
Gene Expression Differences between High-Growth Populus Allotriploids and Their Diploid Parents

Abstract
:1. Introduction
2. Materials and Methods
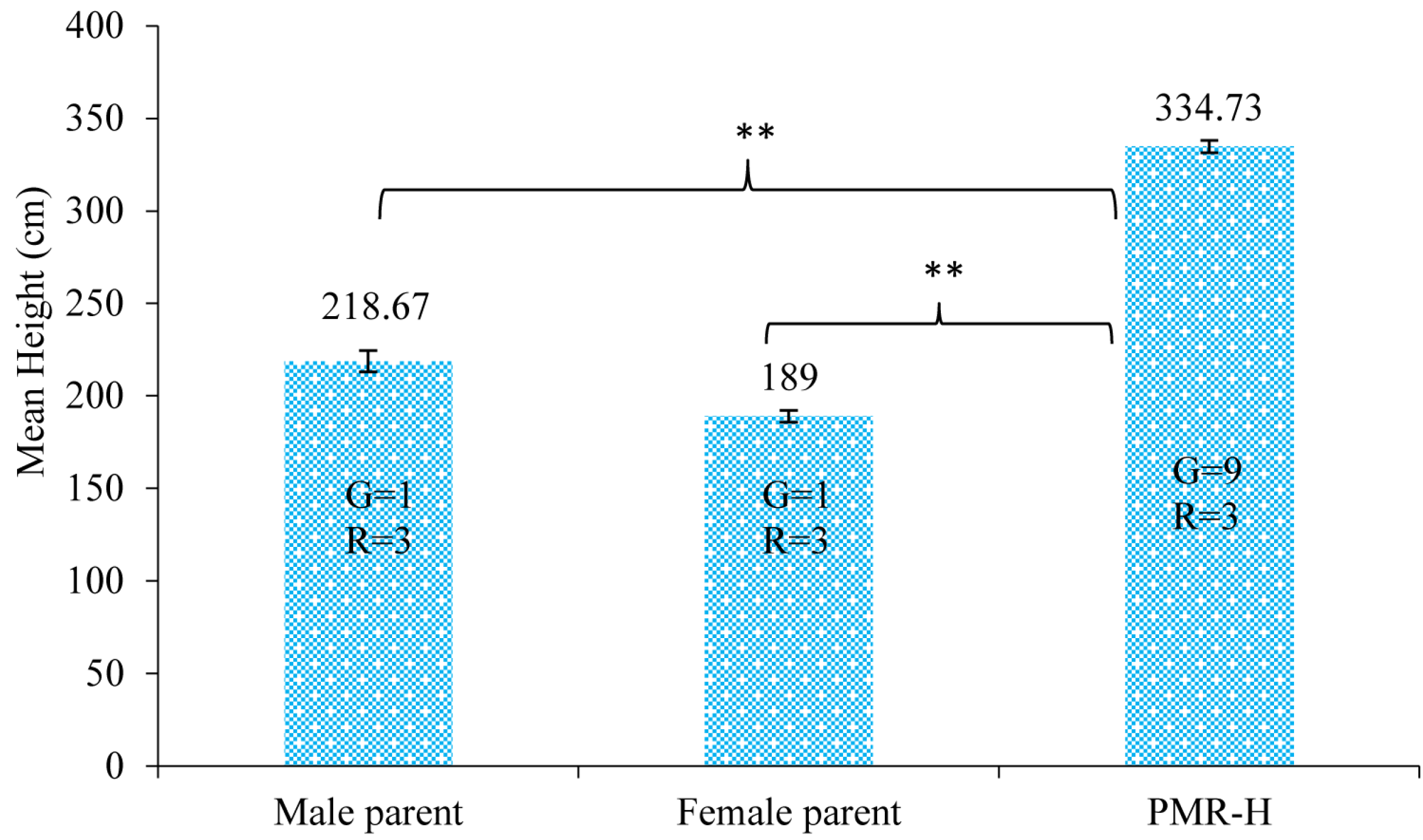
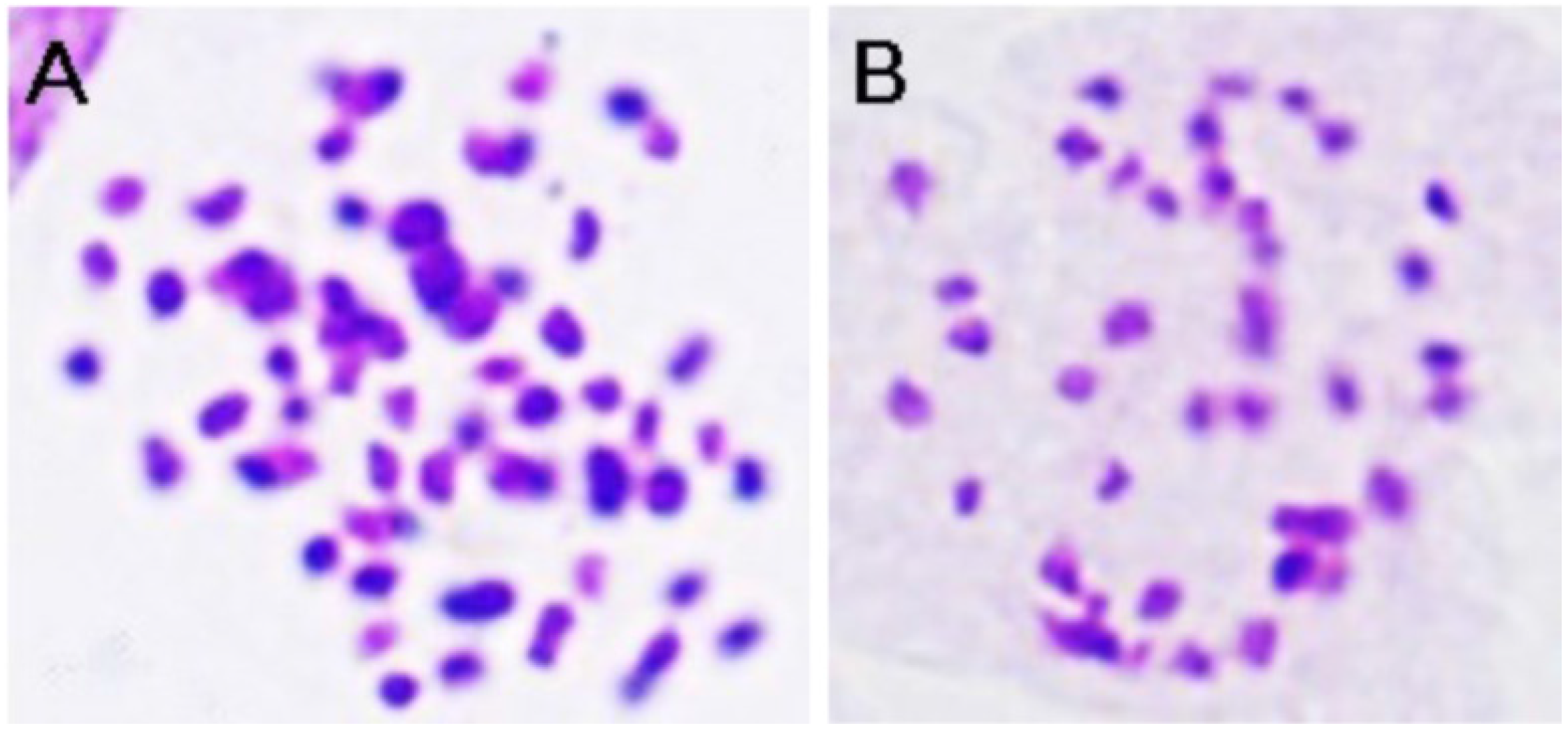
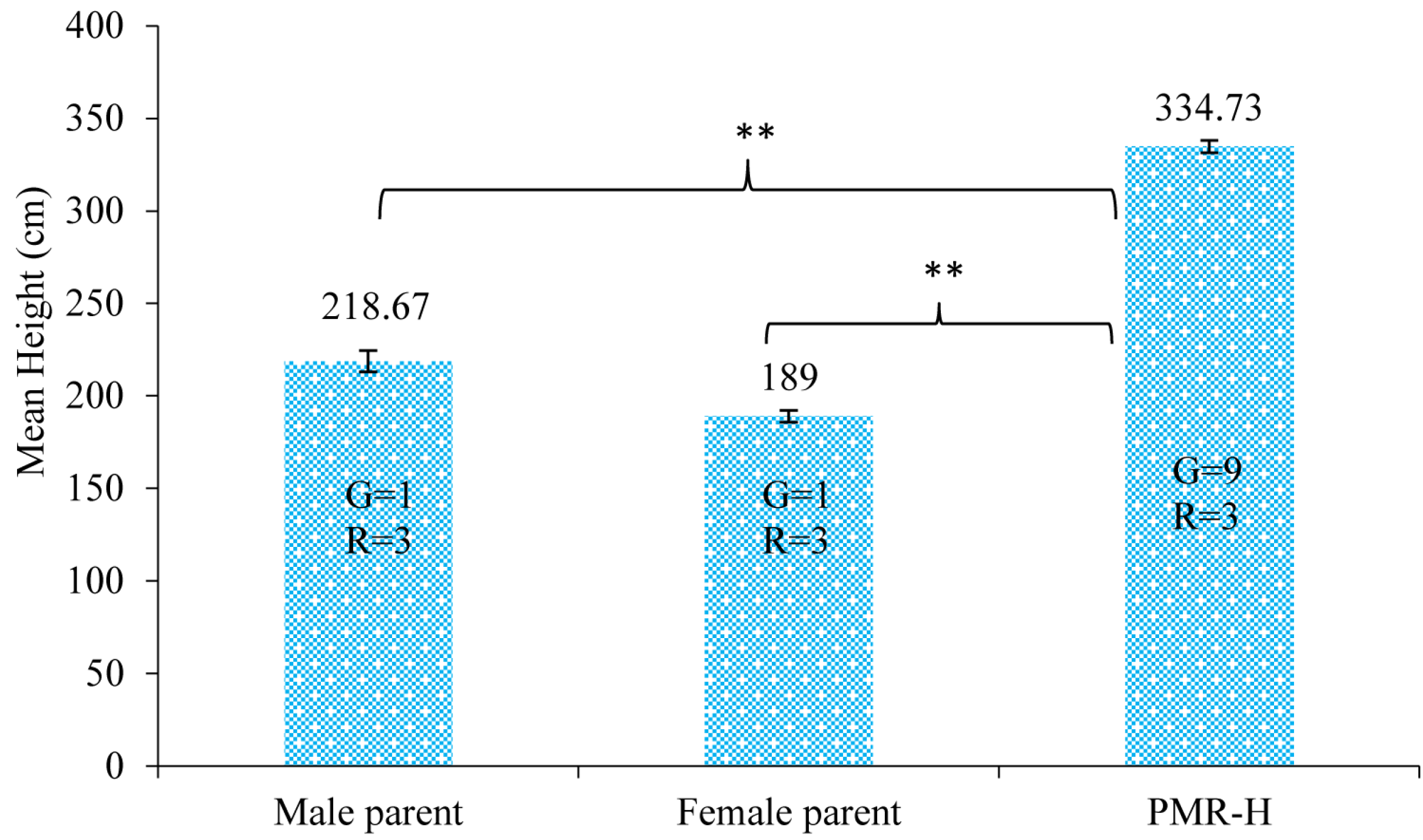
2.1. Plant Materials and RNA Preparation
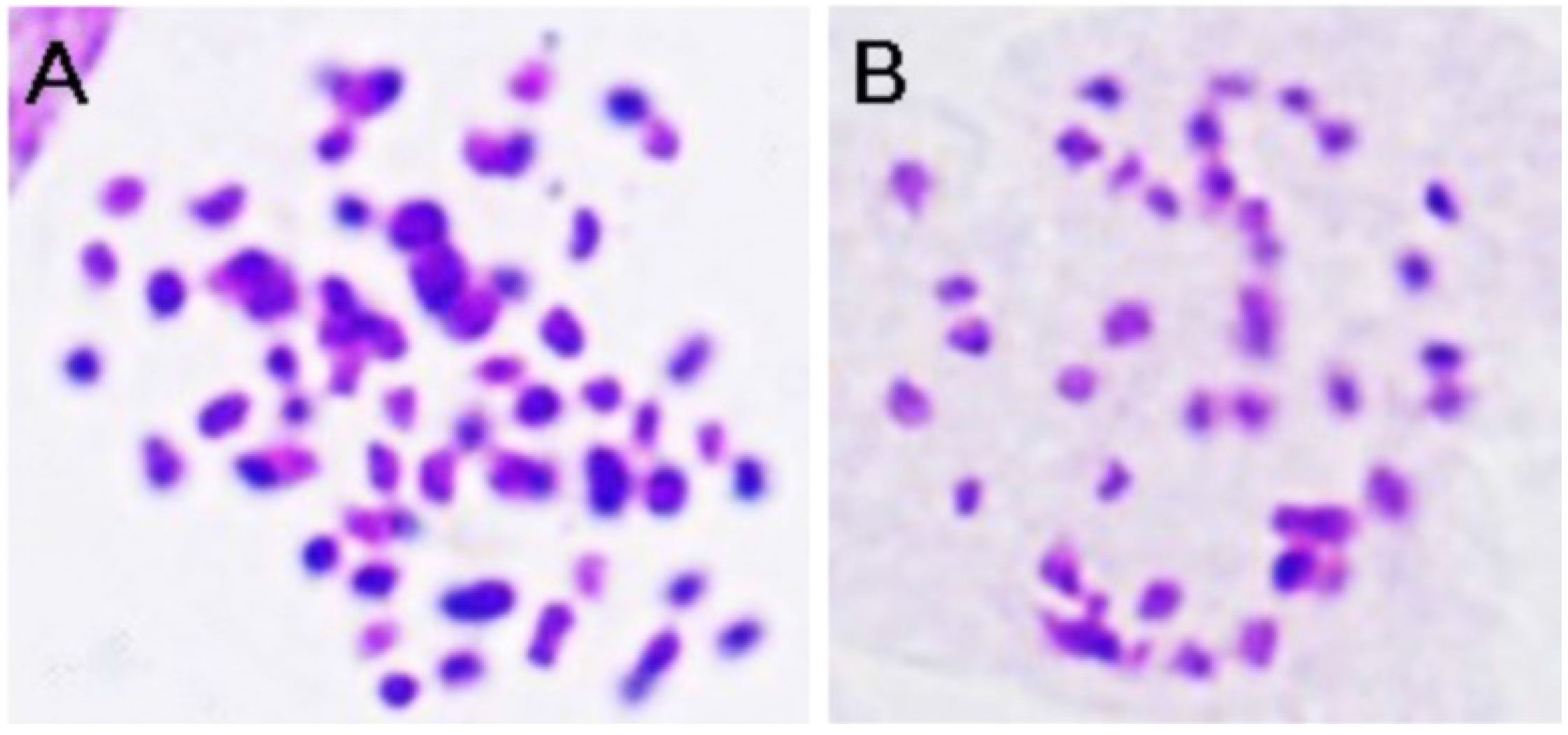
2.2. Library Construction and Illumina RNA-Sequencing
2.3. Mapping of Reads and Differential Gene Expression Analysis
2.4. Gene Annotation
3. Results
3.1. Illumina Sequencing and Sequence Data Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Replicates | Total Reads | Mapped Reads | Mapping Rate | Reads Uniquely Mapped | Unique Mapping Rate |
|---|---|---|---|---|---|---|
| PMR-H | 1 | 30727624 | 25702386 | 83.6% | 24393689 | 94.9% |
| PMR-H | 2 | 30745554 | 25312468 | 82.3% | 23762732 | 93.9% |
| PMR-H | 3 | 27865492 | 23212997 | 83.3% | 22002904 | 94.8% |
| Average | 29,779,557 | 24,742,617 | 83.1% | 23,386,442 | 94.5% | |
| Maternal | 1 | 34747702 | 28629766 | 82.4% | 26582193 | 92.8% |
| Maternal | 2 | 42295592 | 34608859 | 81.8% | 32055183 | 92.6% |
| Maternal | 3 | 39046676 | 31854903 | 81.6% | 29553915 | 92.8% |
| Average | 38,696,656 | 31,697,842 | 82.0% | 29,397,097 | 92.7% | |
| Paternal | 1 | 43718240 | 37101310 | 84.9% | 34418213 | 92.8% |
| Paternal | 2 | 37805466 | 31331934 | 82.9% | 29146283 | 93.0% |
| Paternal | 3 | 29644804 | 24523634 | 82.7% | 22693090 | 92.5% |
| Average | 37,056,170 | 30,985,626 | 83.5% | 28,752,529 | 92.8% |
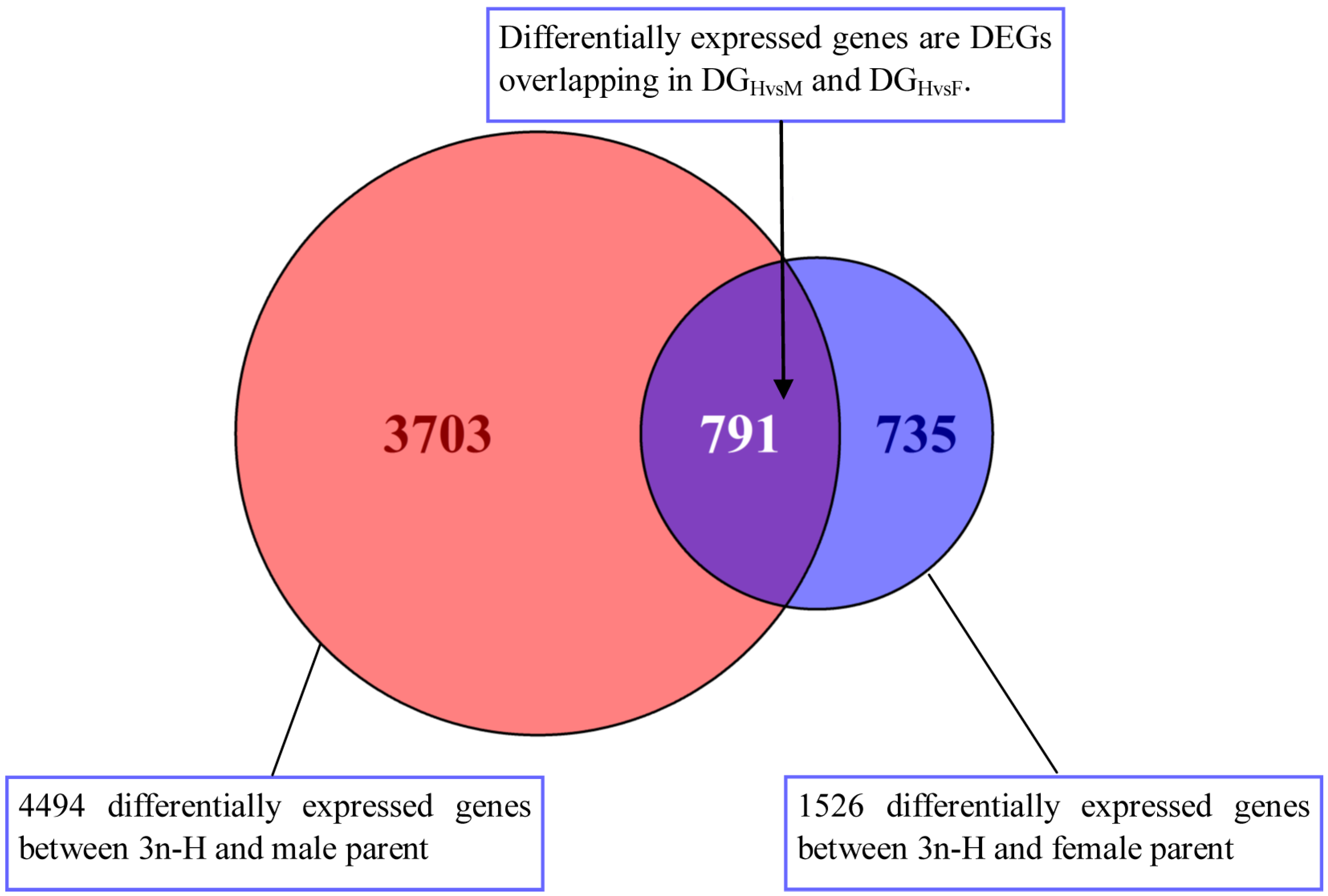
3.2. Differentially Expressed Genes between High-Growth Allotriploid Populus and Their Parents

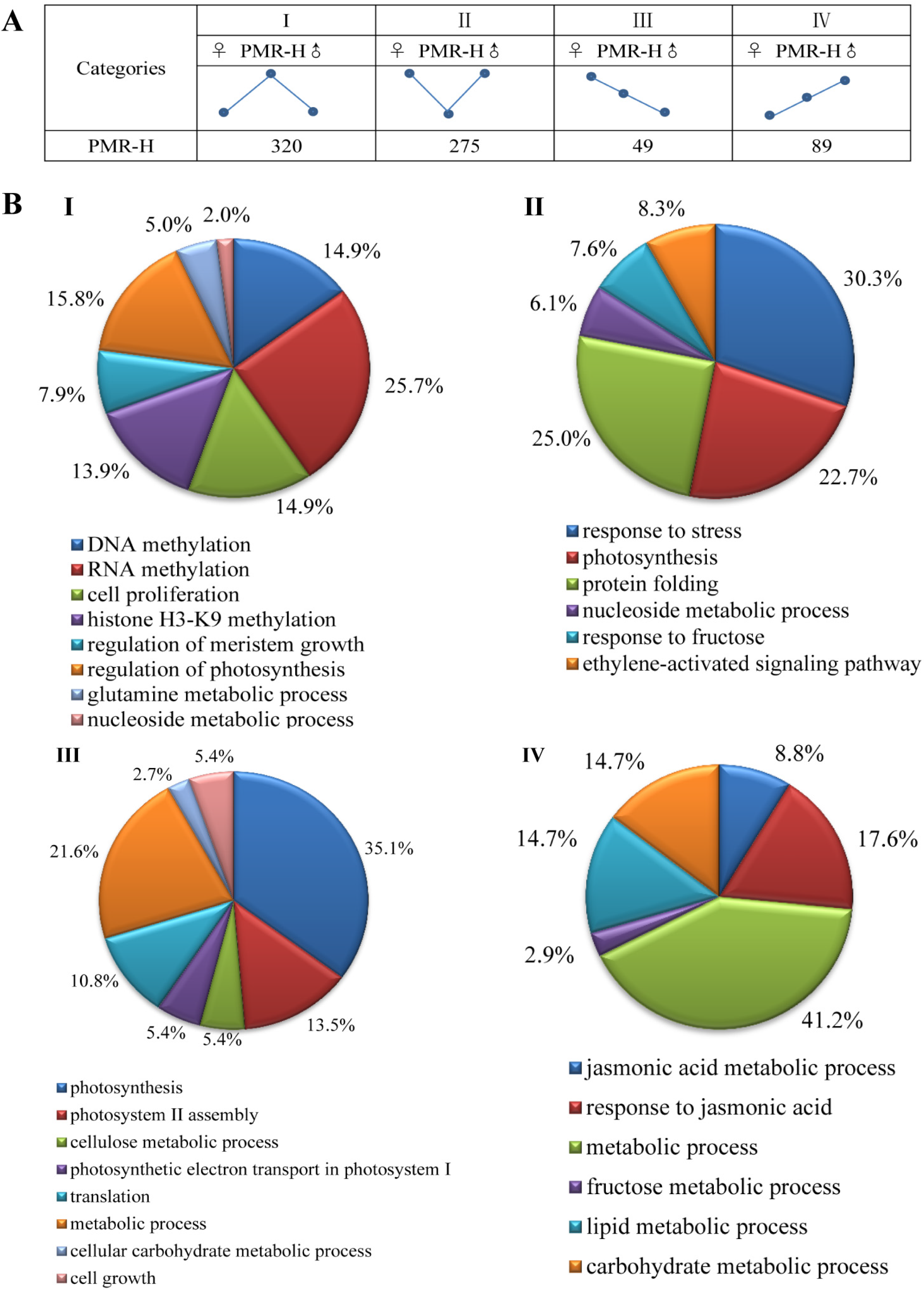

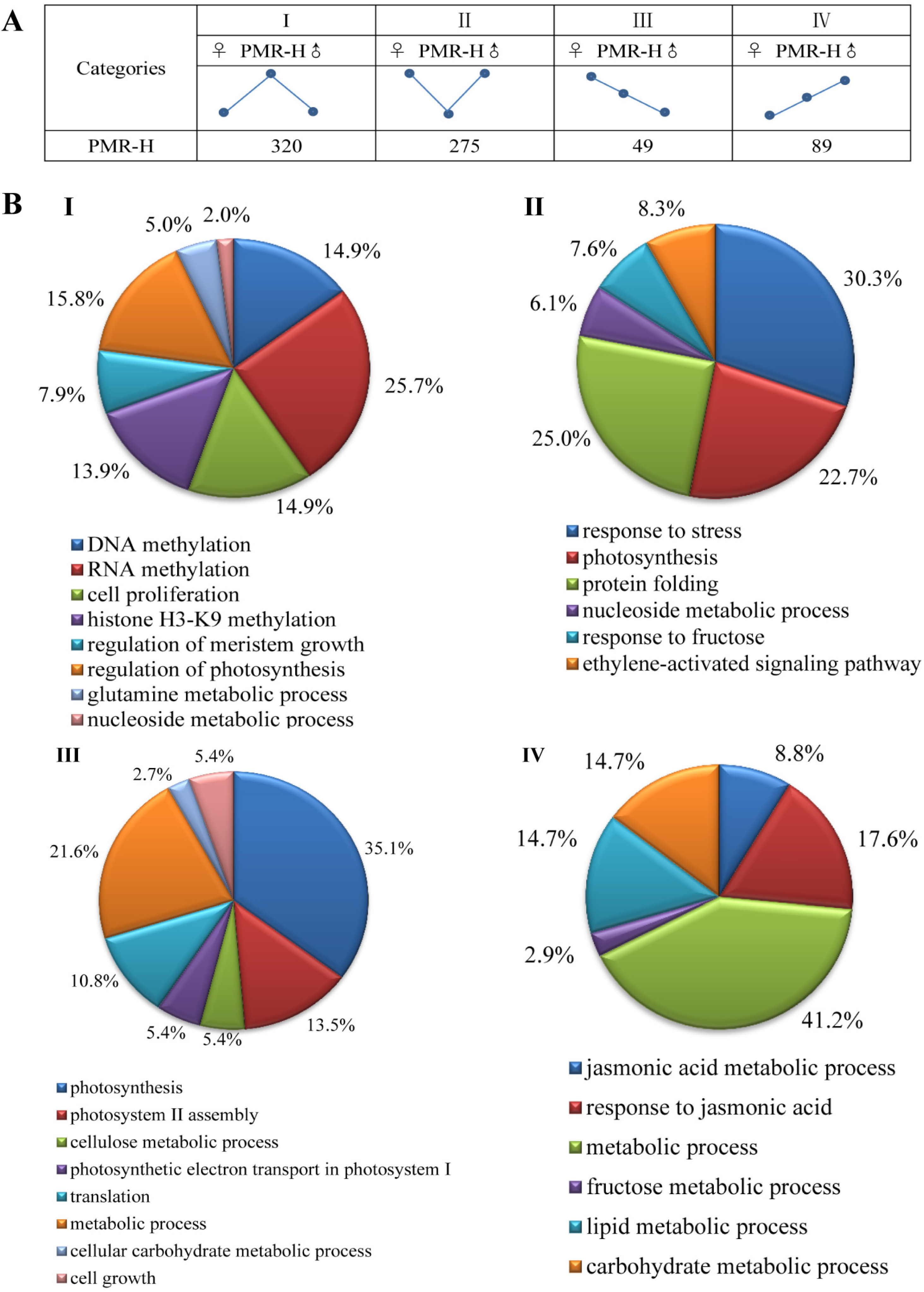
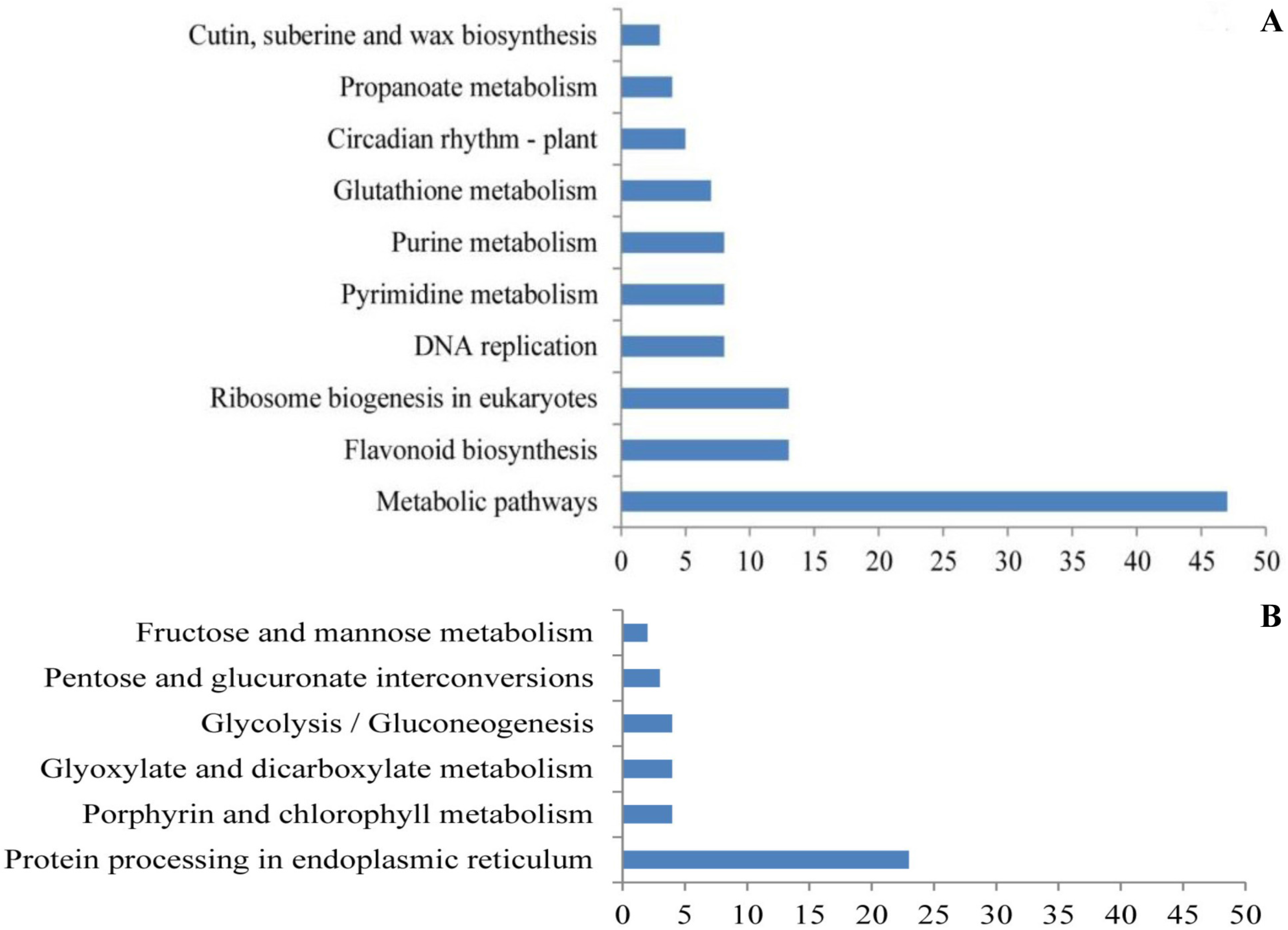
3.3. Non-Additive Genes Expressed in High-Growth Allotriploid Populus Genotypes
| Category | GO Term | Number of DEGs | |
|---|---|---|---|
| Up-Regulated | Down-Regulated | ||
| BP | GO:0015979 Photosynthesis | 5 | 63 |
| GO:0009826 unidimensional cell growth | 2 | 14 | |
| GO:0008283 cell proliferation | 10 | 0 | |
| GO:0006306 DNA methylation | 54 | 0 | |
| GO:0009220 pyrimidine ribonucleotide biosynthetic process | 11 | 0 | |
| GO:0051567 histone H3-K9 methylation | 13 | 0 | |
| GO:0006139 nucleobase-containing compound metabolic process | 6 | 0 | |
| GO:0015994 chlorophyll metabolic process | 3 | 2 | |
| MF | GO:0016168 chlorophyll binding | 10 | 3 |
| GO:0016491 oxidoreductase activity | 8 | 112 | |
| GO:0016747 transferase activity | 23 | 0 | |
| GO:0009055 electron carrier activity | 6 | 44 | |
| GO:0052689 carboxylic ester hydrolase activity | 25 | 4 | |
| CC | GO:0009579 thylakoid | 6 | 46 |
| GO:0009534 chloroplast thylakoid | 32 | 5 | |
| GO:0009535 chloroplast thylakoid membrane | 54 | 3 | |
| GO:0048046 apoplast | 40 | 8 | |
3.4. Cell Differentiation- and Meristem Development-Related Genes are Preferentially Expressed in High-Growth Allotriploid Populus Compared to Their Diploid Parents
| Biological Function | Genes | Poplar Gene Model | TAIR Annotation | FoldChange (3n-H/male Parent) | FoldChange (3n-H/Female Parent) |
|---|---|---|---|---|---|
| Cell proliferation | MCM2 | POPTR_0001s12700 | AT1G44900 | 2.5 | 1.7 |
| MCM3 | POPTR_0004s13660 | AT5G46280 | 2.7 | 1.6 | |
| MCM4 | POPTR_0009s12440 | AT2G16440 | 2.0 | 1.7 | |
| MCM5 | POPTR_0018s12080 | AT2G07690 | 2.1 | 1.6 | |
| MCM6 | POPTR_0001s12380 | AT5G44635 | 2.2 | 1.6 | |
| BARD1 | POPTR_0002s26070 | AT1G04020 | 1.6 | 1.5 | |
| MSH2 | POPTR_0012s05670 | AT3G18524 | 2.2 | 1.6 | |
| HTA3 | POPTR_0005s04250 | AT1G54690 | 1.8 | 1.6 | |
| MSI2 | POPTR_0009s13750 | AT2G16780 | 1.6 | 1.8 | |
| RPA70B | POPTR_0015s06800 | AT5G08020 | 2.3 | 1.8 | |
| SOM1 | POPTR_0019s15030 | AT5G66750 | 2.4 | 2.0 | |
| ATL5 | Potri.014G197100 | AT3G25520 | 1.7 | 1.5 | |
| ATML1 | POPTR_0011s00520 | AT4G21750 | 3.9 | 2.4 | |
| PSK3 | POPTR_0014s00810 | AT1G13590 | 0.6 | 1.8 | |
| MAF5 | POPTR_0003s16840 | AT5G65080 | 0.2 | 0.3 | |
| PSBO | POPTR_0007s12070 | AT5G66570 | 0.6 | 0.7 | |
| CRB | POPTR_0005s01370 | AT1G09340 | 0.6 | 0.6 | |
| Meristem growth | LAX2 | POPTR_0009s13470 | AT2G21050 | 1.7 | 1.6 |
| CYP78A7 | POPTR_0005s08640 | AT5G09970 | 5.5 | 1.9 | |
| JAG | Potri.008G121200 | AT1G68480 | 3.6 | 2.1 | |
| ANT | POPTR_0002s11550 | AT4G37750 | 2.3 | 1.7 | |
| GA20-Oxidase | POPTR_0007s04360 | AT5G07200 | 0.4 | 0.6 | |
| HEN2 | Potri.018G146100 | AT2G06990 | 2.4 | 2.9 | |
| JP630 | POPTR_0010s05000 | AT1G23760 | 1.6 | 1.5 | |
| SPL5 | POPTR_0011s05480 | AT3G15270 | 2.2 | 1.8 | |
| PLP9 | POPTR_0002s05350 | AT3G63200 | 1.7 | 2.1 | |
| RID3 | POPTR_0014s02710 | AT3G49180 | 2.3 | 1.7 | |
| CHR17 | POPTR_0010s02180 | AT5G18620 | 2.2 | 1.7 | |
| EXPA1 | POPTR_0013s05730 | AT1G69530 | 0.3 | 0.6 | |
| ATEXPA10 | POPTR_0010s17440 | AT1G26770 | 0.5 | 0.6 | |
| ATEXPB3 | POPTR_0013s13780 | AT4G28250 | 2.3 | 0.6 | |
| MAN7 | POPTR_0005s12250 | AT5G66460 | 0.5 | 0.6 | |
| LHT7 | Potri.004G181100 | AT4G35180 | 0.5 | 0.7 |
3.5. Auxin-, Gibberellic Acid-, and Jasmonic Acid-Related Genes Are Preferentially Expressed in High-Growth Allotriploid Populus Progeny Genotypes Compared to Their Diploid Parents
| Hormone Types | Genes | Poplar Gene Model | TAIR Annotation | Log2 Ratio (PMR-H/male Parent) a | Log2 Ratio (PMR-H/Female Parent) |
|---|---|---|---|---|---|
| Auxin | CHS | POPTR_0003s17540 | AT5G13930 | 0.86 | 1.21 |
| JP630 | POPTR_0010s05000 | AT1G23760 | 0.68 | 0.59 | |
| TT7 | POPTR_0013s07050 | AT5G07990 | 0.69 | 0.60 | |
| DOT3 | POPTR_0007s06310 | AT5G10250 | 1.43 | 0.96 | |
| PDR12 | POPTR_0001s14650 | AT1G15520 | 1.38 | 1.01 | |
| SAUR78 | POPTR_0630s00200 | AT1G72430 | 1.04 | 1.28 | |
| SAUR51 | POPTR_0004s17150 | AT1G75580 | −1.25 | 1.19 | |
| SAUR29 | POPTR_0004s17180 | AT3G03820 | −1.31 | −1.16 | |
| MYB78 | POPTR_0010s15970 | AT5G49620 | 1.42 | 1.24 | |
| AAE18 | POPTR_0001s04380 | AT1G55320 | 0.70 | 1.15 | |
| LAX2 | POPTR_0009s13470 | AT2G21050 | 0.72 | 0.70 | |
| ARF18 | POPTR_0002s17350 | AT3G61830 | 0.64 | 0.61 | |
| ARF2 | POPTR_0003s16210 | AT5G62000 | 1.03 | 0.63 | |
| NAP9 | POPTR_0012s01280 | AT5G02270 | −2.01 | 20 | |
| GRXS13 | POPTR_0017s04860 | AT1G03850 | −1.16 | −0.97 | |
| MI-1-P | POPTR_0007s05810 | AT4G39800 | −0.69 | −0.80 | |
| PBP1 | POPTR_0004s02630 | AT5G54490 | −1.23 | 1.21 | |
| GDSL-like | POPTR_0001s19220 | AT3G16370 | −0.94 | −0.62 | |
| Gibberellin | XERICO | POPTR_0014s16830 | AT2G04240 | −0.97 | −0.87 |
| EXPA1 | POPTR_0010s17440 | AT1G26770 | −1.00 | −0.73 | |
| F-box protein SNE | POPTR_0014s16360 | AT2G42660 | 0.64 | 1.12 | |
| FKBP-like | POPTR_0015s08290 | AT2G43560 | −0.94 | 1.41 | |
| GAST1 | POPTR_0006s04300 | AT3G10185 | −1.85 | −1.02 | |
| ATHB22 | POPTR_0002s10330 | AT4G24660 | 0.62 | 0.71 | |
| YAP169 | POPTR_0007s04360 | AT5G07200 | −1.18 | −0.61 | |
| Jasmonic Acid | tMYB70 | POPTR_0009s09890 | AT2G23290 | 1.82 | 1.33 |
| WRKY40 | POPTR_0016s14490 | AT1G80840 | 0.64 | 1.32 | |
| MYB78 | POPTR_0010s15970 | AT5G49620 | 1.42 | 1.24 | |
| ANS | POPTR_0003s11900 | AT4G22880 | 1.61 | 1.30 | |
| PDR12 | POPTR_0001s14650 | AT1G15520 | 1.38 | 1.01 | |
| PBP1 | POPTR_0004s02630 | AT5G54490 | −1.23 | 1.21 | |
| SOT18 | POPTR_0003s18750 | AT1G74090 | −0.73 | 0.91 | |
| SOT17 | POPTR_0003s18820 | AT1G18590 | −1.42 | 20.00 | |
| ACL5 | POPTR_0001s29530 | AT5G19530 | −1.42 | 2.39 | |
| NAC032 | POPTR_0007s04780 | AT1G77450 | −2.22 | −1.32 |
3.6. Non-Additive Transcription Factor Gene Expression in High-Growth Populus Allotriploids
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jiao, Y.; Wickett, N.J.; Ayyampalayam, S.; Chanderbali, A.S.; Landherr, L.; Ralph, P.E.; Tomsho, L.P.; Hu, Y.; Liang, H.; Soltis, P.S.; et al. Ancestral polyploidy in seed plants and angiosperms. Nature 2011, 473, 97–100. [Google Scholar] [CrossRef]
- Chen, Z.J. Molecular mechanisms of polyploidy and hybrid vigor. Trends Plant Sci. 2010, 15, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Comai, L. The advantages and disadvantages of being polyploid. Nat. Rev. Genet. 2005, 6, 836–846. [Google Scholar] [CrossRef] [PubMed]
- Buijtenen, J.P.; Joranson, P.N.; Einspahr, D.W. Diploid versus triploid aspen as pulpwood sources with reference to growth, chemical, physical and pulping differences. Tappi 1958, 41, 170–175. [Google Scholar]
- Einspahr, D.W. Production and utilization of triploid hybrid aspen. Iowa State J. Res. 1984, 58, 401–409. [Google Scholar]
- Nilsson-Ehle, H. Note regarding the gigas form of Populus. tremula found in nature. Hereditas 1936, 21, 372–382. [Google Scholar]
- Zhu, Z.T.; Kang, X.Y.; Zhang, Z.Y. Studies on selection of natural triploids of Populus. tomentosa. Sci. Silave Sin. 1998, 34, 22–31. [Google Scholar]
- Lu, M.; Zhang, P.D.; Kang, X.Y. Induction of 2n female gametes in Populus. adenopoda Maxim by high temperature exposure during female gametophyte development. Breed. Sci. 2013, 63, 96–103. [Google Scholar]
- Dong, C.B.; Suo, Y.J.; Kang, X.Y. Assessment of the genetic composition of triploid hybrid Populus using SSR markers with low recombination frequencies. Can. J. For. Res. 2014, 44, 692–699. [Google Scholar] [CrossRef]
- Yang, C.C. Study on Morphogenesis for Height Growth and Heterosis of Clones in Black Poplar Seedling Stage. Ph.D. Thesis, Chinese Academy of Forestry, Beijing, China, 2010. [Google Scholar]
- Shen, Y.; Zhao, Q.; Zou, J.; Wang, W.; Gao, Y.; Meng, J.; Wang, J. Characterization and expression patterns of small RNAs in synthesized Brassica hexaploids. Plant Mol. Biol. 2014, 85, 287–299. [Google Scholar] [CrossRef]
- He, D.; Mathiason, K.; Fennell, A. Auxin and cytokinin related gene expression during active shoot growth and latent bud paradormancy in Vitis riparia grapevine. J. Plant Physiol. 2012, 169, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Stuurman, J.; Jäggi, F.; Kuhlemeier, C. Shoot meristem maintenance is controlled by a GRAS-gene mediated signal from differentiating cells. Genes 2002, 16, 2213–2218. [Google Scholar] [CrossRef]
- Laux, T.; Mayer, K.F.; Berger, J.; Jürgens, G. The WUSCHEL gene is required for shoot and floral meristem integrity in Arabidopsis. Development 1996, 122, 87–96. [Google Scholar] [PubMed]
- Leubner-Metzger, G. Functions and regulation of beta-1,3-glucanases during seed germination, dormancy release and after-ripening. Seed Sci. Res. 2003, 13, 17–34. [Google Scholar] [CrossRef]
- Brand, U.; Fletcher, J.C.; Hobe, M.; Meyerowitz, E.M.; Simon, R. Dependence of stem cell fate in Arabidopsis on a feedback loop regulated by CLV3 activity. Science 2000, 289, 617–619. [Google Scholar] [CrossRef] [PubMed]
- Schoof, H.; Lenhard, M.; Haecker, A.; Mayer, K.F.; Jürgens, G.; Laux, T. The stem cell population of Arabidopsis shoot meristems in maintained by a regulatory loop between the CLAVATA and WUSCHEL genes. Cell 2000, 100, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kang, X.Y.; Li, D.L.; Chen, H.W.; Zhang, P.D. Induction of diploid eggs with colchicine during embryo sac development in Populus. Silvae Genet. 2009, 59, 40–48. [Google Scholar]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Shultz, R.W.; Lee, T.J.; Allen, G.C.; Thompson, W.F.; Hanley-Bowdoin, L. Dynamic localization of the DNA replication proteins MCM5 and MCM7 in plants. Plant Physiol. 2009, 150, 658–669. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R.; Mariconti, L.; Rossignol, P.; Perennes, C.; Cella, R.; Bergounioux, C. Two E2F sites in the Arabidopsis MCM3 promoter have different roles in cell cycle activation and meristematic expression. J. Biol. Chem. 2002, 277, 32978–32984. [Google Scholar] [CrossRef]
- Dresselhaus, T.; Srilunchang, K.O.; Leljak-Levanic, D.; Schreiber, D.N.; Garg, P. The fertilization-induced DNA replication factor MCM6 of maize shuttles between cytoplasm and nucleus, and is essential for plant growth and development. Plant Physiol. 2006, 140, 512–527. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Li, Q.; Zhu, Y.X. Mutation of Arabidopsis BARD1 causes meristem defects by failing to confine WUSCHEL expression to the organizing center. Plant Cell 2008, 20, 1482–1493. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, H.; Zhang, Y.; Hattori, S.; Omae, M.; Shimizu-Sato, S.; Oikawa, T.; Qian, Q.; Nishimura, M.; Kitano, H.; Xie, H.; et al. LAX PANICLE2 of rice encodes a novel nuclear protein and regulates the formation of axillary meristems. Plant Cell 2011, 23, 3276–3287. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, M.; Ohta, D. Diversification of P450 genes during land plant evolution. Annu. Rev. Plant Biol. 2010, 61, 291–315. [Google Scholar] [CrossRef] [PubMed]
- Ohno, C.K.; Reddy, G.V.; Heisler, M.G.; Meyerowitz, E.M. The Arabidopsis JAGGED gene encodes a zinc finger protein that promotes leaf tissue development. Development 2004, 131, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, Y.; Fischer, R.L. Plant organ size control: AINTEGUMENTA regulates growth and cell numbers during organogenesis. Proc. Natl. Acad. Sci. USA 2000, 18, 942–947. [Google Scholar] [CrossRef]
- Hisamatsu, T.; King, R.W.; Helliwell, C.A.; Koshioka, M. The involvement of gibberellin 20-oxidase genes in phytochrome-regulated petiole elongation of Arabidopsis. Plant Physiol. 2005, 138, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.H.; Li, D.M.; Yin, M.H.; Li, X.B.; Zhang, M.; Wang, Y.J.; Dong, J.; Zhao, J.; Luo, M.; Luo, X.Y.; et al. Gibberellin 20-oxidase promotes initiation and elongation of cotton fibers by regulating gibberellin synthesis. J. Plant Physiol. 2010, 167, 829–837. [Google Scholar] [CrossRef]
- Bou-Torrent, J.; Galstyan, A.; Gallemí, M.; Cifuentes-Esquivel, N.; Molina-Contreras, M.J.; Salla-Martret, M.; Jikumaru, Y.; Yamaguchi, S.; Kamiya, Y.; Martínez-García, J.F. Plant proximity perception dynamically modulates hormone levels and sensitivity in Arabidopsis. J. Exp. Bot. 2014, 65, 2937–2947. [Google Scholar] [CrossRef] [PubMed]
- Golisz, A.; Suganom, M.L.; Fujiim, Y. Microarray expression profiling of Arabidopsis thaliana L. in response to allelochemicals identified in buckwheat. J. Exp. Bot. 2008, 59, 3099–3109. [Google Scholar] [CrossRef] [PubMed]
- Péret, B.; Swarup, K.; Ferguson, A.; Seth, M.; Yang, Y.; Dhondt, S.; James, N.; Casimiro, I.; Perry, P.; Syed, A.; et al. AUX/LAX genes encode a family of auxin influx transporters that perform distinct functions during Arabidopsis development. Plant Cell 2012, 24, 2874–2885. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.G.; Ellis, C.; Devoto, A. The jasmonate signal pathway. Plant Cell 2002, 14, S153–S164. [Google Scholar] [PubMed]
- Gomi, K.; Ogawa, D.; Katou, S.; Kamada, H.; Nakajima, N.; Saji, H.; Soyano, T.; Sasabe, M.; Machida, Y.; Mitsuhara, I.; et al. A mitogen-activated protein kinase NtMPK4 activated by SIPKK is required for jasmonic acid signaling and involved in ozone tolerance via stomatal movement in tobacco. Plant Cell Physiol. 2005, 46, 1902–1914. [Google Scholar] [CrossRef]
- Tominaga-Wada, R.; Wada, T. Regulation of root hair cell differentiation by R3 MYB transcription factors in tomato and Arabidopsis. Front. Plant Sci. 2014, 5, 91. [Google Scholar] [CrossRef] [PubMed]
- Brandt, R.; Cabedo, M.; Xie, Y.; Wenkel, S. Homeodomain leucine-zipper proteins and their role in synchronizing growth and development with the environment. J. Integr. Plant Biol. 2014, 56, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.C.; Betzner, A.S.; Huttner, E.; Oakes, M.P.; Tucker, W.Q.; Gerentes, D.; Perez, P.; Smyth, D.R. AINTEGUMENTA, an APETALA2-like gene of Arabidopsis with pleiotropic roles in ovule development and floral organ growth. PlantCell 1996, 8, 155–168. [Google Scholar]
- Gaeta, R.T.; Yoo, S.Y.; Pires, J.C.; Doerge, R.W.; Chen, Z.J.; Osborn, T.C. Analysis of gene expression in resynthesized Brassica napus allopolyploids using Arabidopsis 70mer oligo microarrays. PLoS One 2009, 4, e4760. [Google Scholar] [CrossRef] [PubMed]
- Yoo, M.J.; Szadkowski, E.; Wendel, J.F. Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity 2013, 110, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Auger, D.L.; Gray, A.D.; Ream, T.S.; Kato, A.; Coe, E.H., Jr.; Birchler, J.A. Non-additive gene expression in diploid and triploid hybrids of maize. Genetics 2005, 169, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Dogra Gray, A.; Auger, D.L.; Birchler, J.A. Genomic dosage effects on heterosis in triploid maize. Proc. Natl. Acad. Sci. USA 2013, 10, 2665–2669. [Google Scholar] [CrossRef]
- Washburn, J.D.; Birchler, J.A. Polyploids as a “model system” for the study of heterosis. Plant Reprod. 2013, 27, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.C.; Witucka-Wall, H.; Becher, M.; Blacha, A.; Boudichevskaia, A.; Dörmann, P.; Fiehn, O.; Friedel, S.; von Korff, M.; Lisec, J.; et al. Heterosis manifestation during early Arabidopsis seedling development is characterized by intermediate gene expression and enhanced metabolic activity in the hybrids. Plant J. 2012, 71, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Qi, B.; Huang, W.; Zhu, B.; Zhong, X.; Guo, J.; Zhao, N.; Xu, C.; Zhang, H.; Pang, J.; Han, F.; et al. Global transgenerational gene expression dynamics in two newly synthesized allohexaploid wheat (Triticum aestivum) lines. BMC Biol. 2012, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; Chen, Z.J. Genomic and expression plasticity of polyploidy. Curr. Opin. Plant Biol. 2010, 13, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tian, L.; Lee, H.S.; Wei, N.E.; Jiang, H.; Watson, B.; Madlung, A.; Osborn, T.C.; Doerge, R.W.; Comai, L.; et al. Genomewide nonadditive gene regulation in Arabidopsis allotetraploids. Genetics 2006, 172, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Shaked, H.; Kashkush, K.; Ozkan, H.; Feldman, M.; Levy, A.A. Sequence elimination and cytosine methylation are rapid and reproducible responses of the genome to wide hybridization and allopolyploidy in wheat. Plant Cell 2001, 13, 1749–1759. [Google Scholar] [CrossRef] [PubMed]
- Riechmann, J.L.; Meyerowitz, E.M. The AP2/EREBP family of plant transcription factors. Biol. Chem. 1998, 379, 633–646. [Google Scholar] [PubMed]
- Nagano, Y. Several features of the GT-factor trihelix domain resemble those of the Myb DNA-binding domain. Plant Physiol. 2000, 124, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Wang, F.; Hou, X.L.; Wang, Z.; Huang, Z.N. Genome-wide fractionation and identification of WRKY transcription factors in Chinese cabbage (Brassica rapa ssp. pekinensis) reveals collinearity and their expression patterns under abiotic and biotic stresses. Plant Mol. Biol. Rep. 2014, 32, 781–795. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, S.; Zhu, X.; Liao, T.; Li, Y.; Yao, P.; Suo, Y.; Zhang, P.; Wang, J.; Kang, X. Gene Expression Differences between High-Growth Populus Allotriploids and Their Diploid Parents. Forests 2015, 6, 839-857. https://doi.org/10.3390/f6030839
Cheng S, Zhu X, Liao T, Li Y, Yao P, Suo Y, Zhang P, Wang J, Kang X. Gene Expression Differences between High-Growth Populus Allotriploids and Their Diploid Parents. Forests. 2015; 6(3):839-857. https://doi.org/10.3390/f6030839
Chicago/Turabian StyleCheng, Shiping, Xiaohu Zhu, Ting Liao, Yun Li, Pengqiang Yao, Yujing Suo, Pingdong Zhang, Jun Wang, and Xiangyang Kang. 2015. "Gene Expression Differences between High-Growth Populus Allotriploids and Their Diploid Parents" Forests 6, no. 3: 839-857. https://doi.org/10.3390/f6030839