
A Scanning Transmission X-ray Microscopy Study of Cubic and Orthorhombic C3A and Their Hydration Products in the Presence of Gypsum


Abstract
:1. Introduction
2. Experimental and Methods
2.1. Materials
2.2. Experimental
3. Results and Discussion
3.1. Characterization of Pure Phases
3.1.1. Particle Size Distribution
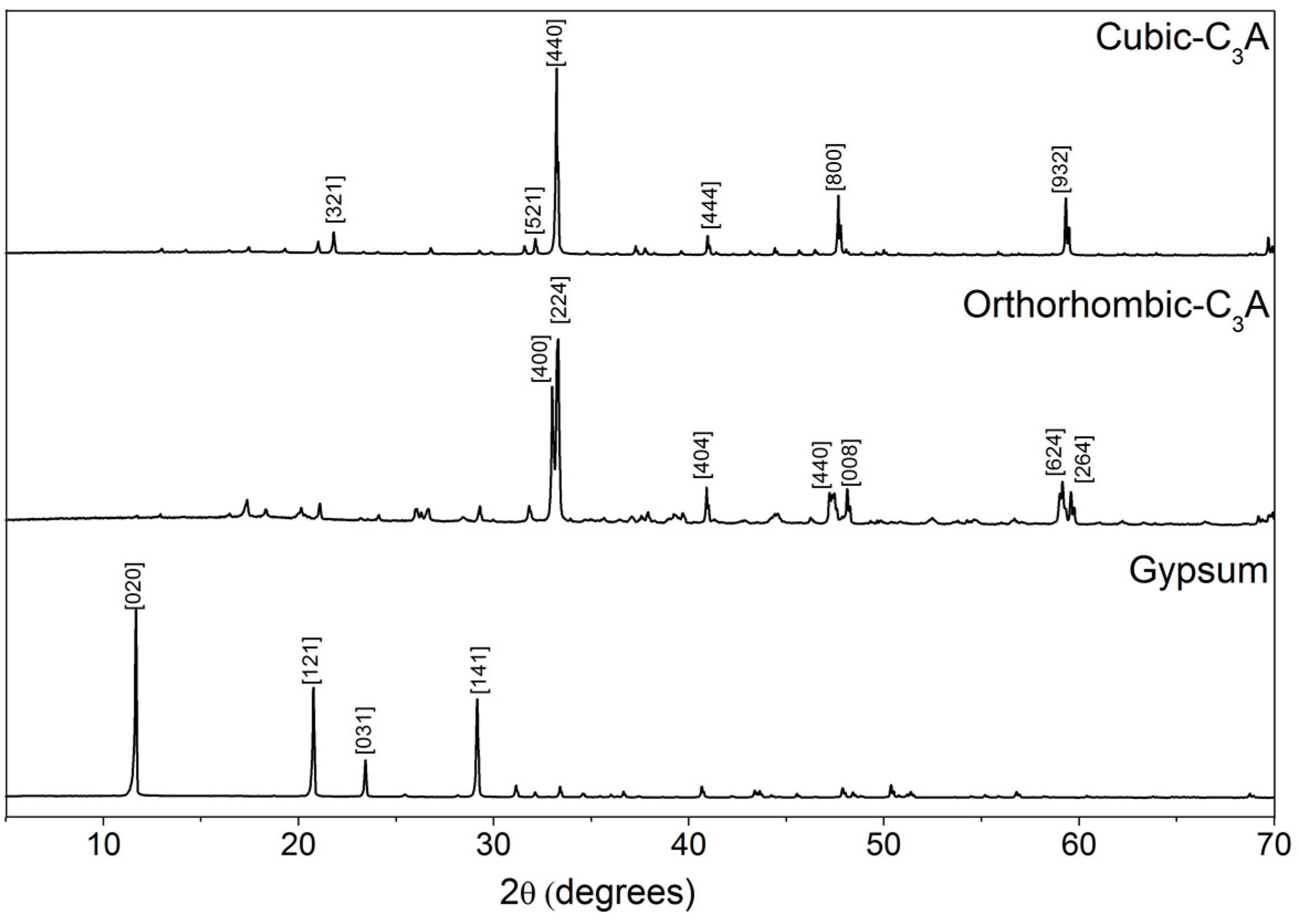
3.1.2. X-ray Diffraction
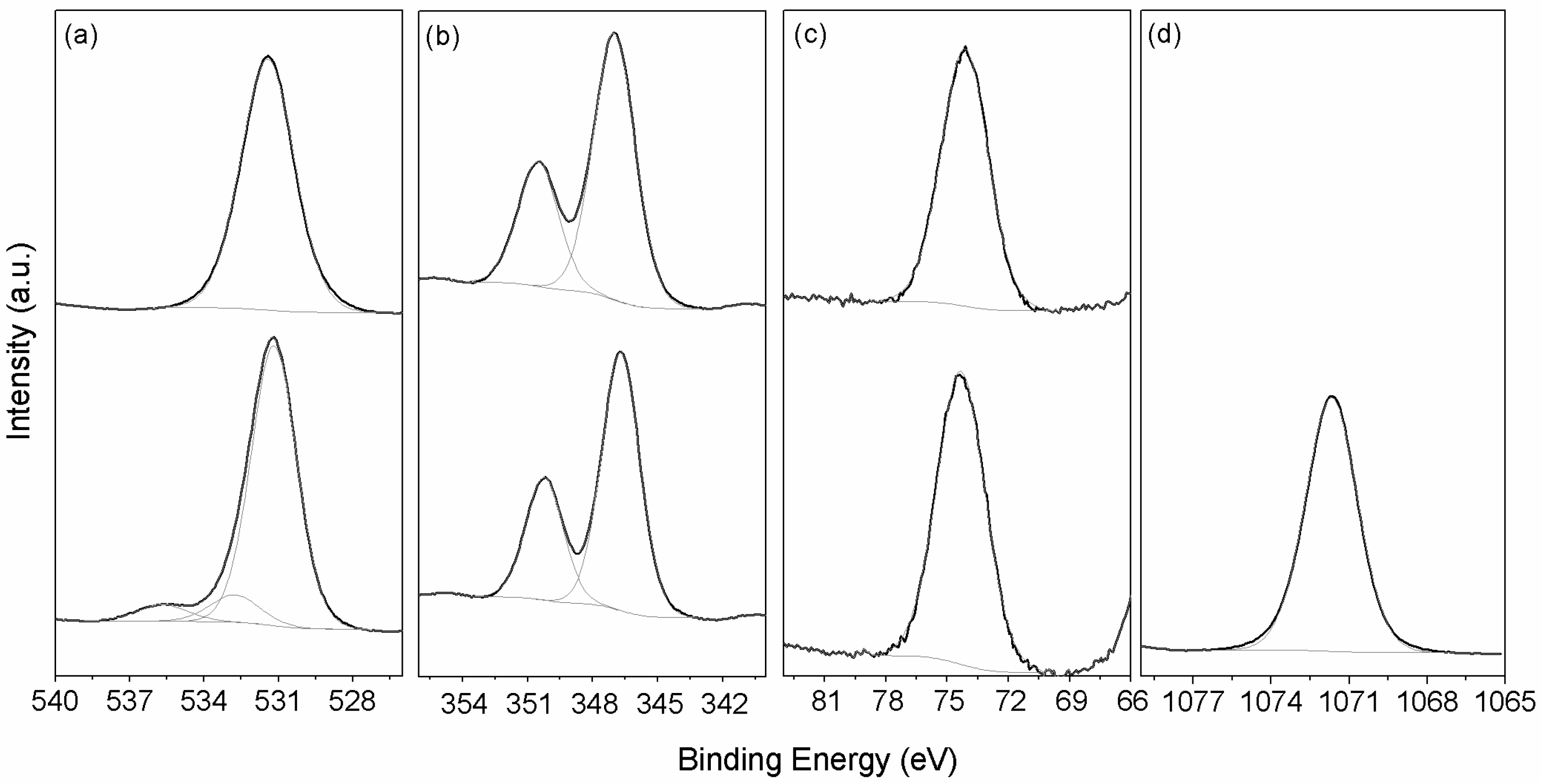
3.1.3. X-ray Photoelectron Spectroscopy (XPS)
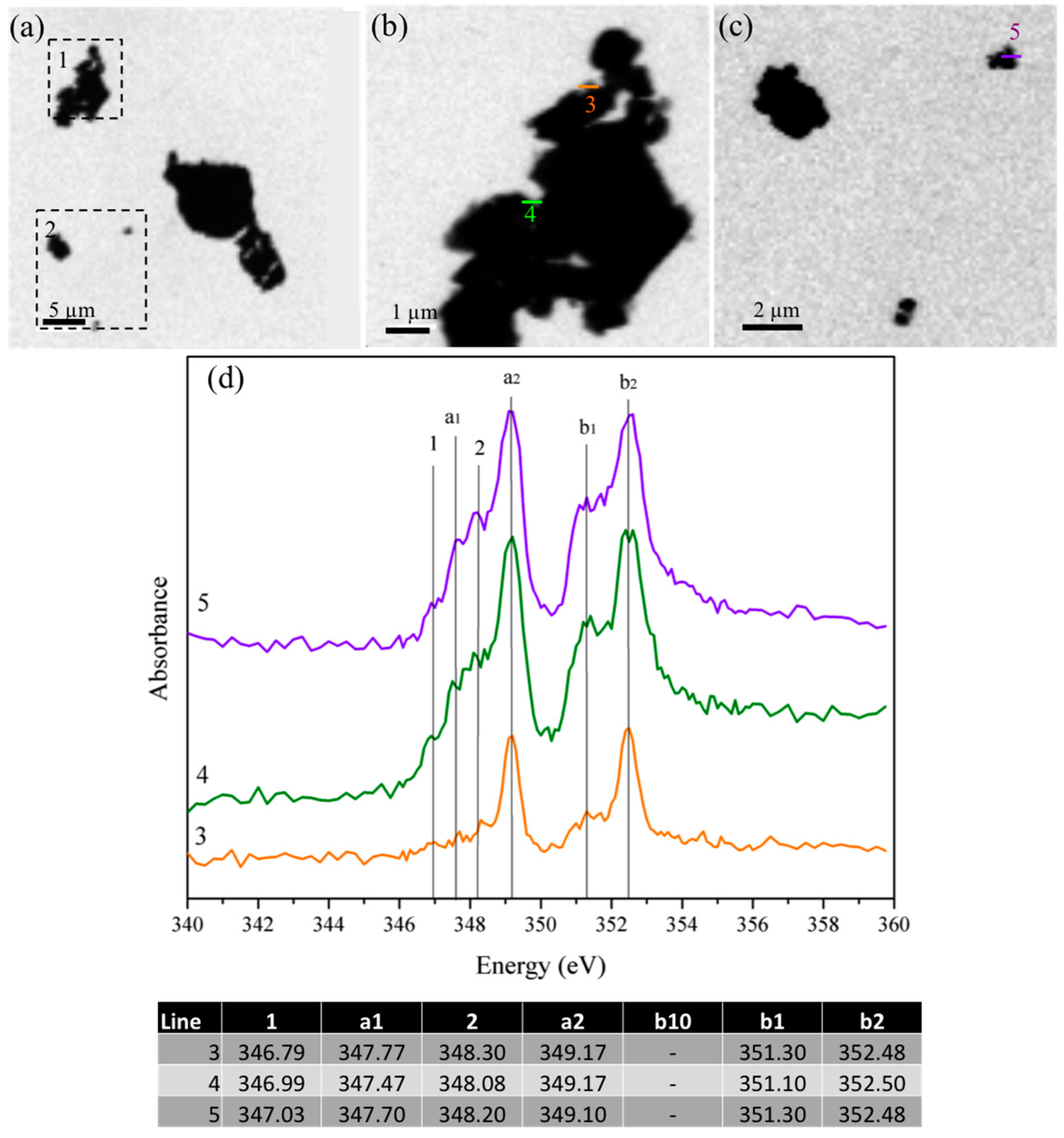
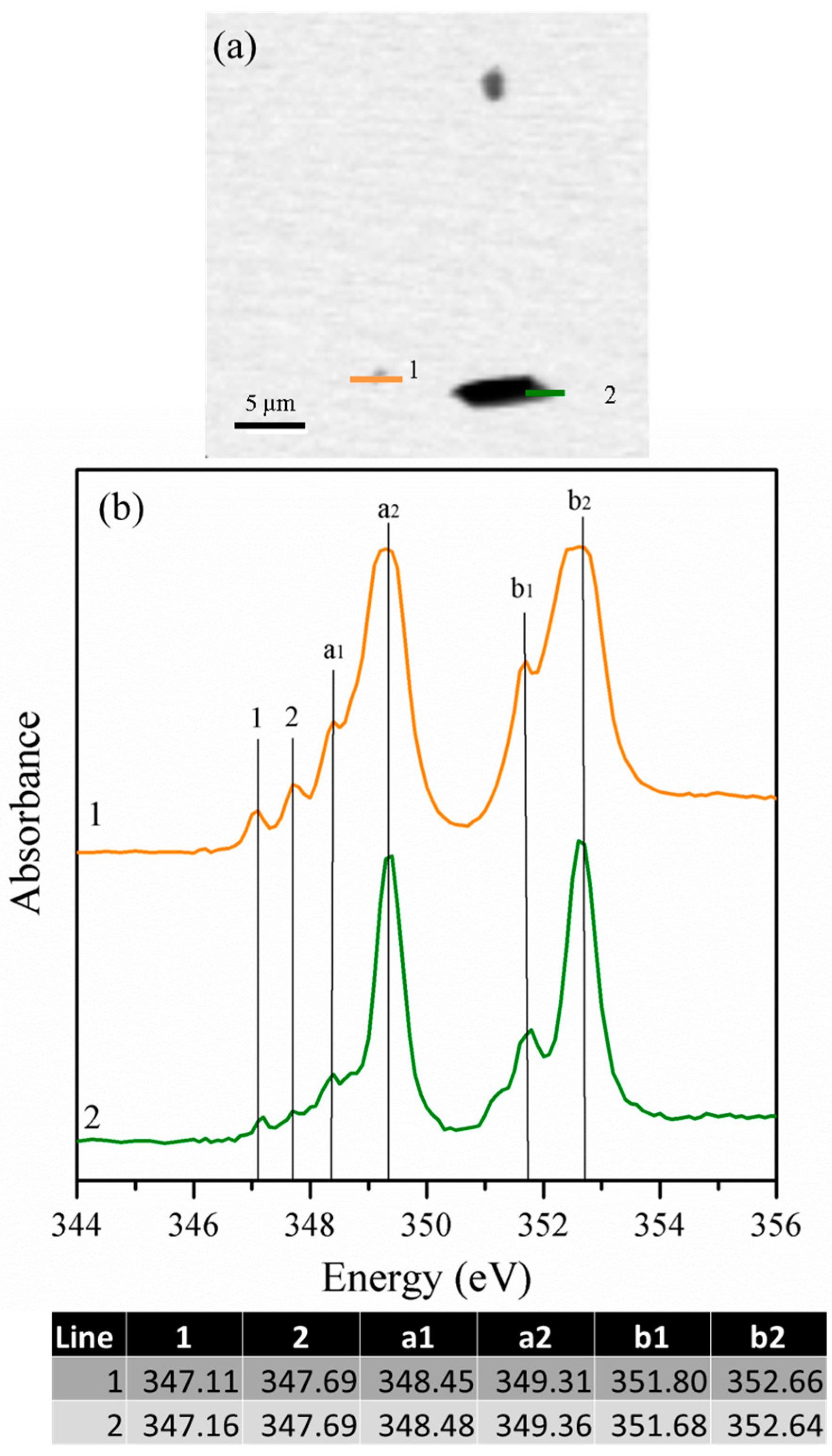
3.1.4. Scanning Transmission X-ray Microscopy
3.2. Hydration Products of C3A in the Presence of Gypsum
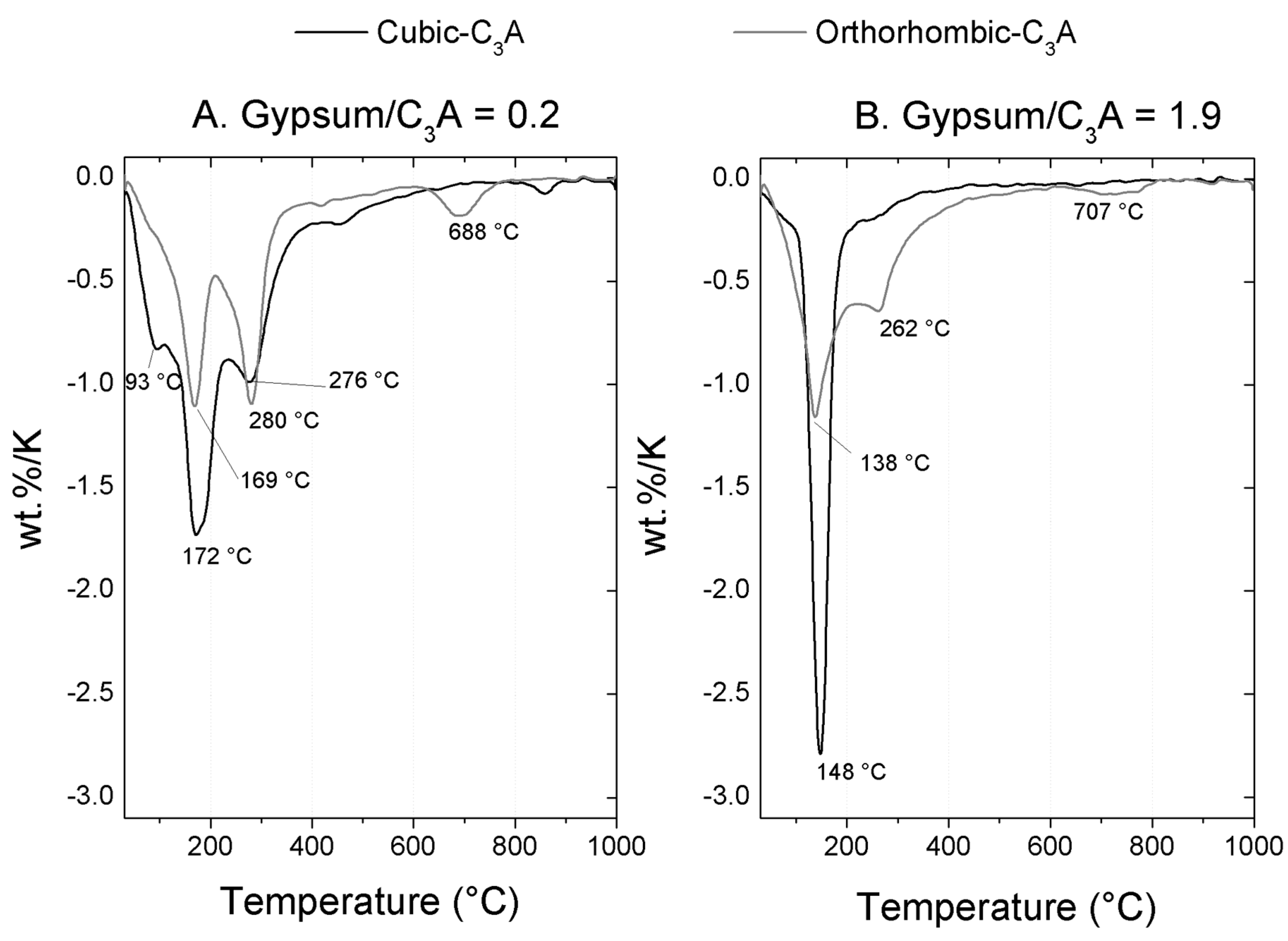
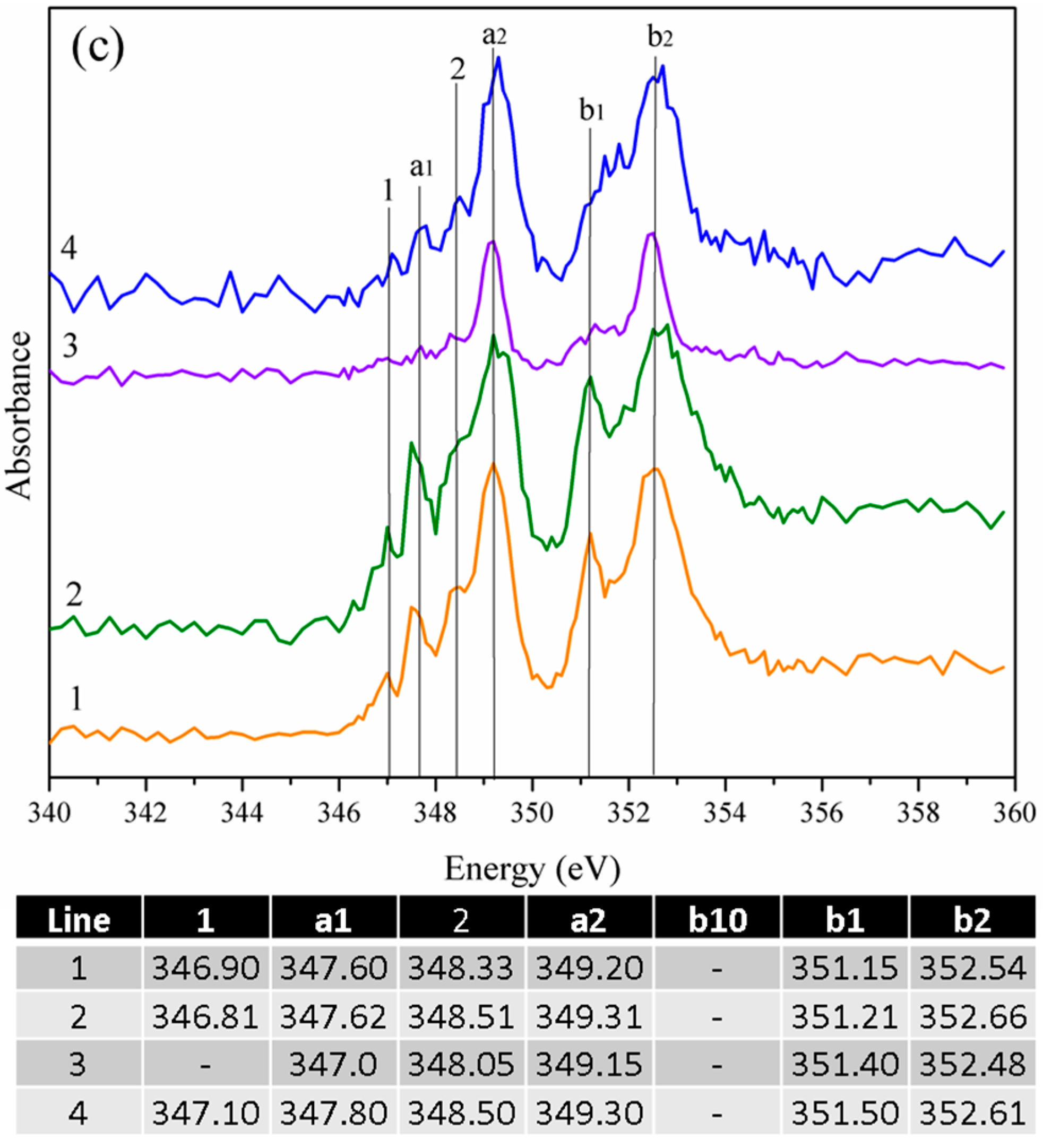
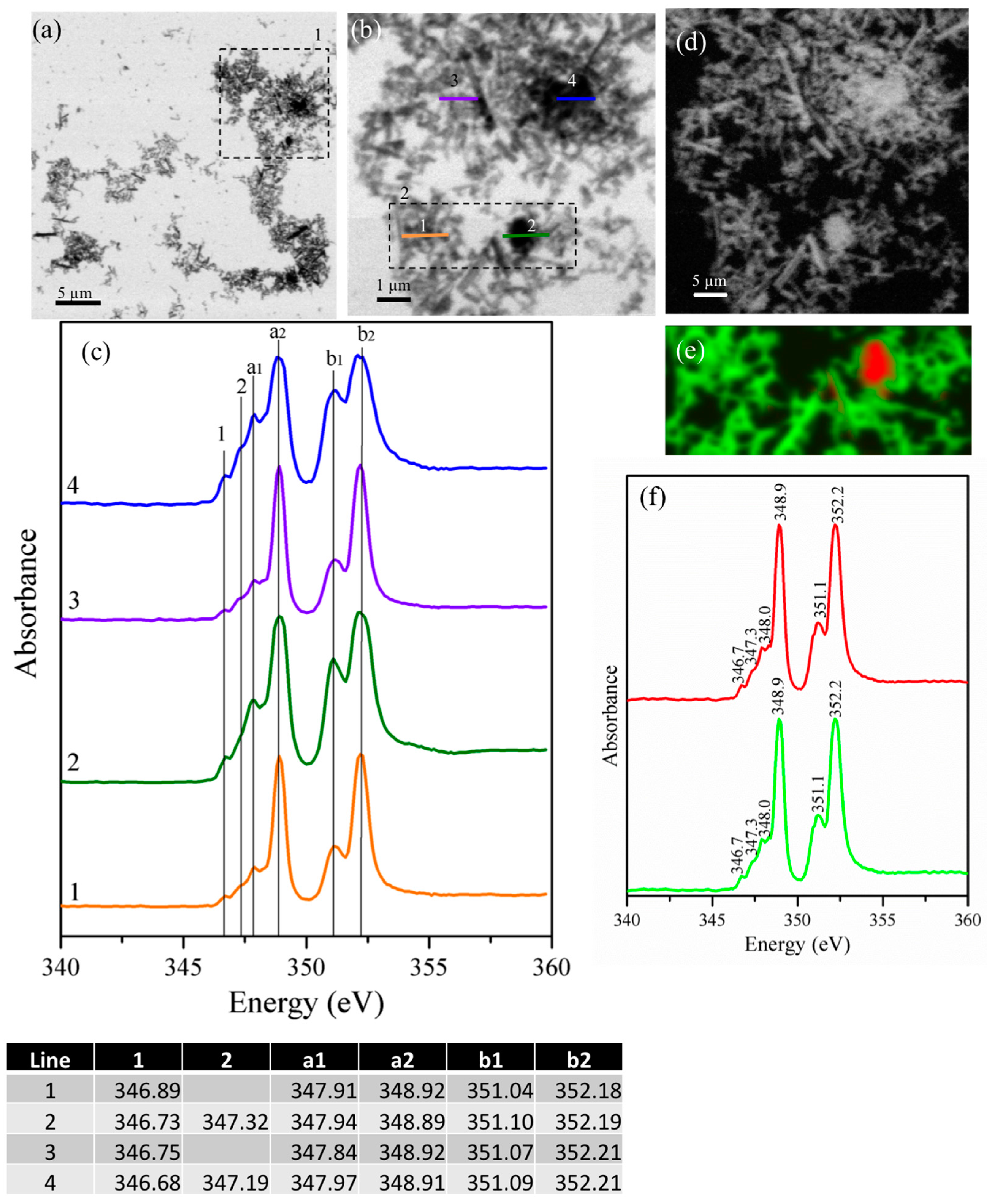
3.2.1. Gypsum/Cubic C3A Blends with Ratios of 0.20 and 1.19
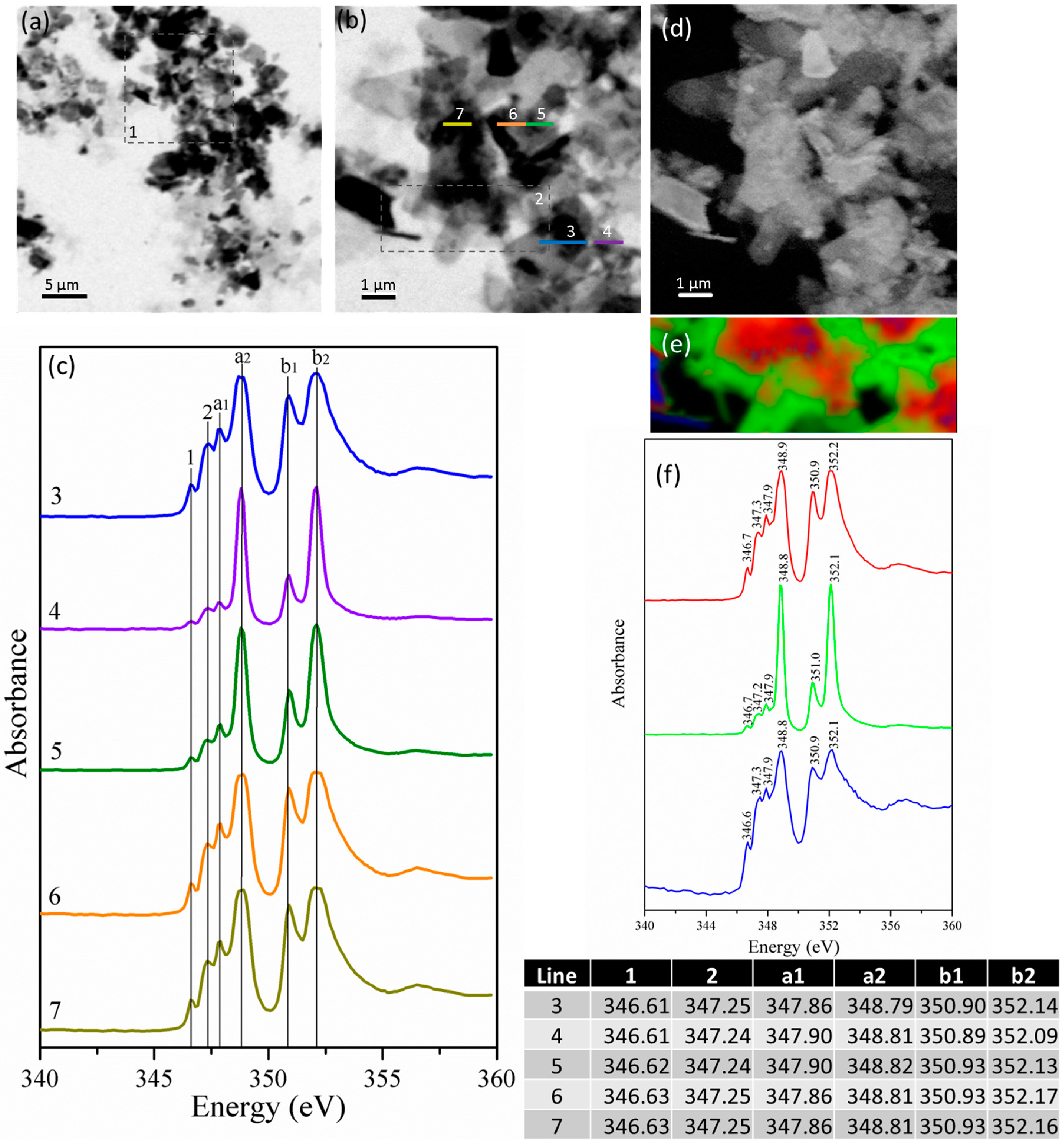
3.2.2. Gypsum/Na-Doped C3A Blend with Ratios of 0.20 and 1.19
4. Final Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A
TGA Analysis
References
- Taylor, H.F.W. Cement Chemistry; Thomas Telford Publishing: London, UK, 1997. [Google Scholar]
- Gobbo, L.; Sant’Agostino, L.; Garcez, L. C3A polymorphs related to industrial clinker alkalies content. Cem. Concr. Res. 2004, 34, 657–664. [Google Scholar] [CrossRef]
- Manzano, H.; Dolado, J.S.; Ayuela, A. Structural, Mechanical, and Reactivity Properties of Tricalcium Aluminate Using First-Principles Calculations. J. Am. Ceram. Soc. 2009, 92, 897–902. [Google Scholar] [CrossRef]
- Takeuchi, T.; Nishi, F.M. Crystal-chemical characterization of the 3CaO·Al2O3 solid-solution series. Z. Krist. 1980, 152, 259–307. [Google Scholar] [CrossRef]
- Ostrowski, C.; Żelazny, J. Solid Solutions of Calcium Aluminates C3A, C12A7 and CA with Sodium Oxide. J. Therm. Anal. Calorim. 2004, 75, 867–885. [Google Scholar] [CrossRef]
- Bullard, J.W.; Jennings, H.M.; Livingston, R.A.; Nonat, A.; Scherer, G.W.; Schweitzer, J.S.; Scrivener, K.L.; Thomas, J.J. Mechanisms of cement hydration. Cem. Concr. Res. 2011, 41, 1208–1223. [Google Scholar] [CrossRef]
- Scrivener, K.L.; Nonat, A. Hydration of cementitious materials, present and future. Cem. Concr. Res. 2011, 41, 651–665. [Google Scholar] [CrossRef]
- Quennoz, A.; Scrivener, K.L. Hydration of C3A–gypsum systems. Cem. Concr. Res. 2012, 42, 1032–1041. [Google Scholar] [CrossRef]
- Breval, E. C3A hydration. Cem. Concr. Res. 1976, 6, 129–137. [Google Scholar] [CrossRef]
- Kirchheim, A.P.; Molin, D.C.D.; Fischer, P.; Emwas, A.; Provis, J.L.; Monteiro, P.J.M. Real-time high-resolution X-ray imaging and nuclear magnetic resonance study of the hydration of pure and Na-doped C3A in the presence of sulfates. Inorg. Chem. 2011, 50, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.; Wistuba, S. Crystal structure refinement and hydration behaviour of doped tricalcium aluminate. Cem. Concr. Res. 2006, 36, 2011–2020. [Google Scholar] [CrossRef]
- Wistuba, S.; Stephan, D.; Raudaschl-Sieber, G.; Plank, J. Hydration and Hydration Products of Two-Phase Portland Cement Clinker Doped with Na2O. 2015. Available online: http://dx.doi.org/10.1680/adcr.2007.19.3.125 (accessed on 5 May 2015).
- Boikova, A.I.; Domansky, A.I.; Paramonova, V.A.; Stavitskaja, G.P.; Nikushchenko, V.M. The influence of Na2O on the structure and properties of 3CaO·Al2O3. Cem. Concr. Res. 1977, 7, 483–492. [Google Scholar] [CrossRef]
- Juenger, M.C.G.; Jennings, H.M. Effects of highly alkalinity on cement pastes. Mater. J. 2001, 98, 251–255. [Google Scholar]
- Kirchheim, A.P.; Fernàndez-Altable, V.; Monteiro, P.J.M.; Molin, D.C.C.D.; Casanova, I. Analysis of cubic and orthorhombic C3A hydration in presence of gypsum and lime. J. Mater. Sci. 2009, 44, 2038–2045. [Google Scholar] [CrossRef]
- Dubina, E.; Plank, J.; Wadsö, L.; Black, L. Impact of environmental moisture on C3A polymorphs in the absence and presence of CaSO4·0.5H2O. Adv. Cem. Res. 2014, 26, 29–40. [Google Scholar] [CrossRef]
- Alonso, M.M.; Puertas, F. Adsorption of PCE and PNS superplasticisers on cubic and orthorhombic C3A. Effect of sulfate. Constr. Build. Mater. 2015, 78, 324–332. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, W.; Liu, G.; Orozco, C.; Monteiro, P.J.M. Effect of Superplasticisers on the Hydration Process, Products and Microstructure of Tricalcium Aluminate Paste in the Presence of Gypsum. 2016. Available online: http://dx.doi.org/10.1680/jadcr.15.00083 (accessed on 15 April 2016).
- Jacobsen, C.; Wirick, S.; Flynn, E.; Zimba, C. Soft X-ray spectroscopy from image sequences with sub-100 nm spatial resolution. J. Microsc. 2000, 197, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.; Chae, S.; Chou, K.W.; Tyliszczak, T.; Monteiro, P.J.M. Effect of polymers on the nanostructure and on the carbonation of calcium silicate hydrates: A scanning transmission X-ray microscopy study. J. Mater. Sci. 2011, 47, 976–989. [Google Scholar] [CrossRef]
- Mendes, A.; Gates, W.P.; Sanjayan, J.G.; Collins, F. NMR, XRD, IR and synchrotron NEXAFS spectroscopic studies of OPC and OPC/slag cement paste hydrates. Mater. Struct. 2011, 44, 1773–1791. [Google Scholar] [CrossRef]
- Monteiro, P.J.M.; Clodic, L.; Battocchio, F.; Kanitpanyacharoen, W.; Chae, S.R.; Ha, J.; Wenk, H.-R. Incorporating carbon sequestration materials in civil infrastructure: A micro and nano-structural analysis. Cem. Concr. Compos. 2013, 40, 14–20. [Google Scholar] [CrossRef]
- Hernández-Cruz, D.; Hargis, C.W.; Bae, S.; Itty, P.A.; Meral, C.; Dominowski, J.; Radler, M.J.; Kilcoyne, D.A.; Monteiro, P.J.M. Multiscale characterization of chemical–mechanical interactions between polymer fibers and cementitious matrix. Cem. Concr. Compos. 2014, 48, 9–18. [Google Scholar] [CrossRef]
- Dähn, R.; Vespa, M.; Tyliszczak, T.; Wieland, E.; Shuh, D.K. Soft X-ray spectromicroscopy of cobalt uptake by cement. Environ. Sci. Technol. 2011, 45, 2021–2027. [Google Scholar] [CrossRef] [PubMed]
- Yoon, I.-H.; Moon, D.H.; Kim, K.-W.; Lee, K.-Y.; Lee, J.-H.; Kim, M.G. Mechanism for the stabilization/solidification of arsenic-contaminated soils with Portland cement and cement kiln dust. J. Environ. Manag. 2010, 91, 2322–2328. [Google Scholar] [CrossRef] [PubMed]
- Geng, G.; Myers, R.J.; Kilcoyne, A.L.; Ha, J.; Monteiro, P.J.M. Ca L2,3-edge near edge X-ray absorption fine structure of tricalcium aluminate, gypsum and calcium (sulfo)aluminate hydrates. Am. Mineral. 2016, 1689–1699. [Google Scholar]
- Gobbo, L.D.A. Aplicação da Difração de Raios-X e Método de Rietveld no Estudo de Cimento Portland; Biblioteca Digital de Teses e Dissertações da Universidade de São Paulo: São Paulo, Brazil, 2009. [Google Scholar]
- Post, J.E.; Bish, D.L. Rietveld Refinement of Crystal Structures Using Powder X-ray Diffraction Data. Mineral. Soc. Am. 1989, 20, 277–308. [Google Scholar]
- Hitchcock, A.P.; Hitchcock, P.; Jacobsen, C.; Zimba, C.; Loo, B.; Rotenberg, E.; Denlinger, J. aXis 2000—Analysis of X-ray Images and Spectra; McMaster University: Hamilton, ON, Canada, 2012. [Google Scholar]
- Hanhan, S.; Smith, A.M.; Obst, M.; Hitchcock, A.P. Optimization of analysis of soft X-ray spectromicroscopy at the Ca 2p edge. J. Electron Spectros. Relat. Phenom. 2009, 173, 44–49. [Google Scholar] [CrossRef]
- Regourd, M.; Chromy, S.; Hjorth, L.; Mortureux, B.; Guinier, A. Polymorphisme des solutions solides du sodium dans l’aluminate tricalcique. J. Appl. Crystallogr. 1973, 6, 355–364. [Google Scholar]
- Mondal, P.; Jeffery, W. The Crystal Structure of Triealeium Aluminate, Ca3AI206. Acta Cryst. 1975, B31, 689–697. [Google Scholar] [CrossRef]
- Lager, G.A.; Armbruster, T.; Faber, J.J. Neutron and X-ray diffraction study of hydrogarnet Ca3Al2(O4H4)3. Am. Mineral. 1987, 72, 756–765. [Google Scholar]
- Nishi, F.; Takeuchi, Y. The Al6 O18 rings of tetrahedra in the structure of Ca8.5 Na Al6 O18. Acta Crystallogr. B 1975, 31, 1169–1173. [Google Scholar] [CrossRef]
- Effenberger, H. Crystal structure and infrared absorption spectrum of synthetic monohydrocalcite, CaCO3·H2O. Mon. Chem./Chem. Mon. 1981, 112, 899–909. [Google Scholar] [CrossRef]
- Moulder, J.F.; Stickle, W.F.; Sobol, P.E.; Bombem, K. Handbook of X-ray Photoelectron Spectroscopy. In A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data; Perkin-Elmer Physical Electronic Division: Eden Prairie, MN, USA, 1992. [Google Scholar]
- Dubina, E.; Plank, J.; Black, L. Impact of water vapour and carbon dioxide on surface composition of C3A polymorphs studied by X-ray photoelectron spectroscopy. Cem. Concr. Res. 2015, 73, 36–41. [Google Scholar] [CrossRef]
- Dubina, E.; Black, L.; Plank, J. Impact of water vapour and carbon dioxide on surface composition of C3A polymorphs studied by X-ray photoelectron spectroscopy. In Proceedings of the 13th International Congress on the Chemistry of Cement (ICCC), Madrid, Spain, 4–8 July 2011.
- Ball, M.C.; Simmons, R.E.; Sutherland, I. Surface composition of anhydrous tricalcium aluminate and calcium aluminoferrite. J. Mater. Sci. 1987, 22, 1975–1979. [Google Scholar] [CrossRef]
- Dubina, E.; Black, L.; Sieber, J.; Plank, R. Interaction of water vapour with anhydrous cement minerals. Adv. Appl. Ceram. 2010, 109, 260–268. [Google Scholar] [CrossRef]
- Dubina, L.B.E.; Plank, J.; Wadsö, L. The effects of prehydration of a combination of cubic C3A with β-hemihydrate on adsorption of BNS superplasticizer. In Proceedings of the 13th ICCC International Congress on the Chemistry of Cement, Madrid, Spain, 4–8 July 2011.
- Dubina, E. The Phenomenon of Cement Ageing on Moist Air: Surface Chemistry, Mechanisms and Effects on Admixture Performance. Available online: http://mediatum.ub.tum.de/node?id=11212512013 (accessed on 26 November 2012).
- Dubina, L.; Sieber, E.; Plank, R.; Black, J. Effects of pre-hydration on hydraulic properties on Portland cement and synthetic clinker phases. Cem. Concr. Sci. 2008, 64, 49–67. [Google Scholar]
- Naftel, S.J.; Sham, T.K.; Yiu, Y.M.; Yates, B.W. Calcium L -edge XANES study of some calcium compounds. J. Synchrotron Radiat. 2001, 8, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Politi, Y.; Metzler, R.A.; Abrecht, M.; Gilbert, B.; Wilt, F.H.; Sagi, I.; Addadi, L.; Weiner, S.; Gilbert, P.U.P.A.; Gilbert, P. Transformation mechanism of amorphous calcium carbonate into calcite in the sea urchin larval spicule. Proc. Natl. Acad. Sci. USA 2008, 105, 17362–17366. [Google Scholar] [CrossRef] [PubMed]
- Minard, H.; Garrault, S.; Regnaud, L.; Nonat, A. Mechanisms and parameters controlling the tricalcium aluminate reactivity in the presence of gypsum. Cem. Concr. Res. 2007, 37, 1418–1426. [Google Scholar] [CrossRef]
- Merlini, M.; Artioli, G.; Cerulli, T.; Cella, F.; Bravo, A. Tricalcium aluminate hydration in additivated systems. A crystallographic study by SR-XRPD. Cem. Concr. Res. 2008, 38, 477–486. [Google Scholar] [CrossRef]
- Meredith, P.; Donald, A.M.; Meller, N.; Hall, C. Tricalcium aluminate hydration: Microstructural observations by in-situ electron microscopy. J. Mater. Sci. 2004, 39, 997–1005. [Google Scholar] [CrossRef]
- Baquerizo, L.G.; Matschei, T.; Scrivener, K.L.; Saeidpour, M.; Wadsö, L. Hydration states of AFm cement phases. Cem. Concr. Res. 2015, 73, 143–157. [Google Scholar] [CrossRef]
- Rashid, S.; Barnes, P.; Bensted, J.; Turrillas, X. Conversion of calcium aluminate cement hydrates re-examined with synchrotron energy-dispersive diffraction. J. Mater. Sci. Lett. 1994, 13, 1232–1234. [Google Scholar] [CrossRef]
- Scrivener, K.L.; Pratt, P.L. Microstructural studies on the hydration of C3A and C4AF independently and in cement paste. Proc. Br. Ceram. Soc. 1984, 35, 201–219. [Google Scholar]
- Lothenbach, B.; Durdziński, P.; de Weerdt, K. Thermogravimetric analysis. In A Practical Guide to Microstructural Analysis of Cementitious Materials; Scrivener, K., Snellings, R., Lothenbach, B., Eds.; CRC Press: Boca Raton, FL, USA, 2015; pp. 178–208. [Google Scholar]




{kind=link}
{kind=link}
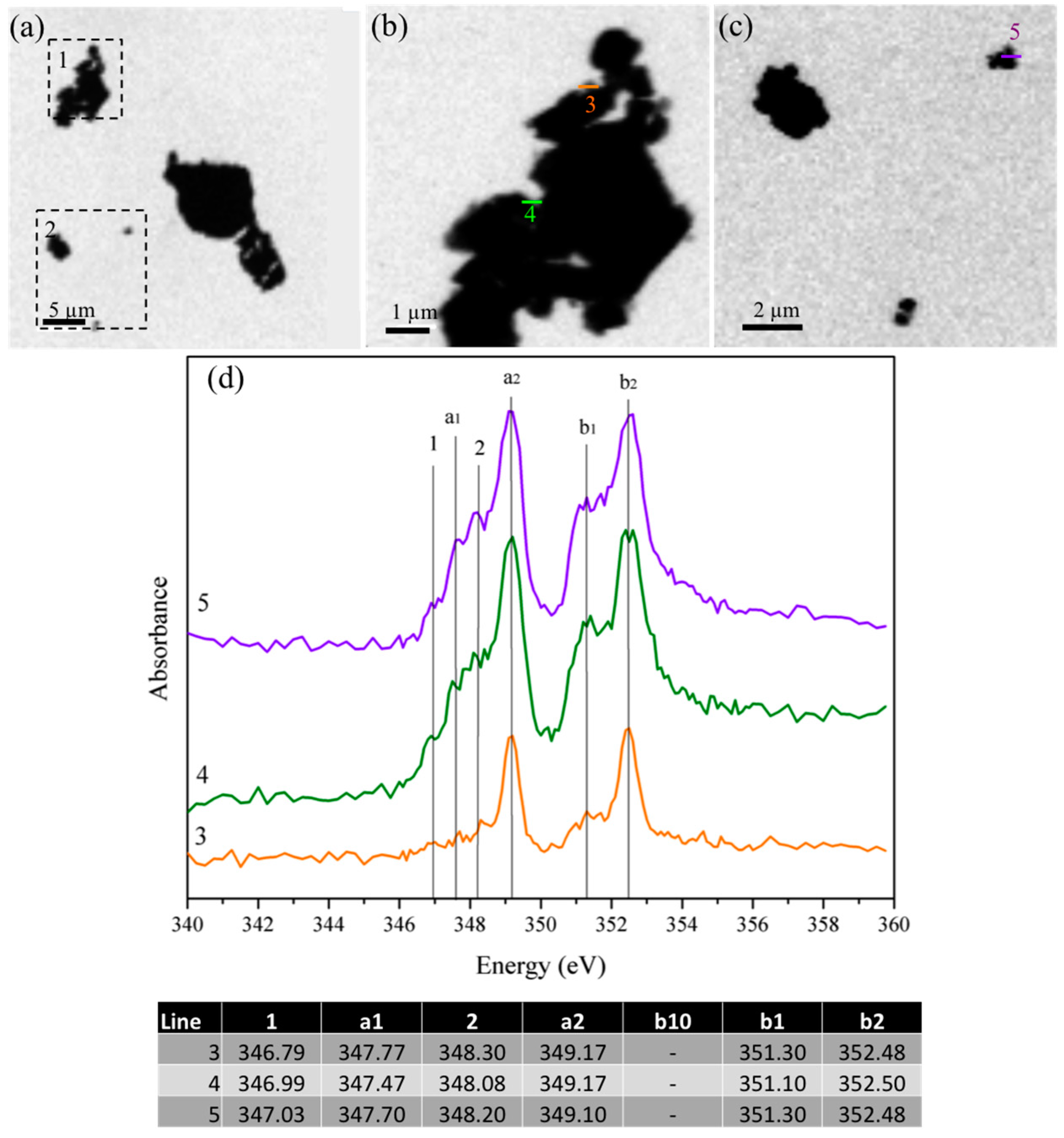{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
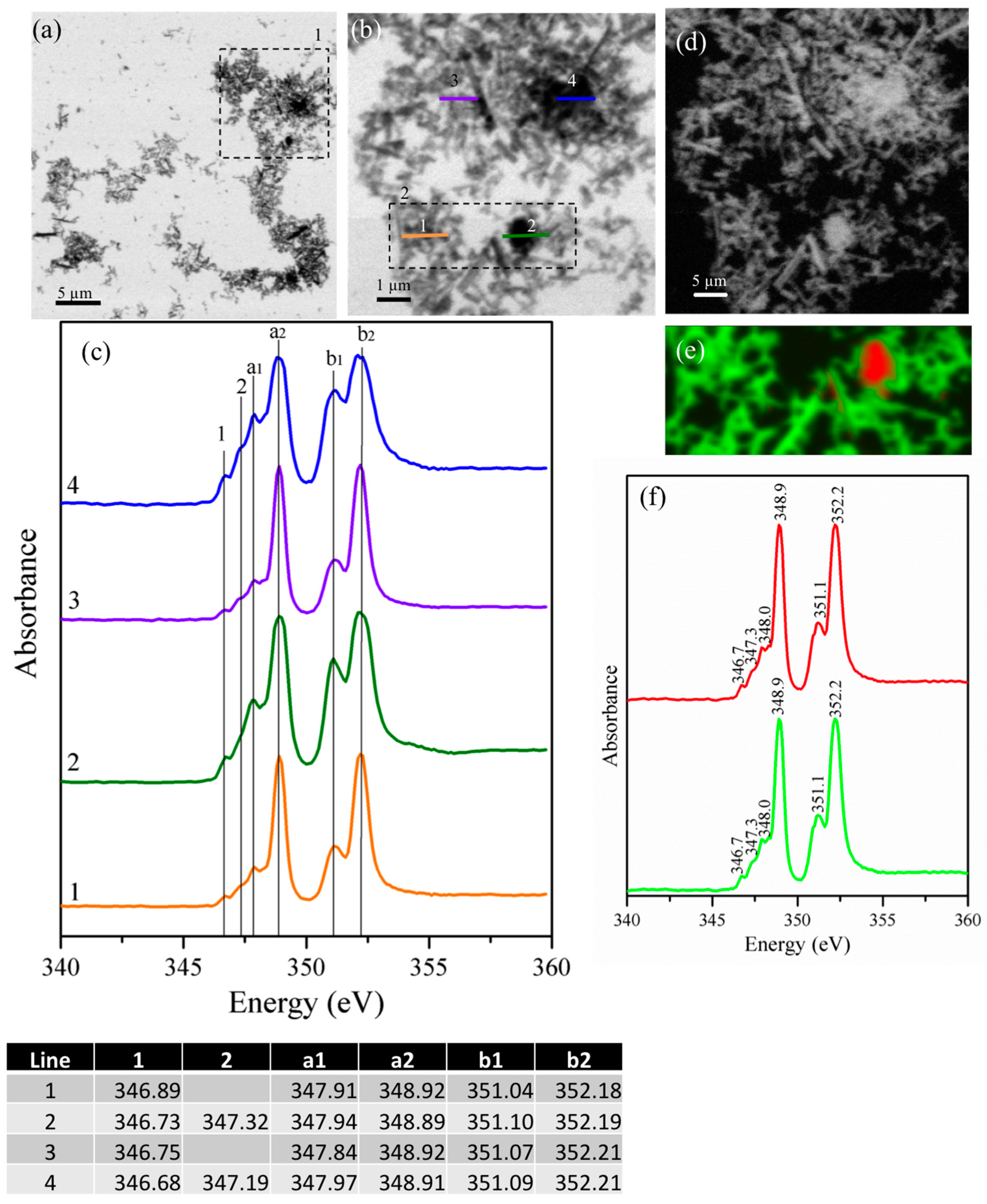{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Start Energy (eV) | End Energy (eV) | Dwell Time (ms) |
|---|---|---|---|
| Wide | 0 | 1200 | 0.1 |
| C 1s | 275 | 295 | 1 |
| O 1s | 525 | 540 | 0.5 |
| Ca 2p | 339 | 359 | 1 |
| Al 2p | 65 | 85 | 1 |
| Al 2s | 108 | 128 | 1 |
| Na 1s | 1065 | 1085 | 1 |
| Name | Cubic C3A | Orthorhombic C3A | ||
|---|---|---|---|---|
| Position | FWHM | Position | FWHM | |
| C 1s | 289.4 | 2.49 | 289.1 | 2.14 |
| O 1s | 528.6 531.4 | 1.31 2.50 | 531.2 532.8 535.7 | 2.16 2.35 2.43 |
| Ca 2p3/2 | 347.0 | 2.26 | 346.7 | 2.06 |
| Ca 2p1/2 | 350.5 | 2.20 | 350.2 | 2.01 |
| Al 2p | 74.1 | 2.69 | 74.3 | 2.76 |
| Na 1s | - | - | 1071.7 | 2.34 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rheinheimer, V.; Chae, S.R.; Rodríguez, E.D.; Geng, G.; Kirchheim, A.P.; Monteiro, P.J.M. A Scanning Transmission X-ray Microscopy Study of Cubic and Orthorhombic C3A and Their Hydration Products in the Presence of Gypsum. Materials 2016, 9, 745. https://doi.org/10.3390/ma9090745
Rheinheimer V, Chae SR, Rodríguez ED, Geng G, Kirchheim AP, Monteiro PJM. A Scanning Transmission X-ray Microscopy Study of Cubic and Orthorhombic C3A and Their Hydration Products in the Presence of Gypsum. Materials. 2016; 9(9):745. https://doi.org/10.3390/ma9090745
Chicago/Turabian StyleRheinheimer, Vanessa, Sejung Rosie Chae, Erich D. Rodríguez, Guoqing Geng, Ana Paula Kirchheim, and Paulo J. M. Monteiro. 2016. "A Scanning Transmission X-ray Microscopy Study of Cubic and Orthorhombic C3A and Their Hydration Products in the Presence of Gypsum" Materials 9, no. 9: 745. https://doi.org/10.3390/ma9090745