Iteration Scheme for Solving the System of Coupled Integro-Differential Equations for Excited and Ionized States of Molecular Systems


Department of Physics, Rostov State Transport University, Narodnogo Opolcheniya Sq., Rostov-on-Don 344038, Russia
*
Author to whom correspondence should be addressed.
Algorithms 2018, 11(1), 1; https://doi.org/10.3390/a11010001
Submission received: 13 November 2017
/
Revised: 12 December 2017
/
Accepted: 19 December 2017
/
Published: 22 December 2017
Abstract
:Investigation of the interaction of electromagnetic radiation with molecular systems provides most of the information on their structure and properties. Interpretation of experimental data is directly determined by the knowledge of the structure of energy levels and its change in the transition of these systems to an excited state. A key task of the methods for calculating the molecular orbitals of excited states is to accurately describe the emerging vacancies of the molecular core, leading to radial relaxation of the electron density. We propose an iterative scheme for solving a system of coupled integro-differential equations for obtaining molecular orbitals of electron configurations with excited/ionized deep and subvalent shells in a single-center representation. The numerical procedure of the iterative scheme is reduced to solving a boundary value problem based on a combination of the three-point difference scheme of Numerov and Thomas algorithm. To increase the rate of convergence of the computational procedure, an accurate account is taken of the behavior of the electron density near the nuclei of the molecular system. The realization of the algorithm of the computational scheme is considered on the example of a diatomic hydrogen fluoride molecule. The energy characteristics of the ground and ionized states of the molecule are estimated, and also the spatial distribution of the electron density is presented for the example of the σ-symmetry shell.
1. Introduction
Theoretical study of the absorption processes [1], photoionization [2], elastic [3], and inelastic [4] scattering of an X-ray photon by a molecular system is a complex problem that requires an exact description of the excited states with emerging vacancies in deep and subvalent electronic shells. Existing methods for calculating wave functions can be classified according to the form of their representation as multicenter (MC) [5] and single-center (SC) [6].
In the MC representation, molecular orbitals (MO) in different regions of space are described with respect to the centers associated with the nuclei of the molecular system. In this approach, the main limitation is related to the choice of the basic set of wave functions affecting the accuracy and amount of calculation [7,8]. In the calculations, the construction of basic functions in the form of a superposition of Slater [9] or Gaussian [10] orbitals are widely used. The most well-known realization of the MC approach is the method of linear combination of atomic orbitals (MO LCAO) [11,12]. Its equations were first written by Roothaan [5]. In this case, the problem of minimizing the energy functional is reduced to finding the coefficients for the expansion of MO in basic atomic orbitals. A fairly complete overview on the use of the MO LCAO presented in [13], shows that the main drawback of the method is the principal difficulties in obtaining excited states of molecular systems caused by the problem of choosing the optimal basis set.
Another approach is to extend the methods for calculating the wave functions of atomic electrons to molecular systems that have been developed in the SC method of MO calculation [6,14,15]. The SC approach is based on the MO representation in the form of an expansion in the basis of functions centered relative on one point of space. The application of the SC method has a number of advantages, especially when studying electron-photon interaction processes in many-electron systems [15,16,17]. From the computational point of view, the simplicity of using SC in comparison with MC is due to the developed theory and methods of atomic calculations. The transition to molecular systems is determined by introducing summation over the orbital angular momenta, since one-electron atomic wave function is formally the SC component of the MO decomposition. This approach avoids the problems associated with the redundancy of the basis, or at least controls them, in contrast to the MC approach. In addition, the absence of multicenter integrals makes it possible to substantially simplify the procedure for performing calculations.
Historically, the first way to implement the SC method was to represent the partial harmonics of the SC expansion in the form of a linear combination of Slater-type orbitals and directly minimize the expressions for the total energy [6,11]. In a different approach, described in detail in [15,16], another variant of the SC method is proposed, which reduces to solving the secular equation. In this case, both the discrete function and the continuous spectrum function are used as basis functions, which are a solution of the Hartree-Fock equations. This realization of the SC approach is not free from the problems of choosing optimal basis sets, as well as from the slow convergence of the SC expansion series of the MO. Moreover, it has a strong influence on the energy of the state of the partial SC harmonics of the MO decomposition with large orbital moment values under low contribution of these harmonics to the function norm. The last two drawbacks can also be attributed to the SC method in general.
The application of the variational principle for obtaining the wave functions of electrons in the case of an atom leads to the well-known system of integro-differential Hartree-Fock equations. In molecular SC systems, the representation allows to obtain a system of integro-differential equations connecting different partial harmonics of MO. This approach is most accurate due to the elimination of the problem of selecting basic sets for describing the MO. In works [17,18,19,20,21,22], this method was realized when calculating the wave function of an excited photoelectron of a discrete and continuous spectrum on the basis of a system of coupled integro-differential equations. Herewith, despite the application of the widely known method of solving the differential equations of Numerov [23], the developed approach has a number of drawbacks related to the use of the approximation of the frozen core, and also the restriction of the SC expansion of the MO to the harmonics. It makes an essential contribution to the expansion of the wave function, due to its slow convergence. In particular, the accuracy of describing vacancies in the excited and ionized states restricts the application of the approximations of “Z + 1” and “frozen core”. In calculating the weakly excited states of molecular systems, the delta-SCF [24] method based on the DFT theory has been developed, requiring the search for a suitable exchange-correlation potential for an accurate description of the electron-electron interaction. At the same time, the application of the method is limited when solving a wide class of problems of elastic and inelastic X-ray photon scattering, as well as photoexcitation processes, which is due to the need to obtain highly-excited electronic states. At the same time, the slow convergence of the MO in the SC representation has a significant effect on the accuracy of the calculated values. According to the estimate made in [18], in order to describe the one-electron energies of MO molecules with ligands of the second period of the Mendeleyev periodic table with an accuracy of ~3%, one must take into account the partial harmonics with orbital angular momentum l ≥ 50 in the SC expansion of MO. The increase in the number of terms of the SC expansion leads to an increase in the system of differential equations, which causes more computational capacity giving an insignificant contribution of the obtained higher harmonics of the expansion to the observed quantities, but important for the energy characteristics of the systems [22]. In [17,25,26] the problem of slow convergence of the SC of MO expansion was solved by “effective” accounting of the influence of partial harmonics with large values of the orbital angular momentum, by increasing the participation coefficient of the last from the accounted partial harmonics in the limited SC expansion when calculating the photoelectron MO. At the same time, such an “effective” technique in calculating individual spectroscopic quantities does not allow to correctly describe the electron-electron Coulomb interaction, including the photoabsorption cross-section.
The paper proposes an iterative scheme for calculating the excited and ionized states of molecular systems obtained outside the framework of the widely used approximations of “Z + 1” and “frozen core”. The numerical procedure is based on solving a system of coupled integro-differential equations for all MOs included in the electronic configuration of the state. The higher harmonics of MO are represented as a composition of the wave functions of the electron shells of atoms expanded in a series of spherical harmonics. This ensures the required description accuracy of electron density near the nuclei of the molecular system. The proposed computational procedure is based on a combination of three-point difference Numerov scheme and Thomas algorithm. The theoretical basis and details of the iteration scheme are described. The efficiency of higher harmonics consideration in the MO decomposition is illustrated by the example of calculating the energy characteristics of the main electronic configuration of a hydrogen fluoride molecule with an increase in the ligand charge. The space expansion of electron density is also presented. The energy characteristics of the ionized states of the σ-symmetry molecule are estimated.
2. Materials and Methods
2.1. Basic Equation of Iteration Scheme
Integro-differential equations underlie the iterative scheme of obtaining radial parts of MO. They are the generalization of Hatree-Fock equation first recorded for an atom [27]. The equations were obtained on the basis of minimizing the energy functional in the Born-Oppenheimer approximation [28] for the wave function on N-electron molecular system represented as antisymmetrized product of one-electron MO . After the variation procedure we obtain a set of coupled integro-differential Hartree-Fock equations systems each of which corresponds to a certain MO of molecule electron configuration. In this case, the single-electron MO , in the nonrelativistic SC representation, has the form [6]:
where is the radial and is the angular part of , the symmetry partial harmonic of MO with the fixed (for a linear molecule) value of the , the projections of angular momentum to the quantization axis OZ (molecule axis); is the main quantum number; is the symmetry of MO; and are the spherical coordinates.
Considering Equation (1) the system of coupled integro-differential equations for MO has the form:
where is the local potential, describing the electron interaction of MO with the nuclei of the molecular system and the direct Coulomb interaction with the electrons of other MO; is the nonlocal potential, describing the exchange Coulomb interaction of MO electrons with electrons of other MO.
Local potential has the following form:
where is the charge of molecular nucleus, located at the chosen origin of coordinates, is the potential of crystal field, describing the electron interaction of MO with the nuclei of molecular system; is the potential of the direct Coulomb interaction of MO electrons with the electrons of other MO; and is the single-electron energy of MO.
The expression for the potential of crystal field in Equation (3) has the form:
where is the number of electrons of MO with , the projections of angular momentum to the quantization axis OZ (molecule axis); is the number of ligands in the molecule; is the charge of the α-th ligand; is the spherical harmonic; и are the coordinates of the α-th ligand; and is the matrix element of the spherical harmonic, the calculation methods of which are presented in [28].
The potential, describing the direct Coulomb interaction of electrons with the electrons from other MO (Equation (3)) has the form:
where is the coefficient for the potential describing the direct Coulomb interaction between and MO depending on the quantum number of electron configuration of a molecule; ; and .
Nonlocal potential describing the interchange Coulomb interaction of electrons of MO with the electrons of other MO in Equation (2) has the form:
where is the coefficient for the potential describing interchange Coulomb interaction between and of MO depending on the quantum number of electron configuration of a molecule; ; and .
The radial part of the electronic potential is determined in the expressions of the direct and interchange potentials. It is calculated on the basis of radial parts of partial harmonics of SC expansion of MO:
In order to simplify Equations (6)–(9), the summing is omitted over the μ-projection of angular momentum of the electrons participating in Coulomb interaction.
2.2. The Inclusion of Higher Spherical Harmonics in SC Expansion of MO
To accurately describe the electron density in the vicinity of the molecular system nuclei and solve the problem of slow convergence of the functional series (1), MO for calculating the system of coupled differential equations has the following form:
where is the normalization factor; is the contribution coefficients of the wave function with the main quantum number and symmetry of the -th ligand of the molecular system in the form:
The functions are the wave functions of the electron shells of atoms entering the molecular system as ligands (atoms are not at the origin) expanded in a series of spherical harmonics.
In this case, the partial harmonics of MO in Equation (11) with the values of the orbital angular momentum are obtained by solving the system of Equation (2). The terms of the expansion Equation (11) with orbital angular momenta are obtained on the basis of the linear combination with the contribution coefficients . Such a representation of MO is due to the fact that the shape of the partial spherical harmonics with large values of the orbital angular momentum is determined to within a coefficient by the ligand potential of the molecular system.
The choice of the value of in Equation (11) is a result of a numerical experiment and depends on the nature of the molecular system and the number of partial harmonics involved in the formation of the chemical bond of the compound.
To determine the contribution coefficients in Equation (11) the partial harmonics are linked in using the equation:
That is, radial part of the -th partial harmonic of the molecular orbital is represented as a superposition of radial partial harmonics of ligand functions with weighting coefficients .
Equation (13) allows compiling a system of algebraic equations for determining the contribution coefficients of ligand functions with symmetry in the form of:
Thus, the determined MO is normalized to unity.
2.3. The Expansion of the Wave Functions of the Electron Shells of an Atom in a Series of Spherical Harmonics
The procedure of obtaining the ligand functions used in Section 2.2 is based on the recalculation of the wave functions of the electron shells of atoms entering the molecular system in a new unified system of coordinates.
In the transition to a uniform coordinate system of the wave function of an electron shell related to a certain atom is represented in the form of an expansion in a series of spherical harmonics [29]:
where is the wave function of electron shell in the atomic system of coordinates; are the spherical coordinates of the atomic system; is the coefficient of expansion in spherical harmonics in the molecular spherical coordinates system; is the orbital angular momentum of expansion of the wave function in the molecular coordinates system; is the projection of the orbital angular momentum in the molecular coordinates system; and are the spherical coordinates in the molecular system.
The coefficient of expansion of the series in Equation (15) can be represented in the form [29]:
where are the origin coordinates of the molecular system in the atomic system, are the coefficients of the theta-function representation in the trigonometric form.
Integration in Equation (16) is performed on the basis of Equation (17) and also relations in Equations (18) and (19), which connect the coordinates in different systems. It enables the expansion of the wave function of the electron shells of atoms with respect to the chosen molecular center.
3. Solution Procedure
3.1. Scheme for Numerical Solution for the System of Equations for MO
The numerical procedure of MO finding is reduced to solving a system of coupled differential Equation (2) for the radial parts of partial harmonics of Equation (1).
The Equation (2) in general can be represented in a matrix form:
where are two-dimensional and one-dimensional matrices respectively, each element of which is a real function specified in the coordinate grid; is a one-dimensional matrix of the MO partial harmonics.
Three-point difference Numerov scheme is applied to solve the equation [30,31]:
where ; , is the identity matrix; and is the number of point at the coordinate grid.
The exponential decay of the partial harmonics of MO as () allows to reduce the Cauchy problem for a second order differential equation to the boundary-value problem, using the Thomas algorithm [32] for the solution. This algorithm connects the values of a function at neighboring points in the following way:
Substituting Equation (17) into Equation (16) enables obtaining the following recurrence relations:
Finding a solution is carried out by independent calculation of coefficients sweeping the values in the range of , «on the left» from the selected crosslinking point Z and «on the right» by reversing the values . Thus, an independent set of coefficient values can be obtained «on the left» and «on the right» from the crosslinking point Z of the solutions.
Boundary conditions are used in order to obtain take-off ratios of recurrence relationships (Equation (24)). The conditions are imposed on the radial parts of partial harmonics of MO and are determined by their exponential attenuation if (). Application of such conditions to Equation (24) results in matrix equations
To obtain the starting coefficients of the recurrence relations (Equation (24)), the boundary conditions are applied on the radial parts of the MO partial harmonics, which are due to their exponential attenuation as r → 0 (r → ∞). Applying these conditions to (24) leads to matrix equations in the form:
The iterative solution of Equations (25) and (26) allow restoring the coefficient values , and at all points of the coordinate grid.
The solution of Equation (21) must satisfy the smoothness requirement corresponding to the equality of the first-order derivatives of a function obtained by sweeping the values “on the left” and “on the right” at the crosslinking point :
This condition leads to a system of linear inhomogeneous equations with respect to the value of the partial harmonic at the crosslinking point :
The algorithm for implementing the numerical procedure of solving the system of Equation (21) consists in determining the eigenvalues and corresponding MO of the -type symmetry.
The solution of the homogeneous system of equations obtained from Equation (21) by zeroing out the nonlocal summand is performed at the stage of initialization of the iterative procedure using the initial approximation (see Section 3.2) for MO. This is, in fact, the solution of the Hartree equation, which does not take into account the exchange terms. When the numerical procedure is implemented, the homogeneous Equation (28) is solved, which right-hand side is equal to zero. Moreover, the existence of non-zero solutions is determined by the criterion:
The selection of solutions on the energy scale until reaching the corresponding MO nγ. For example, the symmetry states for diatomic molecules can be selected by the principal quantum number. In this case, the search for highly excited states of the discrete spectrum involves the sequential determination of all lower one-electron energies. The implemented procedure for finding solutions accounts the obtained spectrum of state energies and uses them as “reference points” on the one-electron energy scale of MO within the range of at subsequent iterations of the computational procedure. This approach ensures time saving for calculations and increases the efficiency of the algorithm for finding solutions of Equation (21).
The final set of radial parts of the partial harmonics c is found for MO when solving Equation (21). Following the procedure given in Section 2.2, the partial harmonics with orbital angular momenta are determined by the superposition of ligand functions. The participation coefficients of the ligand wave functions are determined by the solution of the system of algebraic Equation (14). Here, the functions are preformed according to the numerical procedure given in Section 2.3 on the basis of the wave functions of electron shells of atoms, obtained by solving the Hartree-Fock equations.
Equation (21) has a unique solution for any value of one-electron energy when accounting nonlocal terms . Therefore, the selection of solutions is based on the overlap integral, which is obtained according to the expression:
The criterion for selecting solutions for MO at the intermediate iteration step of the computational procedure is the correspondence of the overlap integral of the wave function (Equation (30)) to the value obtained at the first iteration of the matching process.
3.2. Starting Wave Function MO and Stopping Criteria
Initial approximation for the radial parts of MO partial harmonics of molecule electronic configuration is formed on the basis of the procedure described in Section 2.2. The wave functions of the electron shells of the atom located in the origin of the molecular coordinate system are used for the radial parts of MO partial harmonics with . Ligand functions are calculated for the formation of MO partial harmonics with using the procedure described in Section 2.3.
The calculation is based of preliminary obtained wave functions for the electron shells of neutral atoms in the Hartree-Fock approximation.
The iterative scheme for calculating the MO includes two basic computational cycles. The cycle of recalculation of MO wave functions is being performed till the defined accuracy of the found solution of on the i-th step is reached in comparison with the i-1-th of the process. Here, an accuracy estimation of the matching process starting is carried out on each step starting from the second one according to the formula:
The accuracy of determining is used for recalculating the wave function of MO in the new potential when solving Equation (28).
3.3. Summary of the Iterative Scheme
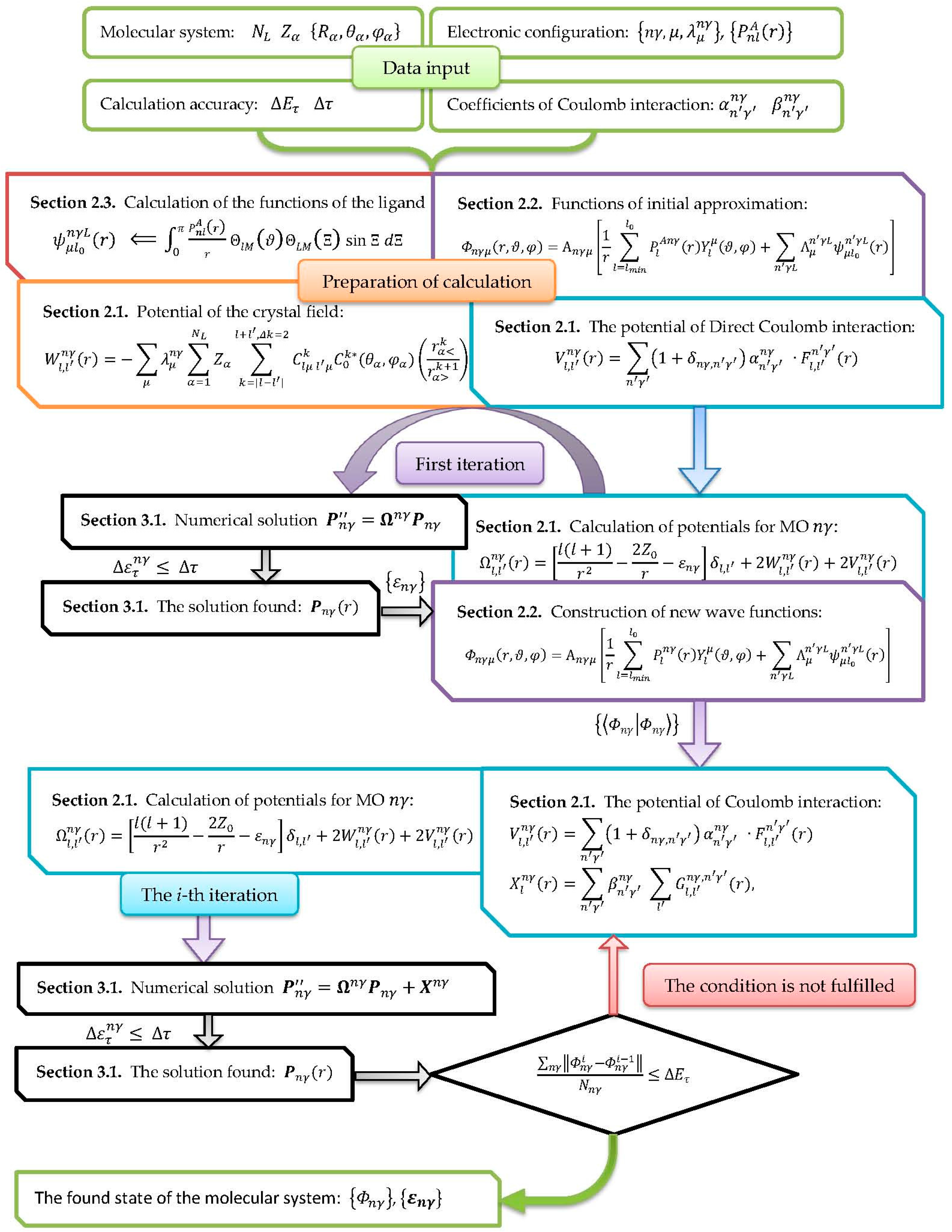
A general iterative scheme for implementing a numerical procedure of calculating the excited and ionized states of molecular systems is represented in Scheme 1.
The construction of the MO wave functions is performed on the preparatory stage according to the Section 2.2. In particular, the ligands functions are formed on the basis of the wave functions of the electron shells of atoms, expanded into series of spherical harmonics (see Section 2.3). The local potential is calculated from Equation (3) for the orbital angular momenta , which includes the potential of crystal field , and direct Coulomb interaction (see Section 2.1).
At the first iteration, a homogeneous system of Equation (21) is solved for every MO with = 0. One-electron energies are searched for in accordance with condition (29) within the interval . There is a direct correspondence between the solution number and the principal quantum number of MO with the chosen symmetry type. The found one-electron energy provides the radial parts of MO with the use of Equation (28). Wave functions of MO with higher harmonics are formed via Equation (11) on the basis of the partial harmonics MO with (see Section 2.2).
Participation coefficient of ligand function is determined by the solution of the system of Equation (14). The MO is normalized to unity for determining in Equation (11). The resulting normalization integrals are used as selection criteria of the solution on the subsequent steps of the iteration scheme. The found single-electron energies allow to determine intervals on the energy scale for searching the energy values of the new iteration, thereby increasing the efficiency of the computational scheme.
Hereafter, an iterative procedure is realized aiming to minimize the energy of molecular system. The criterion of achieving a positive result is a decrease in the deviation of MO wave function calculated in accordance with Equation (31) below a specified value. The cycle of the numerical procedure on the i-th iteration involves recalculation of the electronic potentials and using the MO wave functions obtained on the i-1-st iteration step. The solution of Equation (21) is searched. On the first step, the value is set in accordance with the criterion of Equation (29). The next step is searching the value of in the energy range for which the overlap integral of the given solution coincides with the normalization integral determined at the first iteration of the process.
4. Results and Discussions
The program code realizing the above iterative scheme, as well as the results of calculating the ground and excited states are available at the electronic address [33].
The hydrogen fluoride (HF) molecule was chosen for testing the numerical procedure due to the following circumstances:
- A relatively small number of molecule shells allows the exploration of the possibility of the developed iterative scheme and the procedure for taking into account the higher harmonics in the SC expansion of MO when they are implemented on the ECM with limited CPU time;
- The spectrum of free states and the structure of the wave functions have been thoroughly investigated [34].
In the calculations, the reference system associated with the F atom is chosen. To systemize the one-electron states, the quantum numbers characteristic for molecules of the point symmetry group are used. The MO calculations for the HF molecule are performed for the equilibrium distance between the nuclei of hydrogen atoms and fluorine = 1.733 a.u. [35].
Comparative calculations of the energy characteristics (one-electron MO energies and total energy) of the ground state of the HF molecule are given in Table 1.
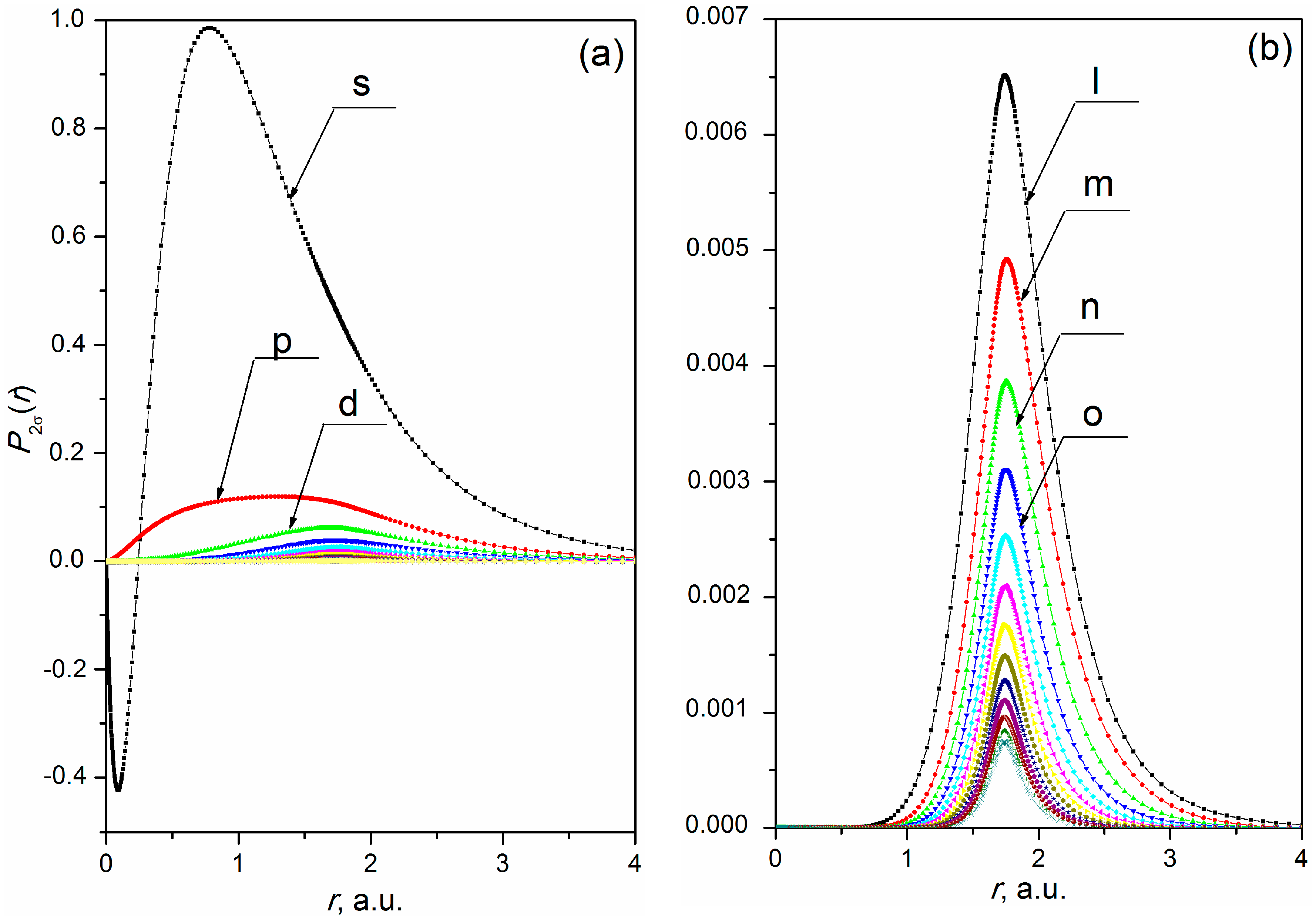
In section (а) of Table 1, the calculation was performed via the MC method of MO LCAO using the General Atomic and Molecular Electronic Structure System [12]. The calculation results in section (b) of Table 1 were obtained in this work using the MO expanded into a series (1) and limited . They show sufficient accuracy which correlate with the calculation methods. The use of higher harmonics of the ligand wave functions for an accurate description of MO in the molecular nuclei region (see Section 2.2) gives an additional contribution to the total energy and regularly changes the energy level of the 1σ-state of the HF molecule. The changes in the energy quantities observed in section (c) of Table 1 are primarily due to the contribution of the 1s state of the H atom, which is a ligand in this case. Construction of MO is determined by the selection of which includes the ligand functions in the MO structure. Figure 1 and Figure 2 illustrate the procedure of determining with respect to the calculation (section (c), Table 1). Figure 1 displays SC expansion of MO 2σ of the ground state of HF molecule (calculation is performed for the SC series (1) with ≤ 60). Figure 2 displays the SC expansion of the wave function of H atom 1s state (the solution of the Hratree-Fock equations), decomposed in the molecular coordinate system (see Section 2.3).
The example of MO 2σ shows (see Figure 1b) that the form of radial parts of the partial harmonics of SC expansion of MO starting with 8 ( is the partial harmonic denoted in the figure) is in good agreement with the form of radial parts of the partial harmonics of SC expansion of the wave function of H atom 1s state (see Figure 2b). It can be concluded that the influence of the ligand field with the charge = 1 on radial parts of the partial harmonics of SC expansion of 2σ–MO with orbital moments 8 is decisive. Thus, in accordance with the procedure proposed above (see Section 2.2) the crosslinking of MO (1) with ligand functions is done starting from 8.
A significant influence of the higher harmonics of the SC expansion on the energy characteristics of the molecular system is observed with the growth of the ligand charge. To estimate this effect, the calculation of the total energy of the electron configuration is performed depending on the charge which varied within the range of = 2 ÷ 5 (instead of case = 1). The results obtained for the total energies are given in Table 2.
The values of the total energies on the basis of MO with SC expansion limited to are given in column (a) of Table 2. The calculation results accounting the ligand functions with expansion up to are given in the column (b) of Table 2.
It is obvious that higher harmonics of the SC expansion of MO begin to significantly affect the energy characteristics of the molecular system. The calculation results of the MO electron state show that this iterative scheme solves the main problem of the SC methods. That is the description of MO behavior in the area of ligand nuclei.
It is also evident that with increasing the ligand charge, the contribution of the higher harmonics of MO, taken into account by this SC approach, increases substantially. Thus, the represented iterative scheme of the SC calculation of MO becomes effective when studying molecules with nonhydrogen ligands. The results of the research show that taking into account the higher harmonics of the expansion of the wave functions of the ligands electron shells is a necessary condition in performing calculations in the SC representation for obtaining accurate energy characteristics of molecular systems.
The possibilities of obtaining the excited states represented by the iterative scheme are considered on the example of ionization of symmetry shells of HF molecule ground state.
Table 3 displays the results of accounting the ligand functions with MO expansion up to . Energy characteristics of the electron configurations: ; are given as the results.
Obtaining the ionized states in the «Z + 1» approximation of MC approach requires a special charge increase of the chosen core of the molecular system, which corresponds to the creation of a vacancy. Such an artificial technique is used in connection with the absence of the possibility of directional ionization of the electron shell. Unified approach to the calculation of any states is in the proposed iterative scheme. It ensures the possibility of ionization of any electronic shell.
5. Conclusions
The iterative scheme proposed in the paper is intended for calculating the wave functions of excited and ionized states of molecular systems. The procedures is based on a numerical solution of a system of coupled integro-differential equations for the radial parts of MO partial harmonics that determine the formation of the chemical bond of the many-electron compound and take into account the higher harmonics of the SC series. The harmonics describe the electron density strongly connected with the molecular system cores. The realized calculation scheme lacks the drawbacks of the existing SC calculation methods associated with the slow convergence of the expansion. The problem of calculating the excited states was overcome. It was connected with the use of “artificial methods” of creating vacancies in electron shells (approximation «Z + 1»). The proposed computation scheme enables performing calculations for the excited states with any number of unfilled electron shells. The calculations carried out for the HF molecule showed good agreement with the results of existing MC approbated software systems.
Acknowledgments
This work is supported by the Grant of the President of Russian Federation No. 14.W01.17.2210-MK of 22–02–2017.
Author Contributions
Anton Kasprzhitskii is responsible for the development, verification, and testing of the numerical procedure of searching one-electron energies and for the calculation of radial parts of partial harmonics of MO of the system of coupled integro-differential equations. Georgy Lazorenko is responsible for the development, testing, and verification of calculation procedures of local and nonlocal electron potentials. He is also responsible for optimizing the procedure of solving the matrix equations, for the procedure of accounting the higher harmonics in MO expansion, and for the implementation of numerical procedures of expansion of wave functions of atom electron shells into the series of spherical harmonics in the new axis of reference. The initial idea of accounting higher harmonics when describing the electron density strongly connected with molecular system nuclei belongs to Victor Yavna. All the authors contributed to the publication of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dutoi, A.D.; Leone, S.R. Simulation of X-ray transient absorption for following vibrations in coherently ionized F2 molecules. Chem. Phys. 2017, 482, 249–264. [Google Scholar] [CrossRef]
- Granados-Castro, C.M.; Ancarani, L.U.; Gasaneo, G.; Mitnik, D.M. A Sturmian Approach to Photoionization of Molecules. In Advances in Quantum Chemistry, 1st ed.; Hoggan, P.E., Ozdogan, T., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 73, pp. 3–57. ISBN 978-0-12-803060-8. [Google Scholar]
- Yavna, V.; Hopersky, A.; Nadolinsky, A.; Yavna, S. Orientation effects in elastic scattering of polarized X-rays by linear molecules. J. Synchrotron Radiat. 2001, 8, 240–242. [Google Scholar] [CrossRef] [PubMed]
- Gel’mukhanov, F.; Gren, H. Resonant inelastic x-ray scattering with symmetry-selective excitation. Phys. Rev. A 1994, 49, 4378–4389. [Google Scholar] [CrossRef] [PubMed]
- Roothaan, C.C.J. Self–Consistent Field Theory for Open Shell of Electronic Systems. Rev. Mod. Phys. 1960, 32, 179–185. [Google Scholar] [CrossRef]
- Bishop, D.M. Single-Center Molecular Wave Function. In Advances in Quantum Chemistry, 1st ed.; Löwdin, P.-O., Ed.; Elsevier: Amsterdam, The Netherlands, 1967; Volume 3, pp. 25–59. ISBN 978-0-12-034803-9. [Google Scholar]
- Pople, J.A. Molecular orbital methods in organic chemistry. Acc. Chem. Res. 1970, 3, 217–223. [Google Scholar] [CrossRef]
- Wilson, S. Electron Correlation in Molecules, 2nd ed.; Dover Publications: Mineola, NY, USA, 2007; pp. 1–304. ISBN 978-0-48-645879-3. [Google Scholar]
- Bunge, C.F.; Barrientos, J.A.; Bunge, A.V.; Cogordan, J.A. Hartree-Fock and Roothaan-Hartree-Fock energies for the ground states of He through Xe. Phys. Rev. A 1992, 46, 3691–3696. [Google Scholar] [CrossRef] [PubMed]
- Huzinaga, S. Gaussian–Type Functions for the Polyatomic Systems. J. Chem. Phys. 1965, 42, 1293–1302. [Google Scholar] [CrossRef]
- Roothaan, C.C.J. New Development in Molecular Orbital Theory. Rev. Mod. Phys. 1951, 23, 69–89. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Lowe, J.P. Quantum Chemistry, 1st ed.; Academic Press: Cambridge, MA, USA, 1978; pp. 1–599. ISBN 978-0-12-457550-9. [Google Scholar]
- Faisal, F.H.M. Electron-Molecular Interactions: I-Single Center Wave-Functions and Potentials. J. Phys. B 1970, 3, 636–640. [Google Scholar] [CrossRef]
- Yavna, V.A.; Nadolinsky, A.M.; Hopersky, A.N. Theoretical study of processes of multiple excitation/ionization in 2s-photoabsorption of the CO molecule. J. Electron Spectrosc. Relat. Phenom. 1998, 94, 49–57. [Google Scholar] [CrossRef]
- Yavna, V.A.; Nadolinsky, A.M.; Demekhin, V.F. Theoretical study of inner shell photoabsorption spectra of simple molecules. J. Electron Spectrosc. Relat. Phenom. 1994, 68, 267–275. [Google Scholar] [CrossRef]
- Demekhin, P.V.; Omel’yanenko, D.V.; Lagutin, B.M.; Sukhorukov, V.L.; Werner, L.; Ehresmann, A.; Schartner, K.-H.; Schmoranzer, H. Investigation of photoionization and photodissociation of an oxygen molecule by the method of coupled differential equations. Opt. Spectrosc. 2007, 103, 318–329. [Google Scholar] [CrossRef]
- Lagutin, B.M.; Migal, Y.F. Single-center method of calculation for clusters and molecules other than hydrides. Theor. Exp. Chem. 1987, 25, 10–17. [Google Scholar] [CrossRef]
- Demekhin, P.V.; Ehresmann, A.; Sukhorukov, V.L. Single center method: A computational tool for ionization and electronic excitation studies of molecules. J. Chem. Phys. 2011, 134, 024113. [Google Scholar] [CrossRef] [PubMed]
- Demekhin, P.V.; Lagutin, B.M.; Petrov, I.D. Theoretical study of angular-resolved two-photon ionization of H2. Phys. Rev. A 2012, 85, 023416. [Google Scholar] [CrossRef]
- Inhester, L.; Burmeister, C.F.; Groenhof, G.; Grubmüller, H. Auger spectrum of a water molecule after single and double core ionization. J. Chem. Phys. 2012, 136, 144304. [Google Scholar] [CrossRef] [PubMed]
- Galitskiy, S.A.; Artemyev, A.N.; Jänkälä, K.; Lagutin, B.M.; Demekhin, P.V. Hartree-Fock calculation of the differential photoionization cross sections of small Li clusters. J. Chem. Phys. 2015, 142, 034306. [Google Scholar] [CrossRef] [PubMed]
- Stern, M.S. Comparison of numerical solutions of the partial-wave Schrödinger differential and integral equations. J. Comput. Phys. 1977, 25, 56–70. [Google Scholar] [CrossRef]
- Gavnholt, J.; Olsen, T.; Engelund, M.; Schiøtz, J. Δ self-consistent field method to obtain potential energy surfaces of excited molecules on surfaces. Phys. Rev. B 2008, 78, 075441. [Google Scholar] [CrossRef]
- Yavna, V.A.; Nadolinsky, A.M.; Demekhin, V.P. Theoretical study of photoabsorption cross sections in the regions of CK- and NK-ionization thresholds of CO and N2 molecules. Opt. Spectrosc. 1990, 69, 1278–1284. [Google Scholar]
- Yavna, V.A.; Nadolinsky, A.M.; Demekhin, V.P. Theoretical study of XANES 2σ-shells CO. Opt. Spectrosc. 1992, 73, 1081–1085. [Google Scholar]
- Cacelli, I. The Hartree–Fock Method. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, 1st ed.; Reedijk, J., Ed.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 1–14. ISBN 978-0-124-09547-2. [Google Scholar]
- Sobel’Man, I.I. Introduction to the Theory of Atomic Spectra, 1st ed.; Pergamon Press: Oxford, UK, 1972; pp. 1–626. ISBN 978-1-483-15972-0. [Google Scholar]
- Varshalovich, D.A.; Moskalev, A.N.; Khersonskii, V.K. Quantum Theory of Angular Momentum, 1st ed.; World Scientific: Singapore, 1988; pp. 1–526. ISBN 978-9-971-50107-5. [Google Scholar]
- Hairer, E.; Nørsett, S.P.; Wanner, G. Solving Ordinary Differential Equations I: Nonstiff Problems, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 1993; pp. 1–528. ISBN 978-3-540-56670-0. [Google Scholar]
- Numerov, B. Note on the numerical integration of d2x/dt2 = f(x, t). Astron. Nachr. 1927, 230, 359–364. [Google Scholar] [CrossRef]
- Ahuja, P. Tridiagonal matrix algorithm (TDMA). In Introduction to Numerical Methods in Chemical Engineering, 1st ed.; Ahuja, P., Ed.; PHI Learning: Delhi, India, 2010; pp. 1–304. ISBN 978-8-120-34018-3. [Google Scholar]
- The Program Code for Calculation the Wave Function of a Molecular Systems. Available online: http://www.rgups.ru/site/assets/files/39146/software_code.zip (accessed on 20 November 2017).
- Yavna, V.A.; Popov, V.A.; Yavna, S.A. Correlation and vibronic effects in K-photoabsorption of HF and HCl. Opt. Spectrosc. 1993, 75, 39–46. [Google Scholar]
- Kane, P.P. Inelastic scattering of X-rays and gamma rays by inner shell electrons. Phys. Rep. 1992, 218, 67–139. [Google Scholar] [CrossRef]
Scheme 1.
Iteration scheme of wave functions of MO of molecular systems electron configurations.

Figure 1.
Radial parts of partial harmonics of SC expansion of 2σ–MO of the ground HF molecule state. Calculation of the ground state is performed for the equilibrium distance ( 1.733 a.u. [35]) between the atom nuclei of H and F: (a) SC expansion of MO into a series (1) c 60; and (b) SC expansion of MO into a series (1) c 8.
Figure 1.
Radial parts of partial harmonics of SC expansion of 2σ–MO of the ground HF molecule state. Calculation of the ground state is performed for the equilibrium distance ( 1.733 a.u. [35]) between the atom nuclei of H and F: (a) SC expansion of MO into a series (1) c 60; and (b) SC expansion of MO into a series (1) c 8.
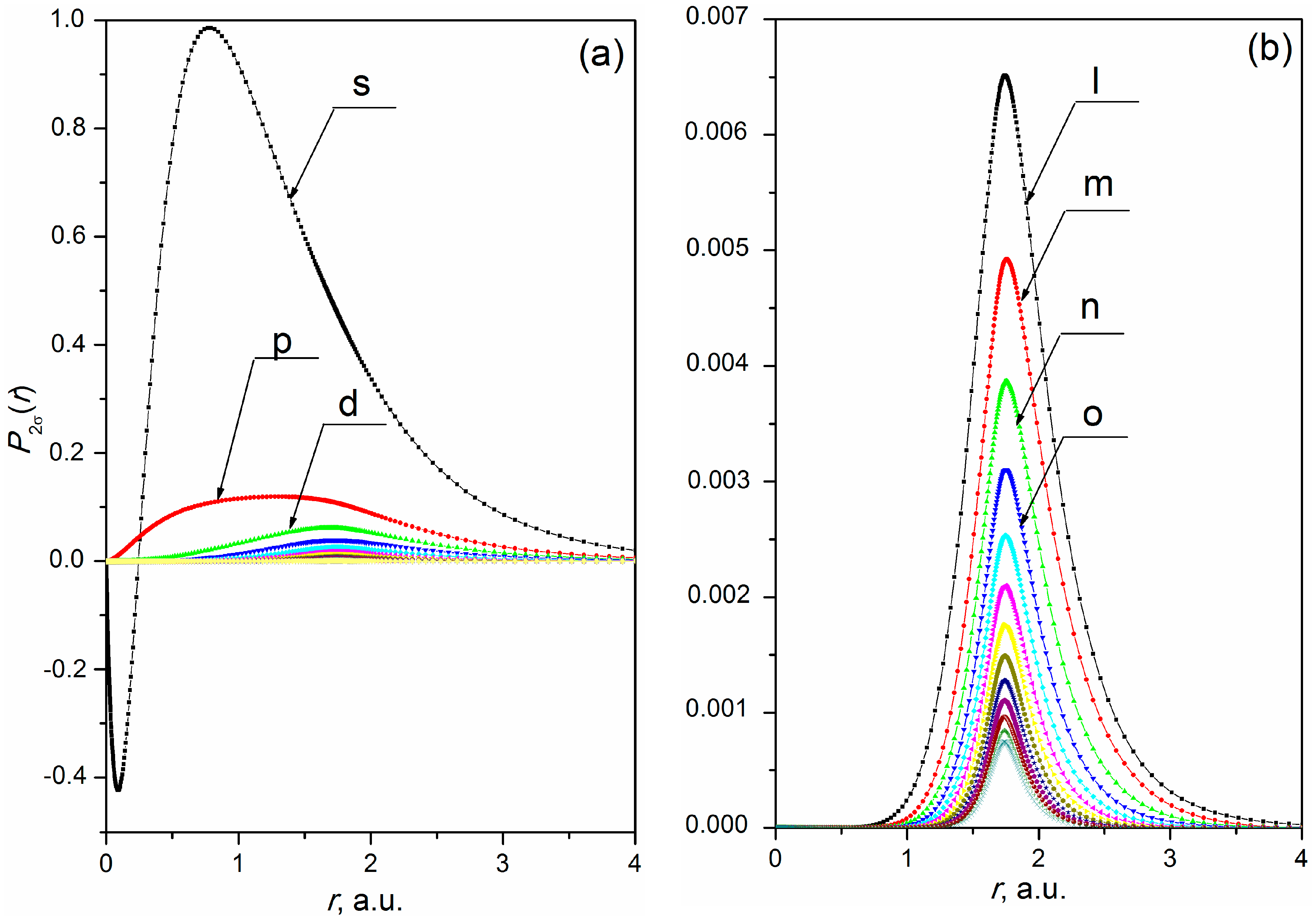
Figure 2.
Radial parts of partial harmonics of SC series of H atom 1s wave function found via a solution of Hatree-Fock equations in the molecular axis of reference: (a) SC expansion into series (1) with 60; and (b) SC expansion into series (1) with 8.
Figure 2.
Radial parts of partial harmonics of SC series of H atom 1s wave function found via a solution of Hatree-Fock equations in the molecular axis of reference: (a) SC expansion into series (1) with 60; and (b) SC expansion into series (1) with 8.


{kind=link}
{kind=link}
{kind=link}
Table 1.
One-electron MO energy and the total energy of the HF molecule (in atomic units).
| Type | 1σ | 2σ | 3σ | 1π | Total Energy |
|---|---|---|---|---|---|
| a | −26.2923 | −1.5990 | −0.7674 | −0.6464 | −100.0577 |
| b | −26.2905 | −1.5995 | −0.7656 | −0.6492 | −100.0594 |
| c | −26.2915 | −1.5995 | −0.7656 | −0.6492 | −100.0616 |
Table 2.
Total energy of the electron state is (in atomic units).
| Total Energy (a) | Total Energy (b) | |
|---|---|---|
| 2 | −101.0902 | −101.0951 |
| 3 | −102.8639 | −102.9189 |
| 4 | −105.1593 | −105.4553 |
| 5 | −107.7773 | −108.5403 |
Table 3.
One-electron energy MO and the total energy of the ionized states of the HF molecule and (in atomic units).
Table 3.
One-electron energy MO and the total energy of the ionized states of the HF molecule and (in atomic units).
| Ionized Shell | 1σ | 2σ | 3σ | 1π | Total Energy |
|---|---|---|---|---|---|
| −30.1430 | −2.3366 | −1.5016 | −1.4186 | −74.6050 | |
| −26.9639 | −2.3036 | −1.3277 | −1.2266 | −98.5775 | |
| −26.9128 | −2.1432 | −1.4587 | −1.2196 | −99.3889 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kasprzhitskii, A.; Lazorenko, G.; Yavna, V. Iteration Scheme for Solving the System of Coupled Integro-Differential Equations for Excited and Ionized States of Molecular Systems. Algorithms 2018, 11, 1. https://doi.org/10.3390/a11010001
AMA Style
Kasprzhitskii A, Lazorenko G, Yavna V. Iteration Scheme for Solving the System of Coupled Integro-Differential Equations for Excited and Ionized States of Molecular Systems. Algorithms. 2018; 11(1):1. https://doi.org/10.3390/a11010001
Chicago/Turabian StyleKasprzhitskii, Anton, Georgy Lazorenko, and Victor Yavna. 2018. "Iteration Scheme for Solving the System of Coupled Integro-Differential Equations for Excited and Ionized States of Molecular Systems" Algorithms 11, no. 1: 1. https://doi.org/10.3390/a11010001
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.