen, J. The emerging race to cure HIV infections. Science 2011, 332, 784–785, 787–789. [Google Scholar] [CrossRef] [PubMed]
- Lakhashe, S.K.; Silvestri, G.; Ruprecht, R.M. No acquisition: A new ambition for HIV vaccine development? Curr. Opin. Virol. 2011, 1, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Talbott, J.R. Size matters: The number of prostitutes and the global HIV/AIDS pandemic. PLoS ONE 2007, 2, e543. [Google Scholar] [CrossRef] [PubMed]
- Baral, S.; Beyrer, C.; Muessig, K.; Poteat, T.; Wirtz, A.L.; Decker, M.R.; Sherman, S.G.; Kerrigan, D. Burden of HIV among female sex workers in low-income and middle-income countries: A systematic review and meta-analysis. Lancet Infect. Dis. 2012, 12, 538–549. [Google Scholar] [CrossRef]
- Fowke, K.R.; Nagelkerke, N.J.; Kimani, J.; Simonsen, J.N.; Anzala, A.O.; Bwayo, J.J.; MacDonald, K.S.; Ngugi, E.N.; Plummer, F.A. Resistance to HIV-1 infection among persistently seronegative prostitutes in Nairobi, Kenya. Lancet 1996, 348, 1347–1351. [Google Scholar] [CrossRef]
- Luo, M.; McLaren, P.J.; Plummer, F.A. (Eds.) Host Genetics and Resistance to HIV-1 infection. In Models of Protection against HIV/SIV; Academic Press, Elsevier Inc.: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Plummer, F.A.; Ball, T.B.; Kimani, J.; Fowke, K.R. Resistance to HIV-1 infection among highly exposed sex workers in Nairobi: What mediates protection and why does it develop? Immunol. Lett. 1999, 66, 27–34. [Google Scholar] [CrossRef]
- Hardie, R.A.; Knight, E.; Bruneau, B.; Semeniuk, C.; Gill, K.; Nagelkerke, N.; Kimani, J.; Wachihi, C.; Ngugi, E.; Luo, M.; et al. A common human leucocyte antigen-DP genotype is associated with resistance to HIV-1 infection in Kenyan sex workers. AIDS 2008, 22, 2038–2042. [Google Scholar] [CrossRef] [PubMed]
- Hardie, R.A.; Luo, M.; Bruneau, B.; Knight, E.; Nagelkerke, N.J.; Kimani, J.; Wachihi, C.; Ngugi, E.N.; Plummer, F.A. Human leukocyte antigen-DQ alleles and haplotypes and their associations with resistance and susceptibility to HIV-1 infection. AIDS 2008, 22, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Lacap, P.A.; Huntington, J.D.; Luo, M.; Nagelkerke, N.J.; Bielawny, T.; Kimani, J.; Wachihi, C.; Ngugi, E.N.; Plummer, F.A. Associations of human leukocyte antigen DRB with resistance or susceptibility to HIV-1 infection in the Pumwani sex worker cohort. AIDS 2008, 22, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Daniuk, C.A.; Diallo, T.O.; Capina, R.E.; Kimani, J.; Wachihi, C.; Kimani, M.; Bielawny, T.; Peterson, T.; Mendoza, M.G.; et al. For protection from HIV-1 infection, more might not be better: A systematic analysis of HIV Gag epitopes of two alleles associated with different outcomes of HIV-1 infection. J. Virol. 2012, 86, 1166–1180. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Sainsbury, J.; Tuff, J.; Lacap, P.A.; Yuan, X.Y.; Hirbod, T.; Kimani, J.; Wachihi, C.; Ramdahin, S.; Bielawny, T.; et al. A genetic polymorphism of FREM1 is associated with resistance against HIV infection in the Pumwani sex worker cohort. J. Virol. 2012, 86, 11899–11905. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.A.; Kimani, J.; Wachihi, C.; Bielawny, T.; Mendoza, L.; Thavaneswaran, S.; Narayansingh, M.J.; Kariri, T.; Liang, B.; Ball, T.B.; et al. HLA class I associations with rates of HIV-1 seroconversion and disease progression in the Pumwani sex worker cohort. Tissue Antigens 2013, 81, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Price, H.; Lacap, P.; Tuff, J.; Wachihi, C.; Kimani, J.; Ball, T.B.; Luo, M.; Plummer, F.A. A TRIM5α exon 2 polymorphism is associated with protection from HIV-1 infection in the Pumwani sex worker cohort. AIDS 2010, 24, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Shea, P.R.; Shianna, K.V.; Carrington, M.; Goldstein, D.B. Host genetics of HIV acquisition and viral control. Annu. Rev. Med. 2013, 64, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Kaul, R.; Rowland-Jones, S.L.; Kimani, J.; Dong, T.; Yang, H.B.; Kiama, P.; Rostron, T.; Njagi, E.; Bwayo, J.J.; MacDonald, K.S.; et al. Late seroconversion in HIV-resistant Nairobi prostitutes despite pre-existing HIV-specific CD8+ responses. J. Clin. Investig. 2001, 107, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med. 2011, 1, a006841. [Google Scholar] [CrossRef] [PubMed]
- HIV Database. Available online: http://www.hiv.lanl.gov/components/sequence/HIV/geo/geo.comp (accessed on 5 October 2017).
- Dowling, W.E.; Kim, B.; Mason, C.J.; Wasunna, K.M.; Alam, U.; Elson, L.; Birx, D.L.; Robb, M.L.; McCutchan, F.E.; Carr, J.K. Forty-one near full-length HIV-1 sequences from Kenya reveal an epidemic of subtype A and A-containing recombinants. AIDS 2002, 16, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Nyamache, A.K.; Muigai, A.W.; Khamadi, S.A. Circulating trends of non-B HIV type 1 subtypes among Kenyan individuals. AIDS Res. Hum. Retrovir. 2013, 29, 400–403. [Google Scholar] [CrossRef] [PubMed]
- Peters, H.O.; Mendoza, M.G.; Capina, R.E.; Luo, M.; Mao, X.; Gubbins, M.; Nagelkerke, N.J.; Macarthur, I.; Sheardown, B.B.; Kimani, J.; et al. An integrative bioinformatic approach for studying escape mutations in human immunodeficiency virus type 1 gag in the Pumwani sex worker cohort. J. Virol. 2008, 82, 1980–1992. [Google Scholar] [CrossRef] [PubMed]
- Arien, K.K.; Vanham, G.; Arts, E.J. Is HIV-1 evolving to a less virulent form in humans? Nat. Rev. Microbiol. 2007, 5, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.G. Resistance and viral subtypes: How important are the differences and why do they occur? Curr. Opin. HIV AIDS 2007, 2, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Kanki, P.J.; Hamel, D.J.; Sankale, J.L.; Hsieh, C.; Thior, I.; Barin, F.; Woodcock, S.A.; Gueye-Ndiaye, A.; Zhang, E.; Montano, M.; et al. Human immunodeficiency virus type 1 subtypes differ in disease progression. J. Infect. Dis. 1999, 179, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Vasan, A.; Renjifo, B.; Hertzmark, E.; Chaplin, B.; Msamanga, G.; Essex, M.; Fawzi, W.; Hunter, D. Different rates of disease progression of HIV type 1 infection in Tanzania based on infecting subtype. Clin. Infect. Dis. 2006, 42, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Yuste, E.; Borderia, A.V.; Domingo, E.; Lopez-Galindez, C. Few mutations in the 5′ leader region mediate fitness recovery of debilitated human immunodeficiency type 1 viruses. J. Virol. 2005, 79, 5421–5427. [Google Scholar] [CrossRef] [PubMed]
- Berkhout, B. Structure and function of the human immunodeficiency virus leader RNA. Prog. Nucleic Acid Res. Mol. Biol. 1996, 54, 1–34. [Google Scholar] [PubMed]
- Lu, K.; Heng, X.; Summers, M.F. Structural determinants and mechanism of HIV-1 genome packaging. J. Mol. Biol. 2011, 410, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Rhim, H.; Park, J.; Morrow, C.D. Deletions in the tRNA(lys) primer-binding site of human immunodeficiency virus type 1 identify essential regions for reverse transcription. J. Virol. 1991, 65, 4555–4564. [Google Scholar] [PubMed]
- Harrich, D.; Hooker, B. Mechanistic aspects of HIV-1 reverse transcription initiation. Rev. Med. Virol. 2002, 12, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Das, A.T.; Klaver, B.; Berkhout, B. Sequence variation of the human immunodeficiency virus primer-binding site suggests the use of an alternative tRNA(lys) molecule in reverse transcription. J. Gen. Virol. 1997, 78 Pt 4, 837–840. [Google Scholar] [CrossRef] [PubMed]
- Mak, J.; Kleiman, L. Primer tRNAs for reverse transcription. J. Virol. 1997, 71, 8087–8095. [Google Scholar] [PubMed]
- Ni, N.; Morrow, C.D. Impact of forced selection of tRNAs on HIV-1 replication and genome stability highlight preferences for selection of certain tRNAs. Virus Res. 2007, 124, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Abbink, T.E.; Berkhout, B. RNA structure modulates splicing efficiency at the human immunodeficiency virus type 1 major splice donor. J. Virol. 2008, 82, 3090–3098. [Google Scholar] [CrossRef] [PubMed]
- Lutzelberger, M.; Reinert, L.S.; Das, A.T.; Berkhout, B.; Kjems, J. A novel splice donor site in the gag-pol gene is required for HIV-1 RNA stability. J. Biol. Chem. 2006, 281, 18644–18651. [Google Scholar] [CrossRef] [PubMed]
- Zeffman, A.; Hassard, S.; Varani, G.; Lever, A. The major HIV-1 packaging signal is an extended bulged stem loop whose structure is altered on interaction with the Gag polyprotein. J. Mol. Biol. 2000, 297, 877–893. [Google Scholar] [CrossRef] [PubMed]
- Miele, G.; Mouland, A.; Harrison, G.P.; Cohen, E.; Lever, A.M. The human immunodeficiency virus type 1 5′ packaging signal structure affects translation but does not function as an internal ribosome entry site structure. J. Virol. 1996, 70, 944–951. [Google Scholar] [PubMed]
- Kumar, S.; Tamura, K.; Nei, M. MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief. Bioinform. 2004, 5, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Abbink, T.E.; Beerens, N.; Berkhout, B. Forced selection of a human immunodeficiency virus type 1 variant that uses a non-self tRNA primer for reverse transcription: Involvement of viral RNA sequences and the reverse transcriptase enzyme. J. Virol. 2004, 78, 10706–10714. [Google Scholar] [CrossRef] [PubMed]
- Das, A.T.; Vink, M.; Berkhout, B. Alternative tRNA priming of human immunodeficiency virus type 1 reverse transcription explains sequence variation in the primer-binding site that has been attributed to APOBEC3G activity. J. Virol. 2005, 79, 3179–3181. [Google Scholar] [CrossRef] [PubMed]
- Kelly, N.J.; Morrow, C.D. Yeast tRNA(Phe) expressed in human cells can be selected by HIV-1 for use as a reverse transcription primer. Virology 2003, 313, 354–363. [Google Scholar] [CrossRef]
- Moore, K.L.; Kosloff, B.R.; Kelly, N.J.; Kirkman, R.L.; Dupuy, L.C.; McPherson, S.; Morrow, C.D. HIV type 1 that select tRNA(His) or tRNA(Lys1,2) as primers for reverse transcription exhibit different infectivities in peripheral blood mononuclear cells. AIDS Res. Hum. Retrovir. 2004, 20, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Djekic, U.V.; Morrow, C.D. Analysis of the replication of HIV-1 forced to use tRNAMet(i) supports a link between primer selection, translation and encapsidation. Retrovirology 2007, 4, 10. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, M.; Eipers, P.G.; Ni, N.; Morrow, C.D. HIV-1 designed to use different tRNAGln isoacceptors prefers to select tRNAThr for replication. Virol. J. 2006, 3, 80. [Google Scholar] [CrossRef] [PubMed]
- McCulley, A.; Morrow, C.D. Nucleotides within the anticodon stem are important for optimal use of tRNA(Lys,3) as the primer for HIV-1 reverse transcription. Virology 2007, 364, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Moore-Rigdon, K.L.; Kosloff, B.R.; Kirkman, R.L.; Morrow, C.D. Preferences for the selection of unique tRNA primers revealed from analysis of HIV-1 replication in peripheral blood mononuclear cells. Retrovirology 2005, 2, 21. [Google Scholar] [CrossRef] [PubMed]
- Strobl, C.; Malley, J.; Tutz, G. An introduction to recursive partitioning: Rationale, application, and characteristics of classification and regression trees, bagging, and random forests. Psychol. Methods 2009, 14, 323–348. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, D.M. FIRM: Formal Inference-Based Recursive Modeling, Release 2; Technical Report; School of Statistics, University of Minnesota: Saint Paul, MN, USA, 1995; p. 546. [Google Scholar]
- Hawkins, D.M. Fitting multiple change-points to data. Comput. Stat. Data Anal. 2002, 37, 323–341. [Google Scholar] [CrossRef]
- Hawkins, D.M.; McKenzie, D.P. A data-based comparison of some recursive partitioning procedures. In Proceedings of the Statistical Computing Section; American Statistical Association: Boston, MA, USA, 1995; pp. 245–252. [Google Scholar]
- Hawkins, D.M.; Musser, B.J. One tree or a forest? Alternative dendrographic models. Comput. Sci. Stat. 1999, 30, 534–542. [Google Scholar]
- Hawkins, D.M.; Musser, B.J. Feature selection with nondeterministic recursive partitioning. In Proceedings of the American Statistical Association, Joint Statistical Meetings, Atlanta, GA, USA, 2–9 August 2001. [Google Scholar]
- Hawkins, D.M.; Young, S.S.; Rusinko, A. Analysis of a large structure-activity data set using recursive partitioning. Quant. Struct. Act. Relat. 1997, 16, 296–302. [Google Scholar] [CrossRef]
- Young, S.S.; Hawkins, D.M. Using recursive partitioning to analyze a large SAR data set. SAR QSAR Environ. Res. 1998, 8, 183–193. [Google Scholar] [CrossRef]
- Young, S.S.; Hawkins, D.M. Using recursive partitioning analysis to evaluate compound selection methods. Methods Mol. Biol. 2004, 275, 317–334. [Google Scholar] [PubMed]
- Douglas, M. Hawkins LLC website. Available online: http://www.douglashawkins.com (accessed on 21 December 2017).
- Finzi, D.; Plaeger, S.F.; Dieffenbach, C.W. Defective Virus Drives Human Immunodeficiency Virus Infection, Persistence, and Pathogenesis. Clin. Vaccine Immunol. 2006, 13, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, J.K.; Kang, S.M.; Morrow, C.D. Construction of a type 1 human immunodeficiency virus that maintains a primer binding site complementary to tRNA(His). J. Virol. 1996, 70, 966–975. [Google Scholar] [PubMed]
- Wakefield, J.K.; Morrow, C.D. Mutations within the primer binding site of the human immunodeficiency virus type 1 define sequence requirements essential for reverse transcription. Virology 1996, 220, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, J.K.; Wolf, A.G.; Morrow, C.D. Human immunodeficiency virus type 1 can use different tRNAs as primers for reverse transcription but selectively maintains a primer binding site complementary to tRNA(3Lys). J. Virol. 1995, 69, 6021–6029. [Google Scholar] [PubMed]
- Zhang, Z.; Kang, S.M.; LeBlanc, A.; Hajduk, S.L.; Morrow, C.D. Nucleotide sequences within the U5 region of the viral RNA genome are the major determinants for an human immunodeficiency virus type 1 to maintain a primer binding site complementary to tRNA(His). Virology 1996, 226, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Z.; Wakefield, J.K.; Kang, S.M.; Morrow, C.D. Nucleotide substitutions within U5 are critical for efficient reverse transcription of human immunodeficiency virus type 1 with a primer binding site complementary to tRNA(His). J. Virol. 1997, 71, 6315–6322. [Google Scholar] [PubMed]
- Kang, S.M.; Zhang, Z.; Morrow, C.D. Identification of a human immunodeficiency virus type 1 that stably uses tRNALys1,2 rather than tRNALys,3 for initiation of reverse transcription. Virology 1999, 257, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Das, A.T.; Klaver, B.; Berkhout, B. Reduced replication of human immunodeficiency virus type 1 mutants that use reverse transcription primers other than the natural tRNA(3Lys). J. Virol. 1995, 69, 3090–3097. [Google Scholar] [PubMed]
- Palmer, M.T.; McPherson, S.; Morrow, C.D. Murine leukemia virus with a primer-binding site complementary to tRNALys,3 adapts to select new tRNAs for replication following extended in vitro culture. Virology 2005, 333, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Palmer, M.T.; Kirkman, R.; Kosloff, B.R.; Eipers, P.G.; Morrow, C.D. tRNA isoacceptor preference prior to retrovirus Gag-Pol junction links primer selection and viral translation. J. Virol. 2007, 81, 4397–4404. [Google Scholar] [CrossRef] [PubMed]
- Alizon, S.; von Wyl, V.; Stadler, T.; Kouyos, R.D.; Yerly, S.; Hirschel, B.; Boni, J.; Shah, C.; Klimkait, T.; Furrer, H.; et al. Phylogenetic approach reveals that virus genotype largely determines HIV set-point viral load. PLoS Pathog. 2010, 6, e1001123. [Google Scholar] [CrossRef] [PubMed]
- Abbink, T.E.; Berkhout, B. HIV-1 reverse transcription initiation: A potential target for novel antivirals? Virus Res. 2008, 134, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Dorman, N.M.; Lever, A.M. Investigation of RNA transcripts containing HIV-1 packaging signal sequences as HIV-1 antivirals: Generation of cell lines resistant to HIV-1. Gene Ther. 2001, 8, 157–165. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HIV-1 Subtype | A1 | A1.KE | A1.UG | B | C | D | D.UG | Total | Ave Seq./ind. | p Value | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | All 142 individuals | No. | 30 | 2162 | 135 | 97 | 263 | 945 | 46 | 3678 | ||
| % | 0.8 | 58.8 | 3.7 | 2.6 | 7.2 | 25.7 | 1.3 | 100 | ||||
| B | Late seroconverter (n = 20) | No. | 0 | 452 | 91 | 0 | 0 | 211 | 46 | 800 | 40 (19–47) | <0.0001 |
| % | 0 | 56.5 | 11.4 | 0 | 0 | 26.4 | 5.8 | 100 | ||||
| Early seroconverter (n = 122) | No. | 30 | 1710 | 44 | 97 | 263 | 734 | 0 | 2878 | 30 (11–75) | ||
| % | 1.0 | 59.4 | 1.5 | 3.4 | 9.1 | 25.5 | 0 | 100 |
| Group | Sequences | Subtypes | Frequency in EC or LSC |
|---|---|---|---|
| PBS-1 | GAGAAAGTTAACAGGGAC, GCGAAAGATAACAGGGAC, GCGAAAGCTAACAGGGAC, GCGAAAGTTAATAGGGA-C, GCGAAAGTTAACAGGGAC, GCGAAAGTTAATAGGGAC, GCGAGAGTTAACAGGGAC, GTGAAAGTTAACAGGGAC, TGGCGCCCGAAGAGGGAC, TGGCGCCCAAACAGGGAC, TGGGCGCCCCAACGGGGAC, TGG-GCCCGAACAGGGAC | A1.KE (1.1%), A1.UG (93.67%), D (3.2%), D.UG (2.1%) | 0/122 EC, 5/12 LSC |
| PBS-4 | TG-GCGCC-GAACAGGGA, TGGCGCCCGAACAGGGTAC, TGGCGCCGGAACAGGGAC, TGGCGCCCGACGTGGGGC, TGGCGCCCGAACCGGGAC, TGGCGCCCGACCAGGGAC, TGGC-CCCGAACAGGAAC, TGGCGCCCGA-CAGGGAC, TG-CGCC-CGAACAGGGAC, TGGCGCCCGTACAGGGAC, TGGCGCCCGAACAGGGTTC, TGGCGCC-GAACAGGGA, TGGCGACCGAACAGGGAC, TGGCGCCCGAAT-GGGAC, TGGCGCCCCAACAGGGAC, TGGCGCCC-AACAGGGAC, TGGCGCCCCGAACAGGGAC, -GCGCCC-GAACAGGGAC, TGGCGCCCGATCAGGGAC, CGGCGCCCGAACAGGGAC, TGGC-GCC-GAACAGGGAC, TGGCGCCCGAACAGG-AC, TG--GCGCCCGAACTGGGA | A1 (2.2%), A1.KE (38%), B (2.2%), C (12.0%), D (42%) | 36/122 EC, 0/20 LSC |
| SD1-1 | AAGGCGAGTAC, GAGGTGAGTAC, CTAGGTGAGTAC, CTAGGTGGGTAC, CTGGTAGGTGC, ACGGTGTTTAC, ATGGTGAGTAC, ACGGTGTGTAC, ACGGTGAATAC | A1.KE (92.4), D (6.2%), D.UG (1.4%) | 0/122 EC, 6/20 LSC |
| SD1-5 | ACGGTAAGTAC, CGGGGGAGTAC, TTGGTGAGTAC, CTGG-TGAGTGC, CCGGTGAGCAC, CTGGGTGAGTAC, CAGGTGAGTGC, CAGGTGAGTAC, CTGGGGAGTAC, GTGGGTGAGTAC, CTGGTGAATAC, CTGGTGAGTGT, CGGGTGAGTAC, ACGGTGAGTGC, CTGGTGAGTGC | A1 (1.4%), A1.KE (90%), B (1.4%), C (2.9%), D (4.3%) | 15/122 EC, 0/20 LSC |
| PS-1 | AGTG, GGAC, CGAG, GGCG, AGGG | A1.KE (50%), A1.UG (16.7%), D (33.3%) | 0/122 EC, 6/20 LSC |
| PS-3 | GGAA, GAAG, GGAT, AGAG, ? | A1.KE (66.8%), A1.UG (1.3%), C (24.1%), D (7.8%) | 30/122, EC, 4/20 LSC |
| Subtypes or PBS | SD or PBS | Seroconverter |
|---|---|---|
| TGGCGCCCGAACAGGGGC TGGCGCCCGAACAGGGTC TGGCGCCCGAATAGGGAC TGGCGCCCGAACAGGAAC TGGCGCCCGCACAGGGAC? (PBS-2) | CTGGTGAGTAC AAGGTGAGTAC ACGGTGTTTAC ACAGTGAGTAC | LSC |
| CAGGTGAGTAC CAGGTGAGTGC | EC | |
| TGGCGCCCGAACAGGGAC TGGCGCCCGAACAGAGAC TGGCGCC-GAACAGGGAC TGGCGCCCGAACGGGGAC TGGC-CCCGAACAGGGAC TGGCCGCCCGAACAGGGAC TG-CGCCCGAACAGGGAC TGGCGCCCGAACTGGGAC (PBS-3) | AAGGCGAGTAC ACGGTGAATAC ATGGTGAGTAC CTAGGTAGGTGC CTAGGTGGGTAC CTGGTAGGTGC GAGGTGAGTAC | LSC |
| ACGGTGAGTGC ACGGTAAGTAC CAGGTGAGTAC CAGGTGAGTGC CCGGTGAGCAC CGGGGGAGTAC CGGGTGAGTAC CTGGTGAGTGT CTGGGGAGTAC CTGGGTGAGTAC CTGG-TGAGTGC CTGGTGAATAC GTGGGTGAGTAC TTGGTGAGTAC | EC | |
| Subtype D | AAGGCGAGTAC ACGGTGAATAC ACGGTGTGTAC ACGGTGTTTAC ATGGTGAGTAC ? | LSC |
| ACGGTAAGTAC ACGGTGAGTGC CCGGTGAGTAC CTAGTGAGTAC CTGGTAAGTAC CTGGTGAATAC CTGGTGAGCAC CTGGCGAGTAC | EC | |
| Subtype D | TGGCGCCCGAACAGGGTC TGGCGCCCCAACGGGGAC TGGCGCCCGAACAGGAAC TGG-GCCCGAACAGGGAC TGGCGCCCAAACAGGGAC TG-CGCCCGAACAGGGAC TGGCCGCCCGAACAGGGAC (D-PBS-1) | LSC |
| TGGCGCCGGAACAGGGAC TGGCGCCCGAACAGGGTAC TGGCGCCCGACGTGGGGC TGGCGACCGAACAGGGAC TGGCGCCCGAACCGGGAC TGGCGCCCGTACAGGGAC TGGC-CCCGAACAGGGAC TGGCCGCCCGATCAGGGAC TG-CGCC-CGAACAGGGAC TGGCGCCCCGAACAGGGAC (D-PBS-3) | EC | |
| A1.KE | CTAGGTGAGTAC CTAGGTGGGTAC CTAGTGAGTAC CTGGTAGGTGC (A1.KE-SD-1) | LSC |
| ACGGTGAGTAC ACGGTGAGTGC CAGGTGAGTAC CAGGTGAGTGC CCGGTGAGTAC CGGGGGAGTAC CTGGGTGAGTAC CTGGTGAGTGT CTGG-TGAGTGC TTGGTGAGTAC ? (A1.KE-SD-4) | EC |
| mlno | PBS1 | SD1 | PS1 | PBS-2/SD-1 | PBS2/A1.UG | PBS-3/SD-1 | PBS-3/SD-2 | SD2 | A1-UG/D.UG | A1.KE/SD1 | D-SD-1 | D-PBS-1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 37 | + | |||||||||||
| 58 | + | + | + | + | + | |||||||
| 290 | + | + | + | |||||||||
| 452 | + | + | + | |||||||||
| 546 | + | + | + | |||||||||
| 768 | + | + | + | + | ||||||||
| 814 | + | + | + | |||||||||
| 825 | ||||||||||||
| 888 | + | + | + | + | + | + | + | |||||
| 890 | + | + | + | + | + | |||||||
| 1072 | + | + | + | |||||||||
| 1102 | + | + | + | + | ||||||||
| 1232 | + | + | ||||||||||
| 1248 | ||||||||||||
| 1250 | + | + | + | + | ||||||||
| 1287 | + | + | + | + | + | |||||||
| 1430 | + | |||||||||||
| 1626 | + | + | + | |||||||||
| 1707 | ||||||||||||
| 1730 |
| mlno | A1.KE | A1.UG | D | D.UG |
|---|---|---|---|---|
| 37 | * | |||
| 58 | * | |||
| 290 | * | |||
| 452 | * | |||
| 546 | * | |||
| 768 | * | * | ||
| 814 | * | |||
| 825 | * | * | ||
| 888 | * | |||
| 890 | * | |||
| 1072 | * | * | ||
| 1102 | * | |||
| 1232 | * | |||
| 1248 | * | |||
| 1250 | * | |||
| 1287 | * | |||
| 1430 | * | * | ||
| 1626 | * | |||
| 1707 | * | |||
| 1730 | * |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sampathkumar, R.; Scott-Herridge, J.; Liang, B.; Kimani, J.; Plummer, F.A.; Luo, M. HIV-1 Subtypes and 5’LTR-Leader Sequence Variants Correlate with Seroconversion Status in Pumwani Sex Worker Cohort. Viruses 2018, 10, 4. https://doi.org/10.3390/v10010004
Sampathkumar R, Scott-Herridge J, Liang B, Kimani J, Plummer FA, Luo M. HIV-1 Subtypes and 5’LTR-Leader Sequence Variants Correlate with Seroconversion Status in Pumwani Sex Worker Cohort. Viruses. 2018; 10(1):4. https://doi.org/10.3390/v10010004
Chicago/Turabian StyleSampathkumar, Raghavan, Joel Scott-Herridge, Binhua Liang, Joshua Kimani, Francis A. Plummer, and Ma Luo. 2018. "HIV-1 Subtypes and 5’LTR-Leader Sequence Variants Correlate with Seroconversion Status in Pumwani Sex Worker Cohort" Viruses 10, no. 1: 4. https://doi.org/10.3390/v10010004
APA StyleSampathkumar, R., Scott-Herridge, J., Liang, B., Kimani, J., Plummer, F. A., & Luo, M. (2018). HIV-1 Subtypes and 5’LTR-Leader Sequence Variants Correlate with Seroconversion Status in Pumwani Sex Worker Cohort. Viruses, 10(1), 4. https://doi.org/10.3390/v10010004