7.2. All Models
Encoding of the case study in
Section 6,
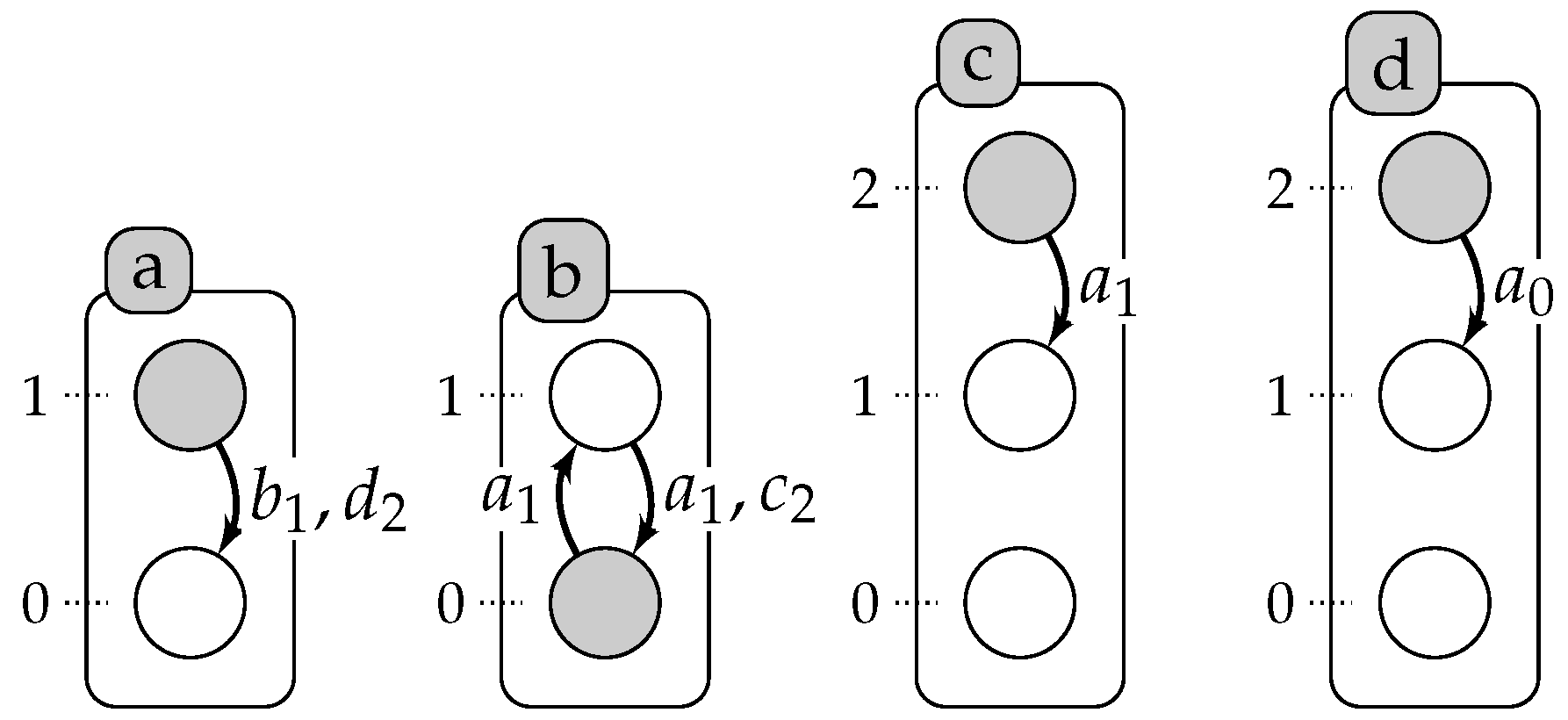
Figure 3 on page 13: We encode the observations as a set of predicates
obs(X,Val,T), where
X is an automaton identifier,
Val a level of this automaton and
T is a time point, such that the automaton
X has the value
Val starting from
T.
1 % All network components with its levels after discretization
2 automatonLevel("a",0..1). automatonLevel("b",0..1). automatonLevel("c",0..1).
3 % Time series data or the observation of the components
4 obs("a",0,0). obs("a",0,1). obs("a",0,2). obs("a",0,3). obs("a",1,3). obs("a",1,4). obs("a",1,4).
5 obs("a",1,5). obs("a",0,5). obs("a",0,6). obs("a",0,7). obs("b",1,0). obs("b",1,1). obs("b",1,2).
6 obs("b",1,3). obs("b",1,4). obs("b",0,4). obs("b",0,5). obs("b",0,5). obs("b",0,6). obs("b",0,7).
7 obs("c",0,0). obs("c",0,1). obs("c",0,2). obs("c",1,2). obs("c",1,3). obs("c",1,4). obs("c",1,5).
8 obs("c",1,6). obs("c",0,6). obs("c",0,7).
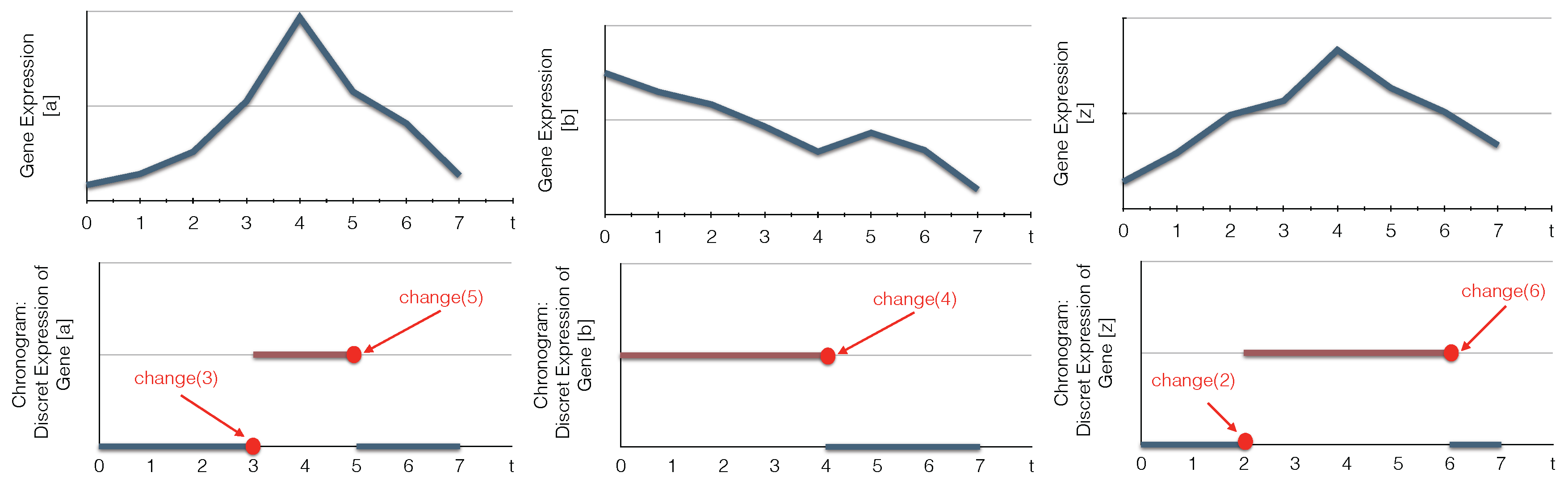
We define in the predicate changeState(X,Val1, Val2, T), the time point T where X changes its level from Val1 to Val2. We admit that at t=0, all components change (lines 11–12). Then, to reduce the complexity of the program, we consider in the predicate time only the time points where the components change their levels (line 18). Similar for the delays, D is a delay when it is equal to the difference between two time steps where some components change their levels (line 19). To compute the chronogram of a component a, we compute the time points where it is constant (i.e., has only one level) between two successive changes of a: T1 and T2 (lines 21–22).
9 % Changes identification
10 % initialization of all changes for each automaton at t=0 (assumption)
11 changeState(X,Val,Val,0) ← obs(X,Val,0).
12 changeState(X,0) ← obs(X,_,0).
13 % Compute all changes of each component according to the observations (chronogram)
14 changeState(X,Val1, Val2, T) ← obs(X, Val1, T), obs(X,Val2,T),obs(X, Val1, T-1), obs(X, Val2, T+1),
15 Val1!=Val2.
16 changeState(X,T) ← changeState(X,_,_,T).
17 % Find all time points where changes occure (reduce complexity)
18 time(T) ← changeState(_,T).
19 delay(D) ← time(T1), time(T2), D=T2-T1, T2>=T1.
20 % Observations processing
21 obs_normalized(X,Val,T1,T2) ← obs(X,Val,T1), obs(X,Val,T2), T1<T2, not existChange(X,Val,T1,T2),
22 time(T1), time(T2).
23 % Verify if X changes its level between two time points T1 and T2
24 existChange(X,Val,T1,T2) ← obs(X,Val,T1), obs(X,Val,T2), obs(X,Val1,T), T>T1, T<T2, Val!=Val1.
25 existChange(X,Val,T1,T2) ← obs(X,Val,T1), obs(X,Val,T2), changeState(X,T), T>T1, T<T2.
26 existChange(X,Val,T1,T2) ← obs(X,Val,T1), obs(X,Val,T2), T1<T2, changeState(X,Val,Val_,T1), Val!=Val_.
We detailed in
Section 3 how to compute the delays. Therefore, the delay is the difference between the time where the change happens (
changeState) and the last change of the components involved in the local transition: the influenced one,
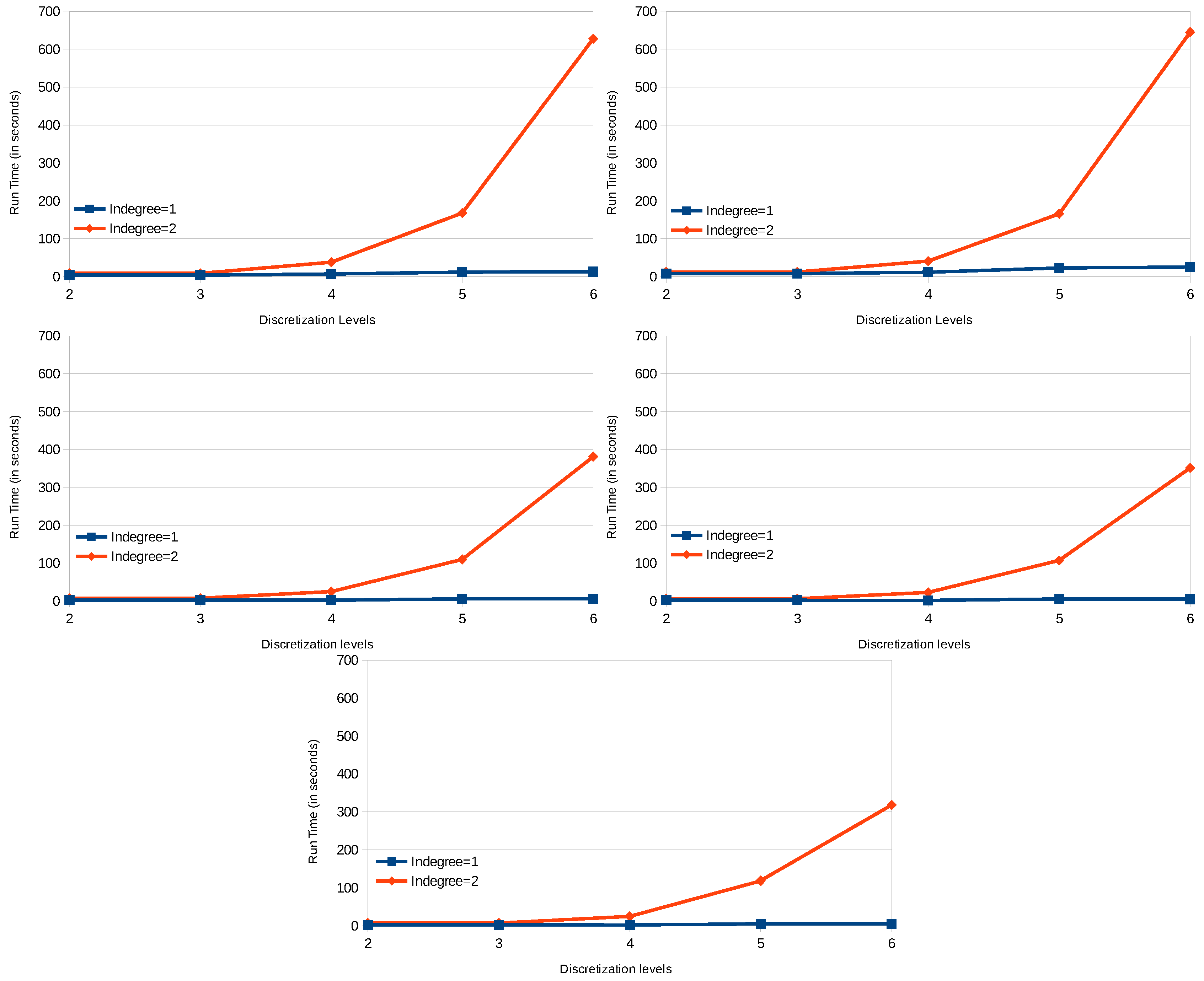
X, and the influencing one(s) (here, we detail only the transition with indegree = 1). Find the last component that has changed, the influenced component
X, the influencing component
Y or one of the components influenced by
X, such that there exists a transition in conflict with the computing one. This last time step of the change
H comparing to the time step
T2 for the component
X having as an influence
Y is computed in the predicate
lastChange(X,Y,H,T2) (lines 29–39). In other words,
H=
.
27 % Find the time step when the transition has started playing
28 % The last change of X such that W is influencing by X and X is influencing by Y
29 lastchange(X,Y,W,Max,T2) ← Max=#max{ T : changeState(Y,T;X,T;W,T), T<T2}, changeState(X, T2),
30 existInfluence(X,Y), existInfluence(W,X), Max>=0.
31 lastChangeAll(X,Y,Max,T2) ← lastchange(X,Y,W,Max,T2), transition(X,_,W,_,_,D, change(T3)), T3<T2,
32 T2-U>D, lastConditionChange(X,Y,U,T2).
33 % Last change between X and its influencing component Y
34 lastConditionChange(X,Y,H,T2) ← H=#max{ T : changeState(Y,T;X,T) , T<T2}, changeState(X, T2),
35 existInfluence(X,Y), H>=0.
36 % Find the time point of the last change: it is the last change of X, or of the component influenced by X
37 % or of the components involved in the transition condition
38 lastChange(X,Y,Max,T2) ← lastChangeAll(X,Y,Max,T2).
39 lastChange(X,Y,H,T2) ← lastConditionChange(X,Y,H,T2), not lastChangeAll(X,Y,H,T2), H!=H, delay(H).
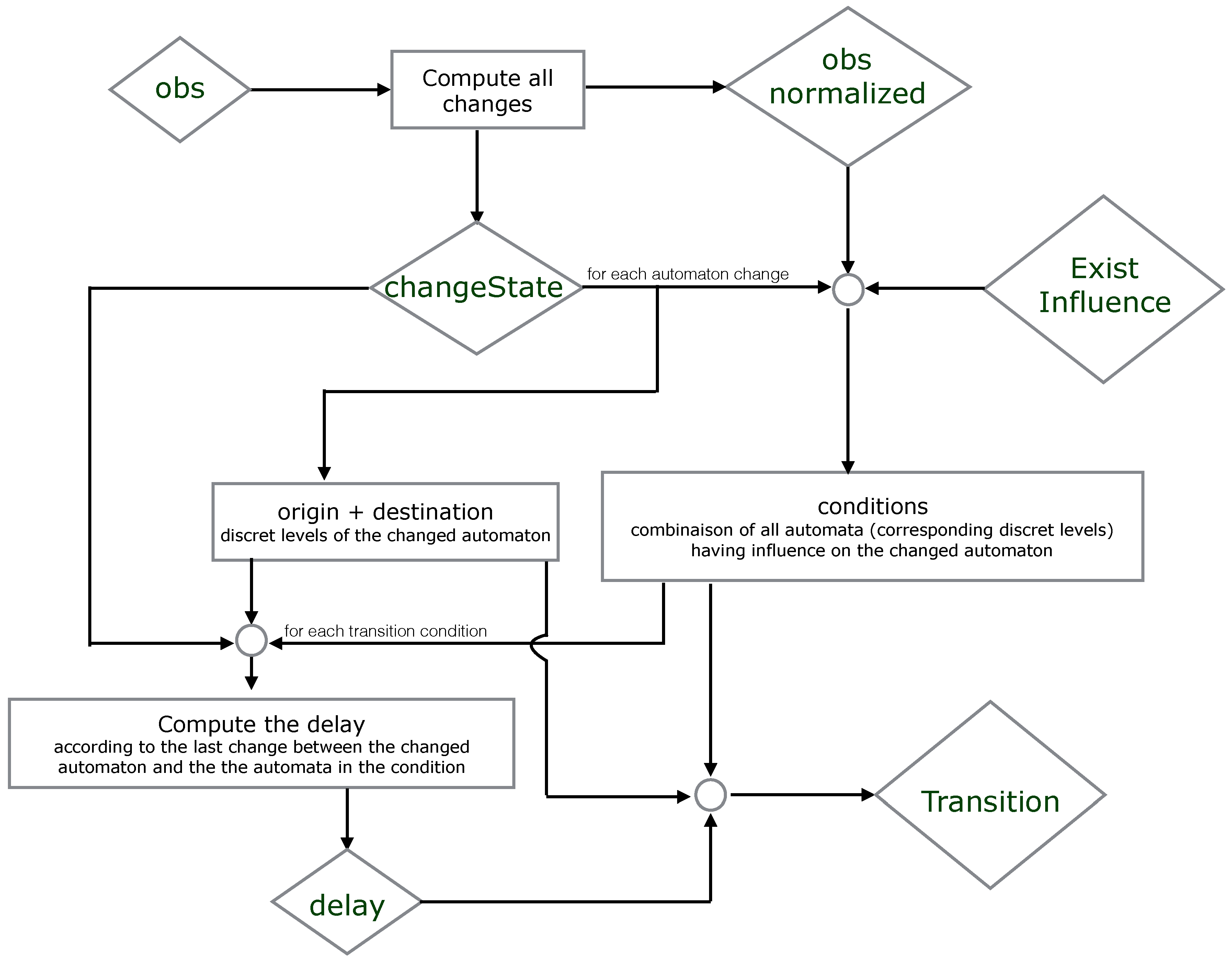
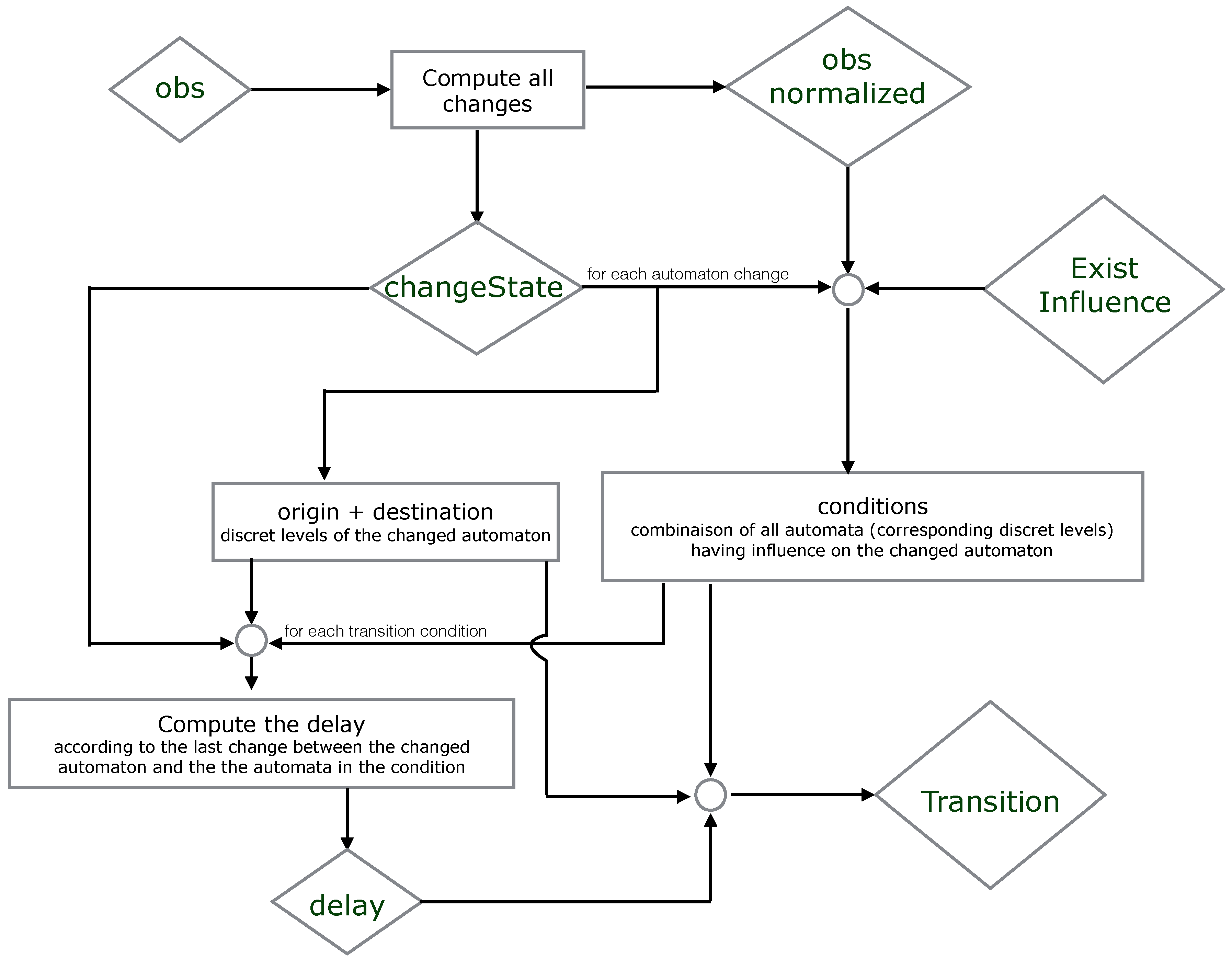
We propose below
Figure 5 where we simplify the explication of the encoding part. It illustrates which resources are necessary to compute the origin, destination, conditions and delay of each timed local transition.
To create as many models as possible that satisfy the given time series data, we add in the head of the rule the brackets { } between the predicates of the transition (see lines 41–43). Then, we make sure that we keep exactly one transition for each change at a step T by component X. We compute this number of different transitions in Tot by the predicate getTransNumber(Tot,X,T) (line 46). Therefore, we eliminate all models that do not satisfy this assumption by the constraints in lines 48–49.
40 % Compute all models with all candidate timed local transitions
41 {transition(Y,Valy,X,Val1,Val2,D, change(T2))} ← obs_normalized(X,Val1,T1,T2),
42 obs_normalized(Y,Valy,T1,T2), changeState(X,Val1,Val2,T2), existInfluence(X,Y),
43 lastChange(X,Y,T1,T2), T2=T1+D, delay(D).
44 transition(Y,Valy,X,Val1,Val2,D) ← transition(Y,Valy,X,Val1,Val2,D, _).
45 % for each change keep only one transition (xOR)
46 getTransNumber(Tot,X,T)← Tot={transition(_,_,X,_,_,_, change(T))}, changeState(X,T), T!=0.
47 % Exactly one transition by change in a model
48 ← getTransNumber(Tot,X,T), changeState(X,T), Tot=0.
49 ← getTransNumber(Tot,X,T), changeState(X,T), Tot>1.
7.4. Refinement According to the Semantics
The following constraint ensures that each output model will respect Definition 14: a component cannot inhibit and activate another one with the same level (line 51).
50 % A component with the same level inhibits and activates the same component
51 ← transition(Y,Valy,X,Val1,Val2,_), transition(Y,Valy,X,Val3,Val4,_), Val1<Val2, Val3>Val4.
Furthermore, according to Definition 14, a component cannot have the same effect on another one with the same level of expression. This property is encoded by the following constraint (line 53).
52 % A component with different levels influence another component with the same effect
53 ← transition(Y,Valy,X,Val1,Val2,_), transition(Y,Valy_,X,Val1,Val2,_), Valy_!=Valy.
According to Definition 12, a transition that is playable at a time point and that is not in conflict with another one should be played (synchronous behavior). If this is not the case, the model is not correct and will be eliminated by the following constraint in lines 68–69.
54 % Last time points in the data
55 timeSeriesSize(Last) ← Last=#max{ T : obs(_,_,T) }.
56 step(0..Last)← timeSeriesSize(Last).
57 % Compute all the obs between all the time points in the data
58 obs_(X,Val,T1,T2) ← obs(X,Val,T1), obs(X,Val,T2), T1<T2, not existsChange(X,Val,T1,T2).
59 existsChange(X,Val,T1,T2) ← obs(X,Val1,T), T>T1, T<T2, Val1!=Val, obs(X,Val,T1), obs(X,Val,T2).
60 % There is a transition with different delays
61 existTransDiffDelays(Y,Valy,X,Val1,Val2,D1,D2) ← transition(Y,Valy,X,Val1,Val2,D1),
62 transition(Y,Valy,X,Val1,Val2,D2), D1!=D2.
63 % There is a transition in conflict with "transition(Y,Valy,X,Val1,Val2,D1)"
64 existTransInConflict(Y,Valy,X,Val1,Val2,D1,T1,T2) ← transition(Y,Valy,X,Val1,Val2,D1),
65 transition(X,Val1,_,_,_,D2,change(T3)), T3>=T2, D2>=D1, T3-D2 <=T2, step(T2),
66 step(T1), T1<T2, D1=T2-T1, obs_(X,Val1,T1,T2), obs_(Y,Valy,T1,T2).
67 % Eliminate all models that do not respect the semantics (Definitions 5–6)
68 ← transition(Y,Valy,X,Val1,Val2,D), obs_(X,Val1,T1,T2), obs_(Y,Valy,T1,T2), step(D),
69 not changeState(X,Val1,Val2,T2), not existTransInConflict(Y,Valy,X,Val1,Val2,D,T1,T2),
70 not existTransDiffDelays(Y,Valy,X,Val1,Val2,D,D2), changeState(X,Val1,Val2,T3),
71 T2!=Max, timeSeriesSize(Max), obs_(X,Val1,T1,T3), T3-T1=D2, delay(D2), D=T2-T1.
7.5. Refinement on the Delays
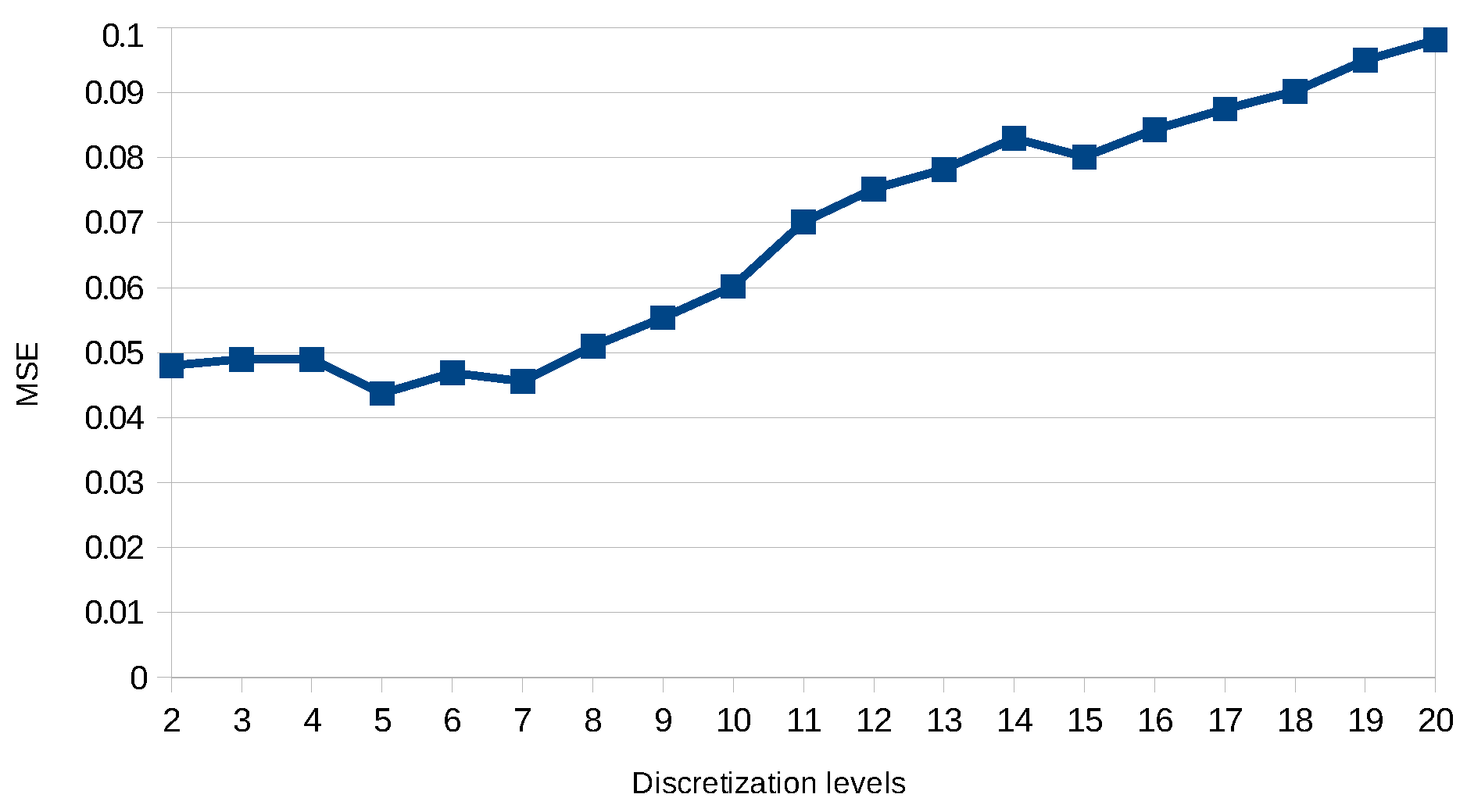
The several refinements of the generated models we propose can be seen as parameters that can be combined or not. For example, the user can specify that for the same transition, if we find different delays, we can take the average value or even specify an interval in which we define the minimum value and the maximum value.
According to Definition 17, in one model, a transition cannot have different delays; so that such models can be eliminated by the following constraint in line 12.
73 % No different delays for the same transition
74 ← transition(Y,Valy,X,Val1,Val2,D1), transition(Y,Valy,X,Val1,Val2,D2), D1!=D2.
Otherwise, we can merge all transitions that only differ by their delay in one transition, but whose delay is equal to the average delay of these transitions. To compute the average delays for a transition in the same model according to Definition 16, we do as follows in ASP. First, we compute the total number of these transitions in Tot by nbreTotTrans(Y,Valy,X,Val1,Val2,Tot) (lines 77–77), where the shared part between all transitions is Y,Valy,X,Val1,Val2; then, the sum of all of these delays in S by the predicate sumDelays (lines 80–81). To find the average, we divide S by Tot (Davg=S/Tot). The new transition after merging is then transAvgDelay(Y,Valy,X,Val1,Val2,Davg) (lines 83–84).
75 % Transitions with the average of delays
76 % Number of the repetition of each transition
77 nbreTotTrans(Y,Valy,X,Val1,Val2,Tot) ← Tot={transition(Y,Valy,X,Val1,Val2,_,_)},
78 transition(Y,Valy,X,Val1,Val2,_).
79 % The sum of the delays for each transition
80 sumDelays(Y,Valy,X,Val1,Val2,S) ← S=#sum{ D: transition(Y,Valy,X,Val1,Val2,D)},
81 transition(Y,Valy,X,Val1,Val2,_), S!=0.
82 % Compute the average delay for each transition
83 transAvgDelay(Y,Valy,X,Val1,Val2,Davg) ← nbreTotTrans(Y,Valy,X,Val1,Val2,Tot),
84 sumDelays(Y,Valy,X,Val1,Val2,S), Davg=S/Tot.
It is also possible to compute the interval of the delays according to Definition 15: [Max, Min] with Max is the maximum value of the delays of this transition, computed by the predicate maxDelay (lines 86–87); Min is the minimum value of the delays computed by the predicate minDelay (lines 88–89). Finally, the generation of a transition after merging is done by the predicate transIntervalDelay in lines 90–91.
85 % Compute the maximum value and the minimum value of the delays of a same transition
86 maxDelay(Y,Valy,X,Val1,Val2,Max) ← Max=#max{ D : transition(Y,Valy,X,Val1,Val2,D)},
87 transition(Y,Valy,X,Val1,Val2,_).
88 minDelay(Y,Valy,X,Val1,Val2,Min) ← Min=#min{ D : transition(Y,Valy,X,Val1,Val2,D)},
89 transition(Y,Valy,X,Val1,Val2,_).
90 transIntervalDelay(Y,Valy,X,Val1,Val2,interval(Min,Max)) ← minDelay(Y,Valy,X,Val1,Val2,Min),
91 maxDelay(Y,Valy,X,Val1,Val2,Max).
According to the Definition 13, we want to find the more frequent transitions in the models. In ASP, the option "
” computes the cautious consequences (intersection of all answer sets) of a logic program (algorithm implementation in ASP). Therefore, we use this option while executing.



{kind=link}
{kind=link}
{kind=link}
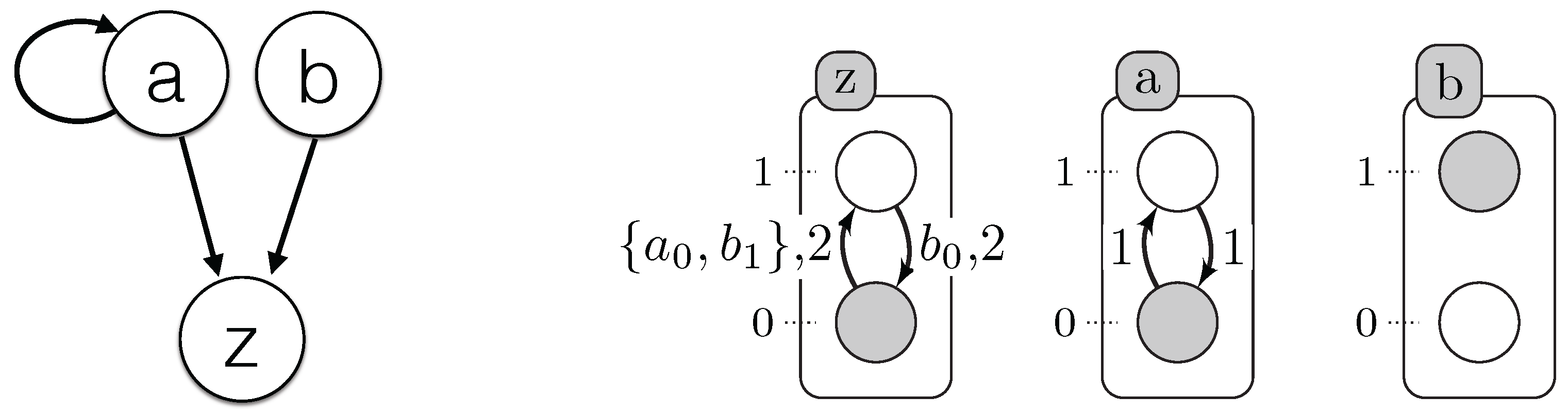{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}