
Influence of Starvation on the Structure of Gut-Associated Bacterial Communities in the Chinese White Pine Beetle (Dendroctonus armandi)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Insect Collection and Gut Dissection
2.2. Bacterial DNA Extraction
2.3. Nested PCR
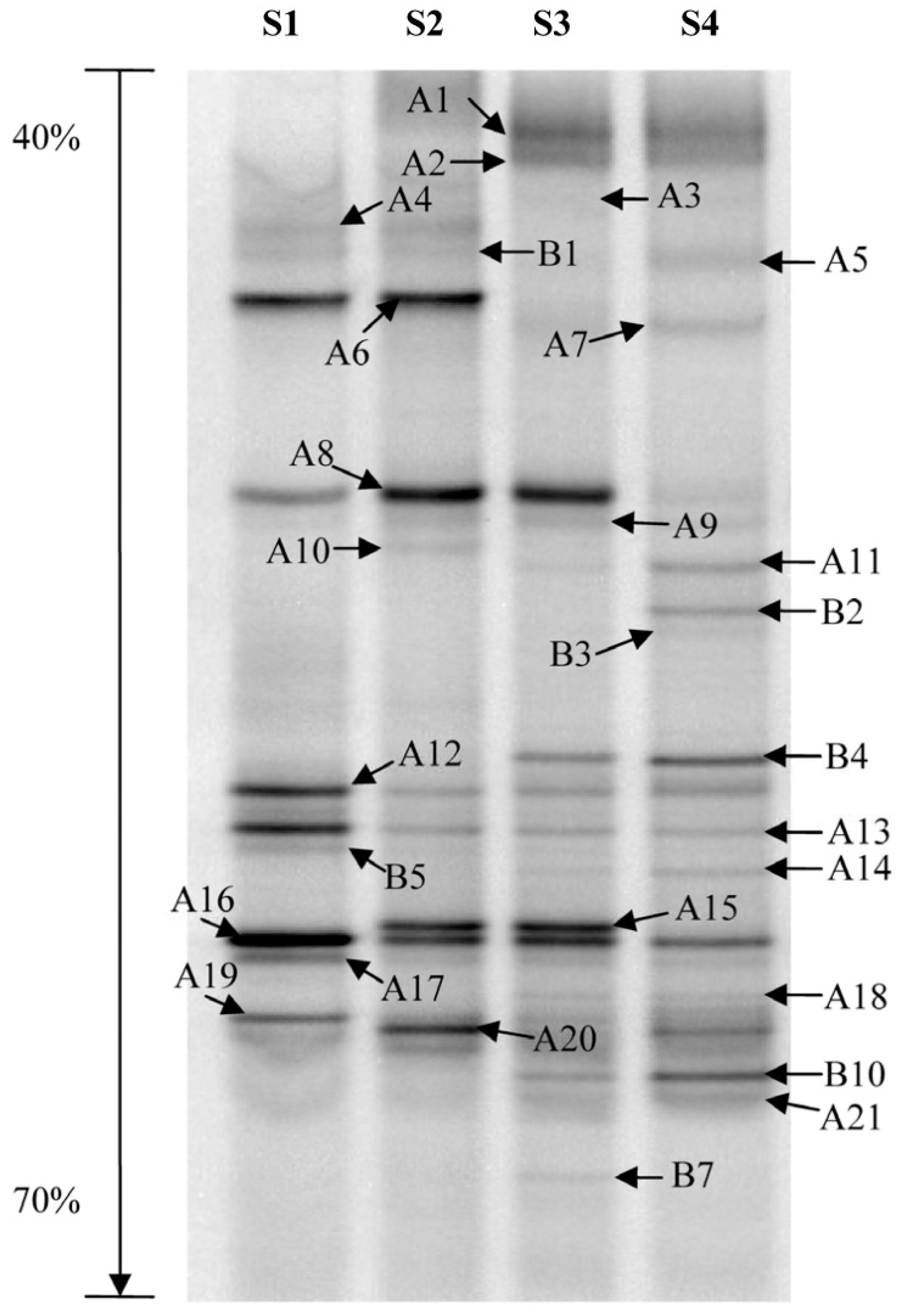
2.4. Denaturing Gradient Gel Electrophoresis (DGGE)
2.5. DGGE Band Profile Analysis
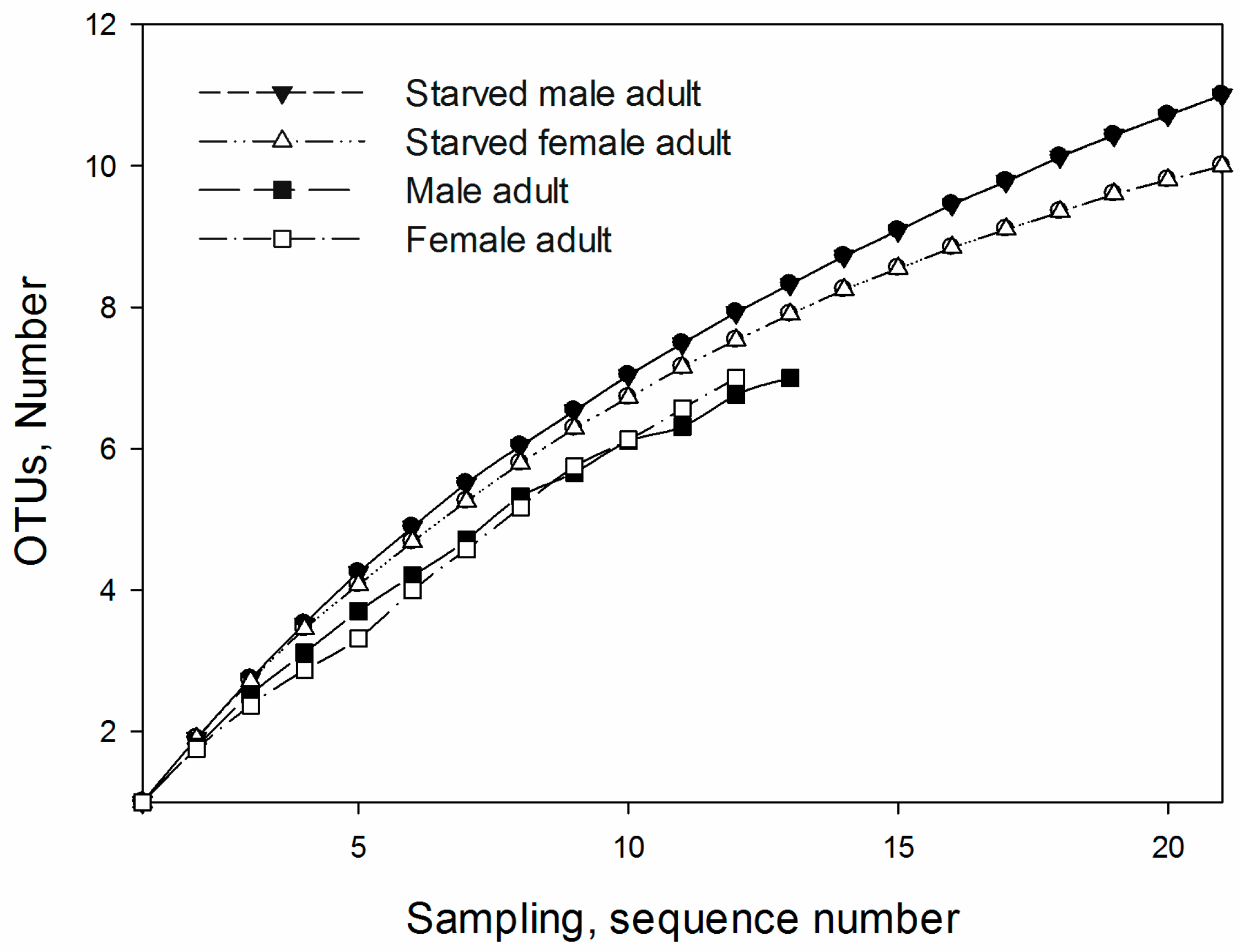
2.6. Operational Taxonomic Units and Richness Estimation
3. Results
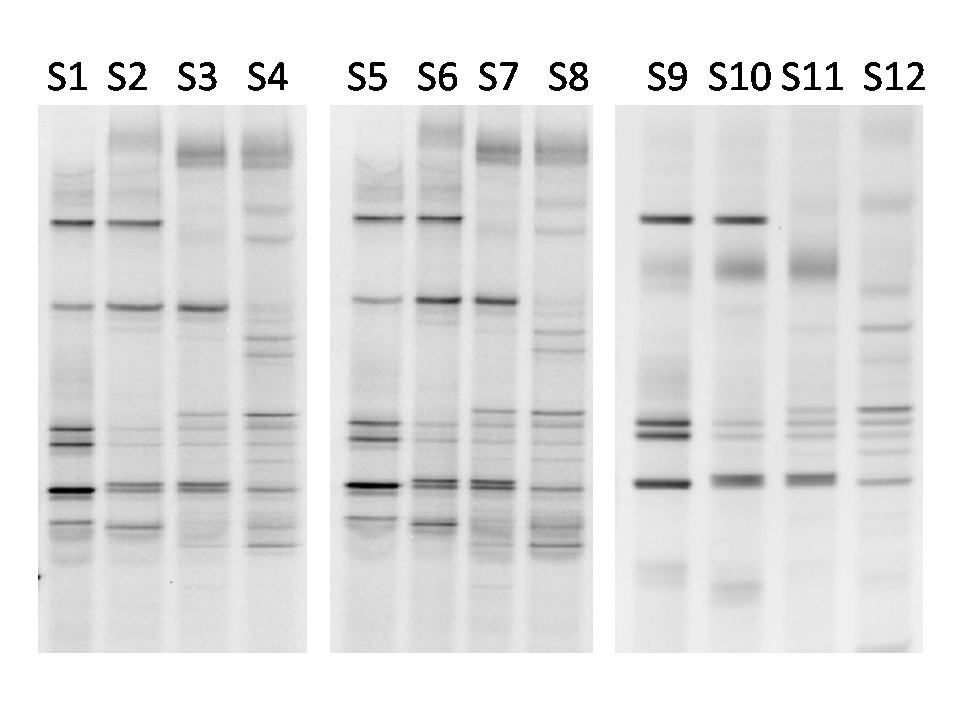
3.1. Bacterial Diversity Analysis
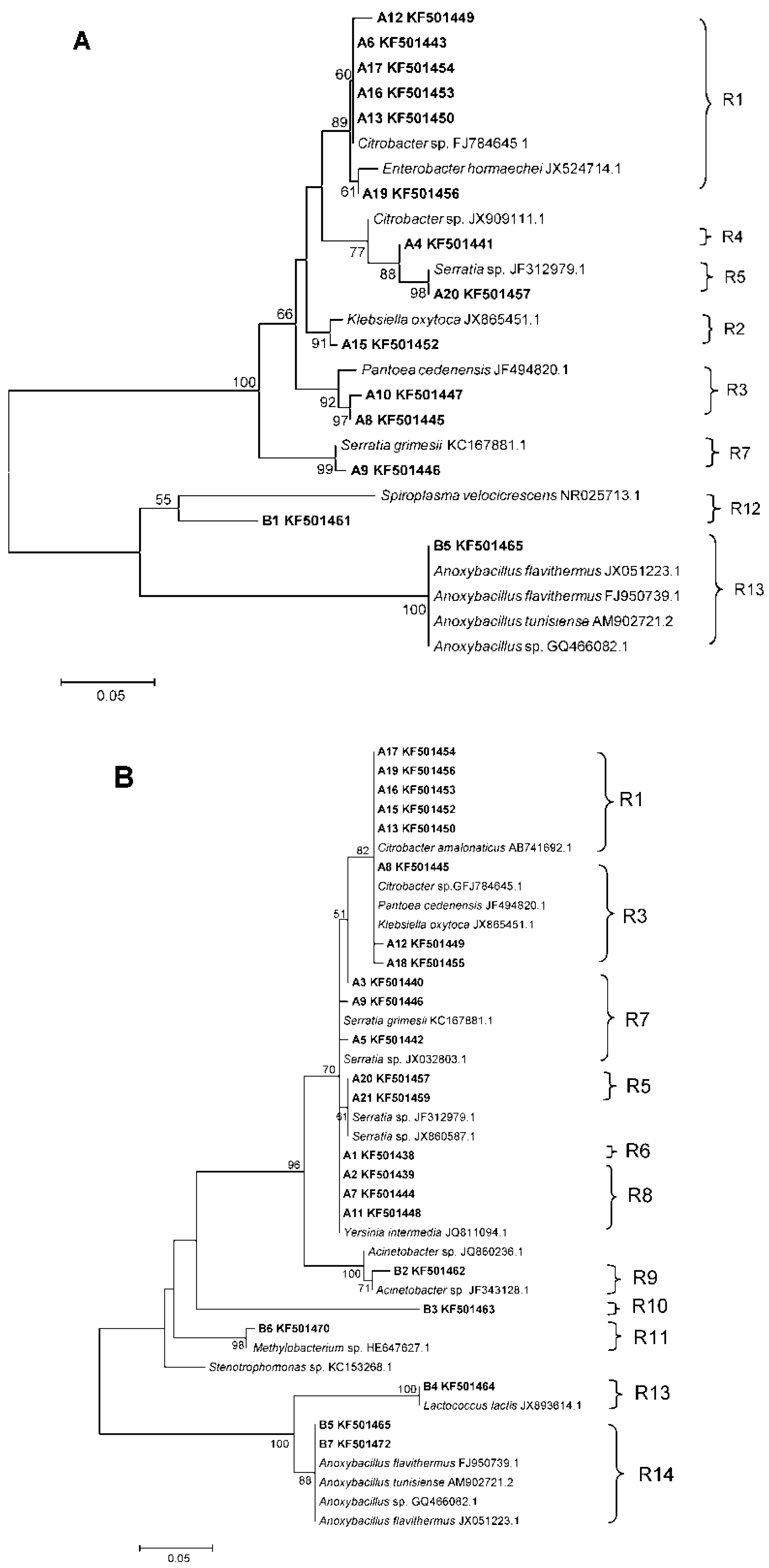
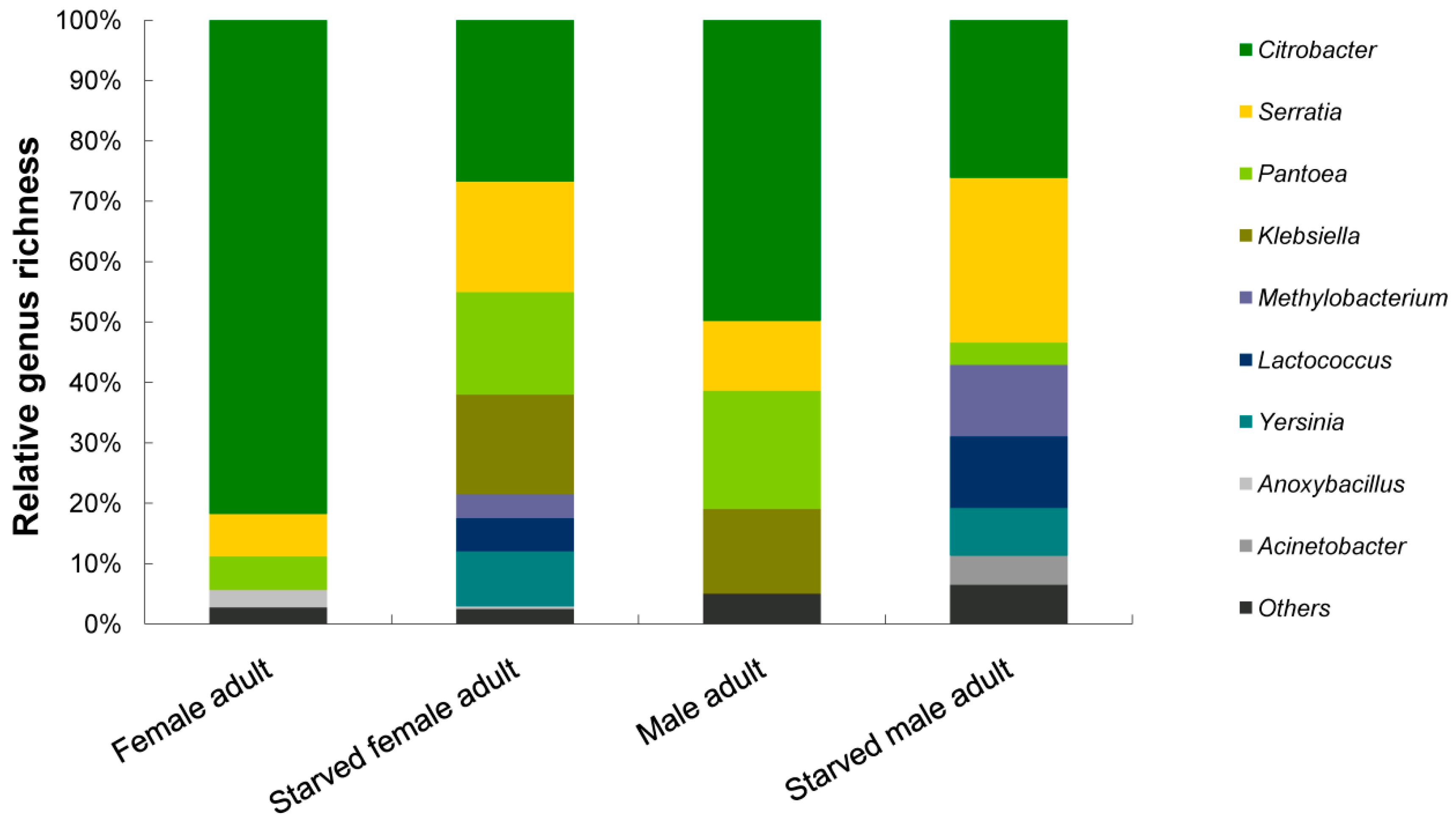
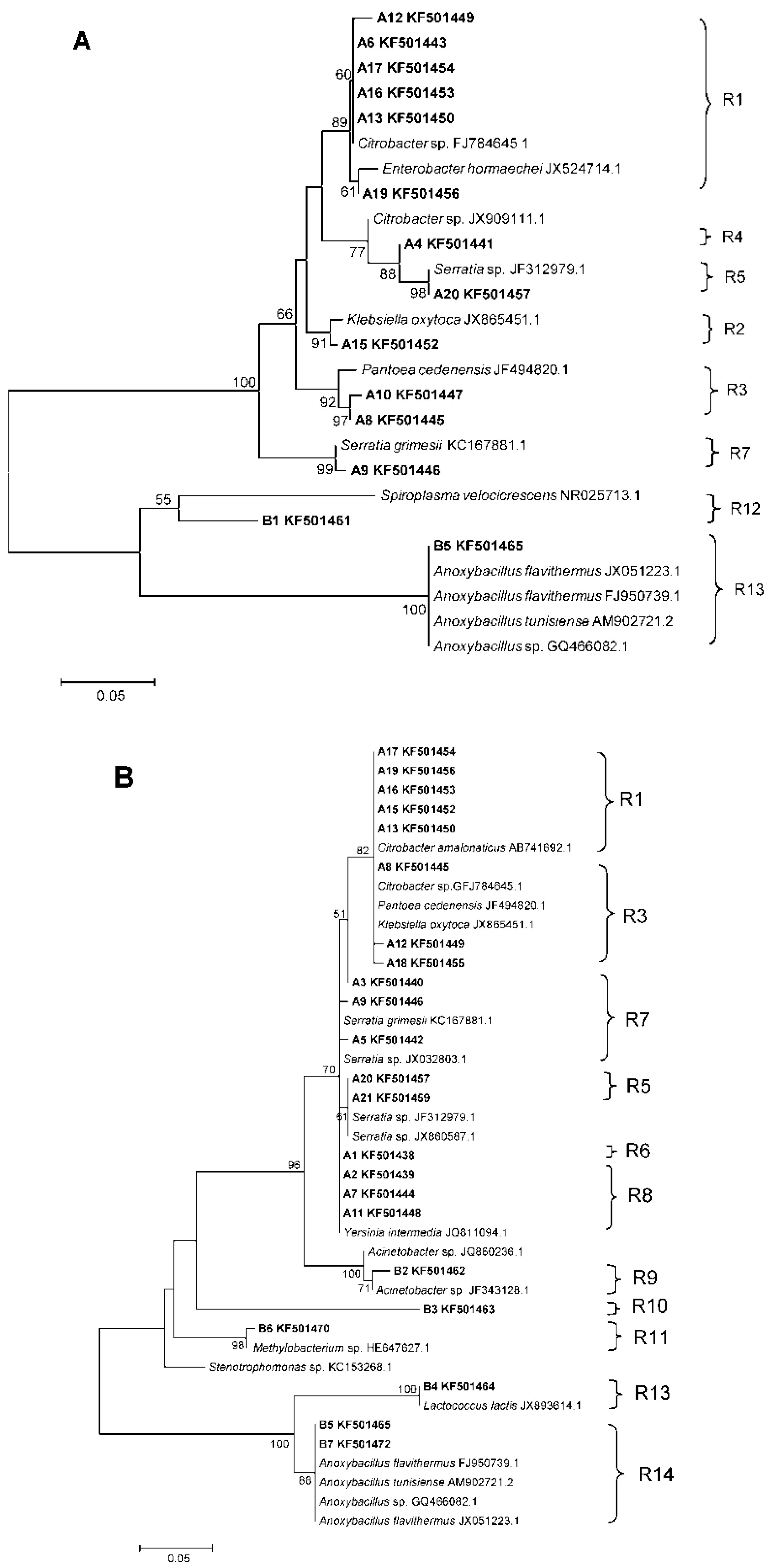
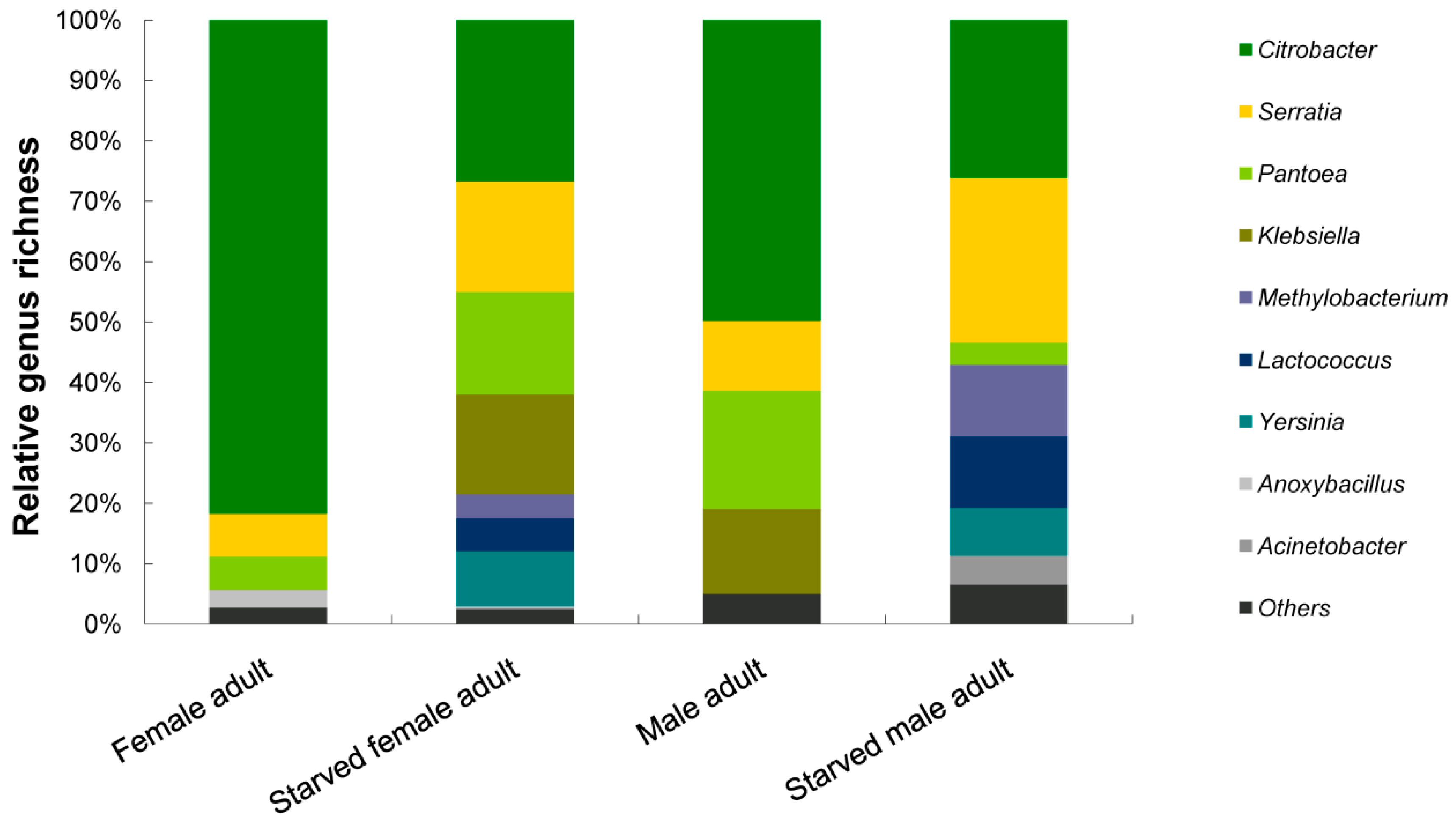
3.2. Phylogenetic Analyses and Dominant Taxa
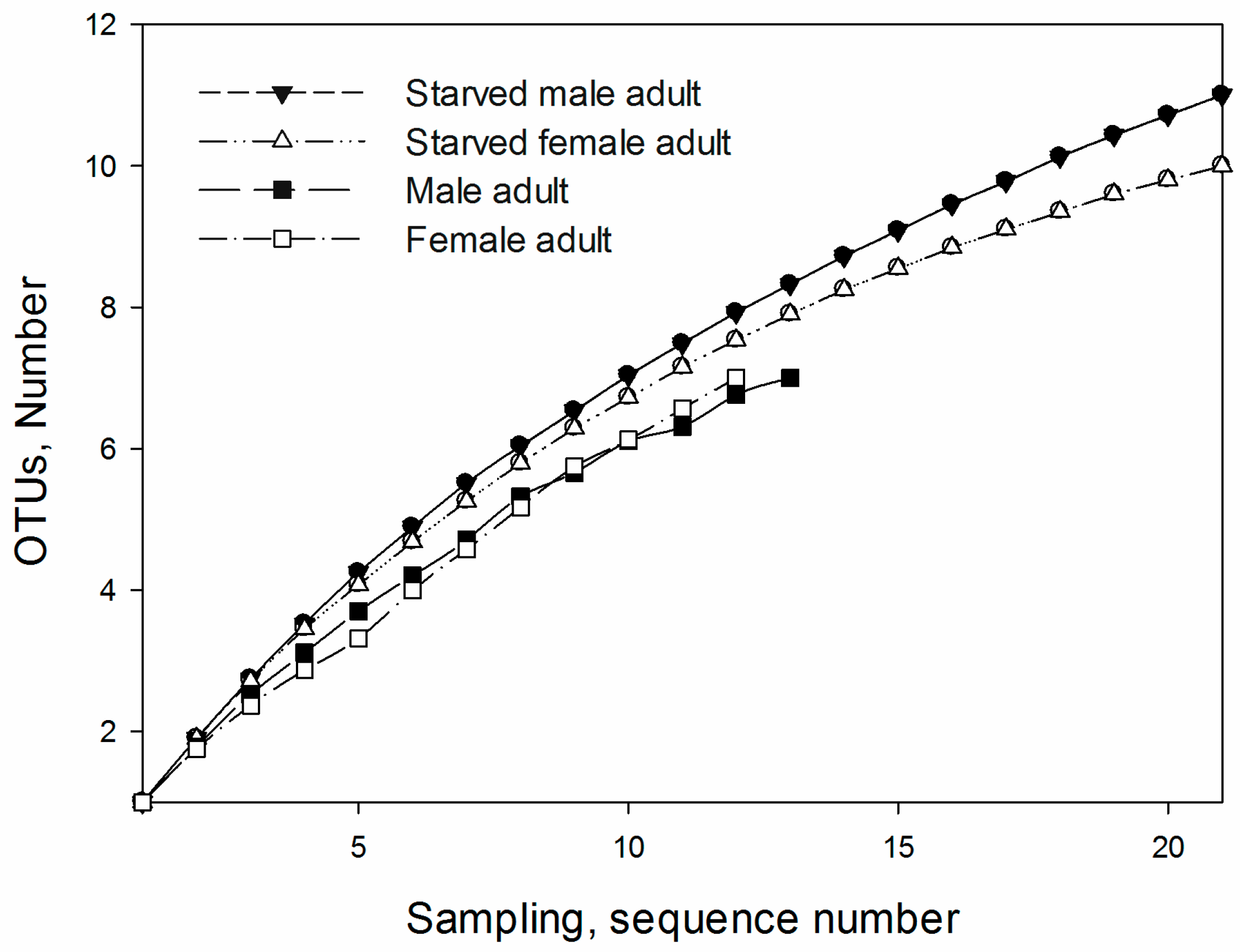
3.3. OTUs
4. Discussion
Bacterial Community Structure
Supplementary material
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Douglas, A.E. The microbial dimension in insect nutritional ecology. Funct. Ecol. 2009, 23, 38–47. [Google Scholar] [CrossRef]
- Xu, L.; Lou, Q.; Cheng, C.; Lu, M.; Sun, J. Gut-Associated bacteria of Dendroctonus valens and their involvement in verbenone production. Microb. Ecol. 2015, 70, 1012–1023. [Google Scholar] [CrossRef] [PubMed]
- Therrien, J.; Mason, C.J.; Cale, J.A.; Adams, A.; Aukema, B.H.; Currie, C.R.; Raffa, K.F.; Erbilgin, N. Bacteria influence mountain pine beetle brood development through interactions with symbiotic and antagonistic fungi: Implications to climate-driven host range expansion. Oecologia 2015, 2, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Audrey-Anne, D.; Amélie, B.; Philippe, C.; Jean-Philippe, B.; Eric, D.; Claude, G. Surveying the endomicrobiome and ectomicrobiome of bark beetles: The case of Dendroctonus simplex. Sci. Rep.-UK 2015, 5, 17190. [Google Scholar]
- Six, D.L.; Klepzig, K.D. Dendroctonus bark beetles as model systems for studies on symbiosis. Symbiosis 2004, 37, 1–26. [Google Scholar]
- Hu, X.; Yu, J.; Wang, C.; Chen, H. Cellulolytic bacteria associated with the gut of Dendroctonus armandi larvae (Coleoptera: Curculionidae: Scolytinae). Forests 2014, 5, 455. [Google Scholar] [CrossRef]
- Morales-Jimenez, J.; Zuniga, G.; Villa-Tanaca, L.; Hernandez-Rodriguez, C. Bacterial community and nitrogen fixation in the red turpentine beetle, Dendroctonus valens Leconte (Coleoptera: Curculionidae: Scolytinae). Microb. Ecol. 2009, 58, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Dillon, R.J.; Dillon, V.M. The gut bacteria of insects: Nonpathogenic interactions. Annu. Rev. Entomol. 2004, 49, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Cardoza, Y.J.; Klepzig, K.D.; Raffa, K.F. Bacteria in oral secretions of an endophytic insect inhibit antagonistic fungi. Ecol. Entomol. 2006, 31, 636–645. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Tang, M. Spatial and temporal dynamics of bark beetles in Chinese white pine in Qinling mountains of Shaanxi province, China. Environ. Entomol. 2007, 36, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wang, C.; Chen, H.; Ma, J. Differences in the structure of the gut bacteria communities in development stages of the Chinese white pine beetle (Dendroctonus armandi). Int. J. Mol. Sci. 2013, 14, 21006–21020. [Google Scholar] [CrossRef] [PubMed]
- Dillon, R.J.; Webster, G.; Weightman, A.J.; Charnley, A.K. Diversity of gut microbiota increases with aging and starvation in the desert locust. Anton. Leeuw. Int. J. G. 2010, 97, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Çelebi, Ö.; Sevim, E.; Sevim, A. Investigation of the internal bacterial flora of Eurygaster integriceps (Hemiptera: Scutelleridae) and pathogenicity of the flora members. Biologia 2014, 69, 1365–1375. [Google Scholar] [CrossRef]
- Fettig, C.J.; Hilszczański, J. Management strategies for bark beetles in conifer forests. In Bark Beetles: Biology and Ecology of Native and Invasive Species; Vega, F.E., Hofstetter, R.W., Eds.; Springer: London, UK, 2015; pp. 555–584. [Google Scholar]
- Delalibera, I.; Handelsman, J.; Raffa, K.F. Contrasts in cellulolytic activities of gut microorganisms between the wood borer, Saperda vestita (Coleoptera: Cerambycidae), and the bark beetles, Ips pini and Dendroctonus frontalis (Coleoptera: Curculionidae). Environ. Entomol. 2005, 34, 541–547. [Google Scholar] [CrossRef]
- Galand, P.E.; Fritze, H.; Yrjala, K. Microsite-Dpendent changes in methanogenic populations in a boreal oligotrophic fen. Environ. Microbiol. 2003, 5, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Lü, D.; Li, Z.; Qin, S.; Ma, H.; Liu, G. Bacterial community structure in the Cerasus sachalinensis kom. rhizosphere based on the polymerase chain reaction-Dnaturing gradient gel electrophoresis (pcr-dgge) method. Afr. J. Biotechnol. 2013, 10, 13430–13438. [Google Scholar]
- Xu, Z.Y.; Tang, M.; Chen, H.; Ban, Y.H.; Zhang, H.H. Microbial community structure in the rhizosphere of Sophora viciifolia grown at a lead and zinc mine of northwest China. Sci. Total Environ. 2012, 435–436. [Google Scholar] [CrossRef] [PubMed]
- Muyzer, G.; Hottenträger, S.; Teske, A.; Wawer, C. Denaturing Gradient Gel Electrophoresis of PCR-Amplified 16S rDNA—A New Molecular Approach to Analyse the Genetic Diversity of Mixed Microbial Communities. In Molecular Microbial Ecology Manual; Akkermans, A.D.L., van Elsas, J.D., De Bruijn, F.J., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1996; pp. 1–23. [Google Scholar]
- Muyzer, G.; de Waal, E.C.; Uitterlinden, A.G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 1993, 59, 695–700. [Google Scholar] [PubMed]
- Cole, J.R.; Wang, Q.; Cardenas, E.; Fish, J.; Chai, B.; Farris, R.J.; Kulam-Syed-Mohideen, A.S.; McGarrell, D.M.; Marsh, T.; Garrity, G.M.; et al. The ribosomal database project: Improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009, 37, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Lee, J.H.; Jung, Y.; Kim, M.; Kim, S.; Kim, B.K.; Lim, Y.W. Eztaxon: A web-Based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int. J. Syst. Evol. Microbiol. 2007, 57, 2259–2261. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. Mega5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Handelsman, J. Introducing dotur, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol. 2005, 71, 1501–1506. [Google Scholar] [CrossRef] [PubMed]
- Chao, A.; Shen, T.-J. Nonparametric estimation of shannon’s index of diversity when there are unseen species in sample. Environ. Ecol. Stat. 2003, 10, 429–443. [Google Scholar] [CrossRef]
- Gotelli, N.J.; Colwell, R.K. Quantifying biodiversity: Procedures and pitfalls in the measurement and comparison of species richness. Ecology letters 2001, 4, 379–391. [Google Scholar] [CrossRef]
- Colwell, R.K.; Mao, C.X.; Chang, J. Interpolating, extrapolating, and comparing incidence-Based species accumulation curves. Ecology 2004, 85, 2717–2727. [Google Scholar] [CrossRef]
- Cocolin, L.; Aggio, D.; Manzano, M.; Cantoni, C.; Comi, G. An application of PCR-Dgge analysis to profile the yeast populations in raw milk. Int. Dairy J. 2002, 12, 407–411. [Google Scholar] [CrossRef]
- Adams, A.S.; Aylward, F.O.; Adams, S.M.; Erbilgin, N.; Aukema, B.H.; Currie, C.R.; Suen, G.; Raffa, K.F. Mountain pine beetles colonizing historical and naive host trees are associated with a bacterial community highly enriched in genes contributing to terpene metabolism. Appl. Environ. Microbiol. 2013, 79, 3468–3475. [Google Scholar] [CrossRef] [PubMed]
- Morales-Jimenez, J.; Vera-Ponce de Leon, A.; Garcia-Dominguez, A.; Martinez-Romero, E.; Zuniga, G.; Hernandez-Rodriguez, C. Nitrogen-Fixing and uricolytic bacteria associated with the gut of Dendroctonus rhizophagus and Dendroctonus valens (Curculionidae: Scolytinae). Microb. Ecol. 2013, 66, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Winder, R.S.; Macey, D.E.; Cortese, J. Dominant bacteria associated with broods of mountain pine beetle, Dendroctonus ponderosae (Coleoptera: Curculionidae, Scolytinae). J. Entomol. Soc. Br. Columbia 2010, 107. [Google Scholar]
- Santo Domingo, J.W.; Kaufman, M.G.; Klug, M.J.; Holben, W.E.; Harris, D.; Tiedje, J.M. Influence of diet on the structure and function of the bacterial hindgut community of crickets. Mol. Ecol. 1998, 7, 761–767. [Google Scholar] [CrossRef]
- Heimpel, A.M. The pH in the gut and blood of the larch sawfly, Pristiphora erichsonii (htg.), and other insects with reference to the pathogenicity of Bacillus cereus fr. And fr. Can. J. Zool. 1955, 33, 99–106. [Google Scholar] [CrossRef]
- French, J.R.; Turner, G.L.; Bradbury, J.F. Nitrogen fixation by bacteria from the hindgut of termites. J. Gen. Microbiol. 1976, 96, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Behar, A.; Yuval, B.; Jurkevitch, E. Enterobacteria-mediated nitrogen fixation in natural populations of the fruit fly Ceratitis capitata. Mol. Ecol. 2005, 14, 2637–2643. [Google Scholar] [CrossRef] [PubMed]
- Lilburn, T.G.; Kim, K.S.; Ostrom, N.E.; Byzek, K.R.; Leadbetter, J.R.; Breznak, J.A. Nitrogen fixation by symbiotic and free-living spirochetes. Science 2001, 292, 2495–2498. [Google Scholar] [CrossRef] [PubMed]
- Yaman, M.; Ertürk, Ö.; Aslan, İ. Isolation of some pathogenic bacteria from the great spruce bark beetle, Dendroctonus micans and its specific predator, Rhizophagus grandis. Folia Microbiol. 2010, 55, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Brand, J.M.; Bracke, J.W.; Markovetz, A.J.; Wood, D.L.; Browne, L.E. Production of verbenol pheromone by a bacterium isolated from bark beetles. Nature 1975, 254, 136–137. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.M.; Tisa, L.S. Friend or foe? A review of the mechanisms that drive Serratia towards diverse lifestyles. Can. J. Microbiol. 2013, 59, 627–640. [Google Scholar] [CrossRef] [PubMed]
- Brand, J.M.; Bracke, J.W.; Markovetz, A.J.; Wood, D.L.; Browne, L.E. Production of verbenol pheromone by a bacterium isolated from bark beetles. Nature 1975, 254, 136–137. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, S.L.; Trunk, K.; English, G.; Fritsch, M.J.; Pourkarimi, E.; Coulthurst, S.J. The opportunistic pathogen Serratia marcescens utilizes type vi secretion to target bacterial competitors. J. Bacteriol. 2011, 193, 6057–6069. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Contreras, M.; Vlisidou, I. The diversity of insect-bacteria interactions and its applications for disease control. Biotechnol. Genet. Eng. Rev. 2008, 25, 203–243. [Google Scholar] [CrossRef]
- Dillon, R.; Vennard, C.; Buckling, A.; Charnley, A. Diversity of locust gut bacteria protects against pathogen invasion. Ecol. Lett. 2005, 8, 1291–1298. [Google Scholar] [CrossRef]
- Sevim, A.; Gökçe, C.; Erbaş, Z.; Ozkan, F. Bacteria from Ips sexdentatus (Coleoptera: Curculionidae) and their biocontrol potential. J. Basic Microbiol. 2012, 52, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Abe, K.; Sato, M. Biological control of an insect pest by gut-colonizing Enterobacter cloacae transformed with ice nucleation gene. J. Appl. Microbiol. 2000, 88, 90–97. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lane | D. armandi sample | S | EH | H′ |
|---|---|---|---|---|
| S1 | Female adult | 12 | 0.854255 | 2.122745 |
| S2 | Male adult | 14 | 0.885312 | 2.336389 |
| S3 | Starved female adult | 22 | 0.858404 | 2.653363 |
| S4 | Starved male adult | 21 | 0.921066 | 2.804207 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, X.; Li, M.; Zhang, F.; Chen, H. Influence of Starvation on the Structure of Gut-Associated Bacterial Communities in the Chinese White Pine Beetle (Dendroctonus armandi). Forests 2016, 7, 126. https://doi.org/10.3390/f7060126
Hu X, Li M, Zhang F, Chen H. Influence of Starvation on the Structure of Gut-Associated Bacterial Communities in the Chinese White Pine Beetle (Dendroctonus armandi). Forests. 2016; 7(6):126. https://doi.org/10.3390/f7060126
Chicago/Turabian StyleHu, Xia, Ming Li, Feiping Zhang, and Hui Chen. 2016. "Influence of Starvation on the Structure of Gut-Associated Bacterial Communities in the Chinese White Pine Beetle (Dendroctonus armandi)" Forests 7, no. 6: 126. https://doi.org/10.3390/f7060126
APA StyleHu, X., Li, M., Zhang, F., & Chen, H. (2016). Influence of Starvation on the Structure of Gut-Associated Bacterial Communities in the Chinese White Pine Beetle (Dendroctonus armandi). Forests, 7(6), 126. https://doi.org/10.3390/f7060126