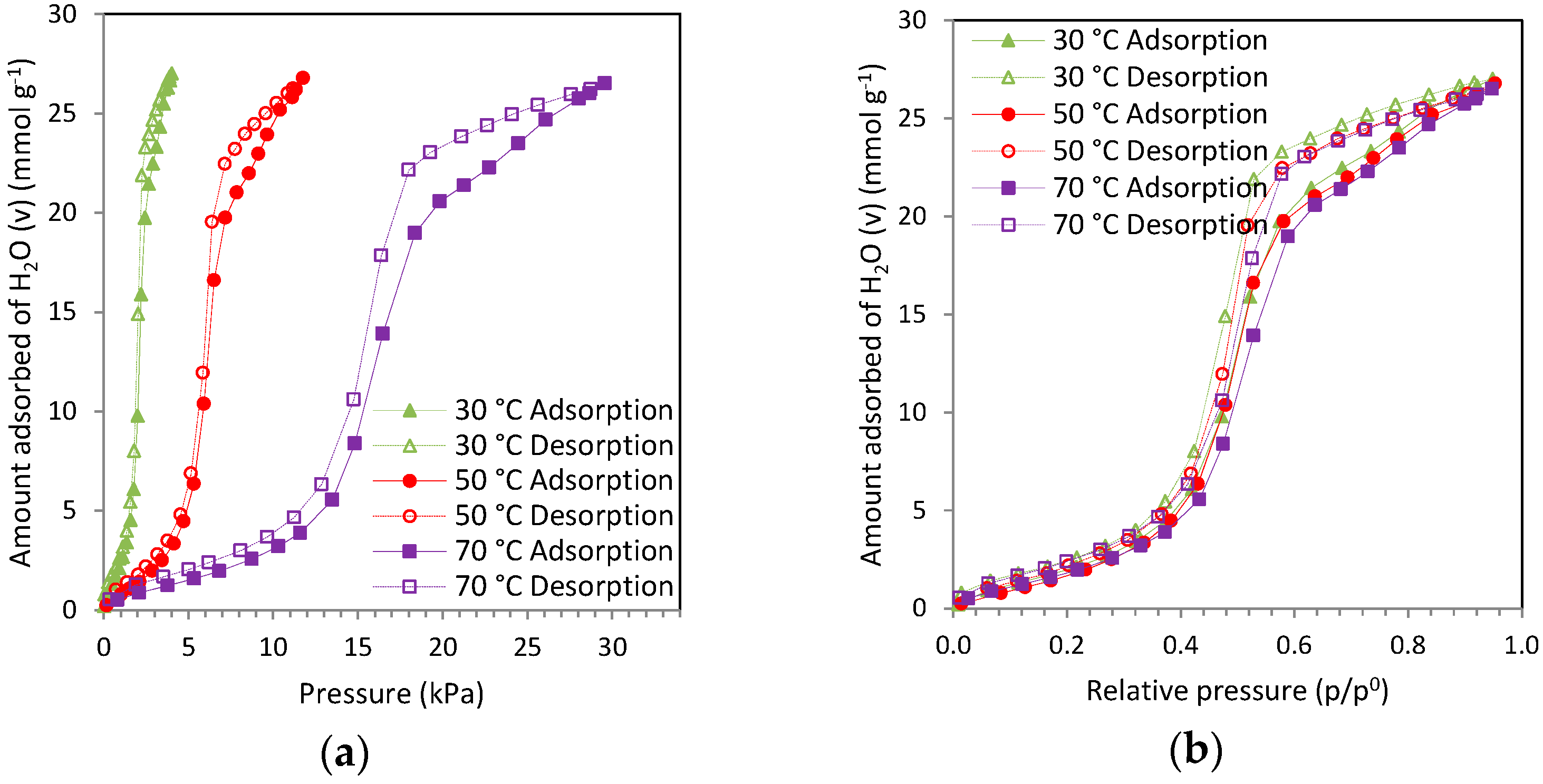
The sorption of water in the pores of activated carbons is known to be mediated by surface chemistry. Several semi-empirical and theoretical water adsorption models incorporating the role of surface chemistry can be found in the literature. Typically these models can predict the concentration of surface functional groups, the molecular size of water clusters, and the water adsorption capacity in the micropores as well as on the surface functional groups, and also estimate equilibrium rate constants. Several of these models such as the Dubinin-Serpinsky (DS) equation [
12], the Dubinin-Astakhov (DA) equation [
13], and the Do-Jumpirom-Do (DJD) equation [
14] were used in this work to describe water vapor adsorption on the carbons at 30 °C.
3.3.1. Dubinin-Serpinsky (DS)
Water adsorption isotherms have been fitted to the Dubinin-Serpinsky (DS) model [
12] (see
Supplementary Materials for clarification on the notation of Equation (1)).
The experimental data were fitted over a suitable range of
p/p0, usually from 0.3 to 0.6. The parameters
A1,
A2,
and A3 of the DS model were adjusted to minimize the sum of square residuals between the experimental adsorption data and the values calculated using DS at the adsorption temperature. As an example, the experimental data and model approximation for RN2, RN2D5, and RN2D10 carbons are shown in
Figure S6 in the Supplementary Materials.
Each sample exhibits a region where the quadratic fitting of the DS equation applies and so the parameter a0 for the carbons studied can be estimated.
Table 6 reports the DS parameter values estimated for the carbons. The model fits reasonably well the experimental data up to intermediate pressures but it should be taken into account that in general, the DS equation is restricted to describe the initial region of the water vapor isotherm (0 <
p/p0 < 0.3).
The number of primary centers, represented by a0, decreases after all the post-treatments, either because of the loss of oxygen surface groups due to heat treatment or due to the unavailability of these groups for water adsorption owing to the acid wash or wet impregnation procedures.
This trend is consistent with the TPD and FTIR analysis for several samples. Nevertheless RN1A shows a lower content of oxygen surface functional groups than that obtained from these techniques. As we have explained above, this clearly suggests that not all of the oxygen content of the samples is available for H
2O adsorption (see
Figure 3).
As suggested by Dubinin,
a0.6 represents the amount of water adsorbed near
p/p0= 0.6 and corresponds in many cases to the monolayer covering the walls of the micropores. Since
a0.6 is close to the true surface of these pores (not to be confused with the total micropore volume W
0), the ratio
a0/
a0.6 represents the fraction of the surface occupied by the primary centers. This ratio, rather than
a0 alone, is a useful parameter for the characterization and comparison of different activated carbons [
41].
It is noteworthy that for the impregnated samples the ratio a0/a0.6 remains constant with respect to the parent sample. Thus the reduction of the number of oxygen functional groups available to water vapor adsorption and the total adsorption capacity decrease in the same way independently of the amine loading for the impregnation, maintaining the proportion of the parent sample.
The DS equation might not be the most suitable model for a detailed description of water adsorption in microporous carbon adsorbents over the whole relative pressure range. Nevertheless, the DS equation showed a fair description of the adsorption of water vapor at low relative pressures thus providing information about the surface oxygen functionalities on the carbons.
3.3.2. Dubinin-Astakhov (DA)
Water adsorption isotherms were fitted to the Dubinin-Astakhov (DA) equation [
13].
The experimental data were fitted over a suitable range of p/p0; usually from 0.4 to 0.95. The optimum value of n for the DA equation was calculated by linear regression and selected as the value of n that minimizes the residual sum of squares.
As an example, the experimental data and model approximation for RN2, RN2D5, and RN2D10 carbons are shown in
Figure S7 in the Supplementary Materials. The model fits reasonably well the experimental data in the pressure range evaluated.
Table 7 reports the estimated DA parameters for all the studied carbons.
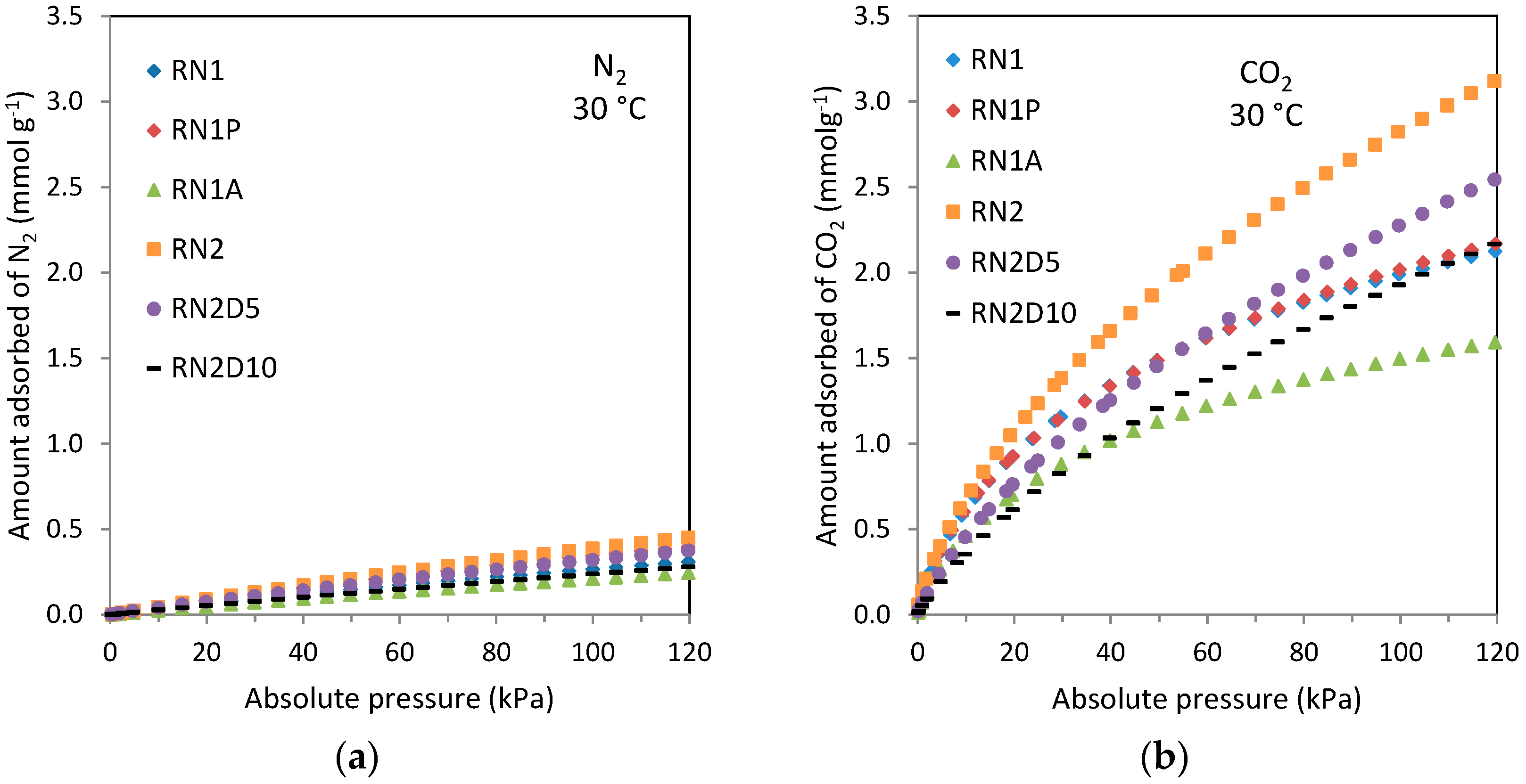
Estimation of the micropore volume by means of water vapor adsorption together with the liquid water density generally yields lower values than when estimated from other gaseous adsorbates (e.g., nitrogen or carbon dioxide). In such a confined space, molecular packing is not as effective as bulk liquid water. Iiyama
et al. showed by an X-ray diffraction technique in a slit-shaped carbon nanospace that the adsorbed water has a more ordered structure. This is thought to be an ice-like structure, with a lower density than liquid water [
42].
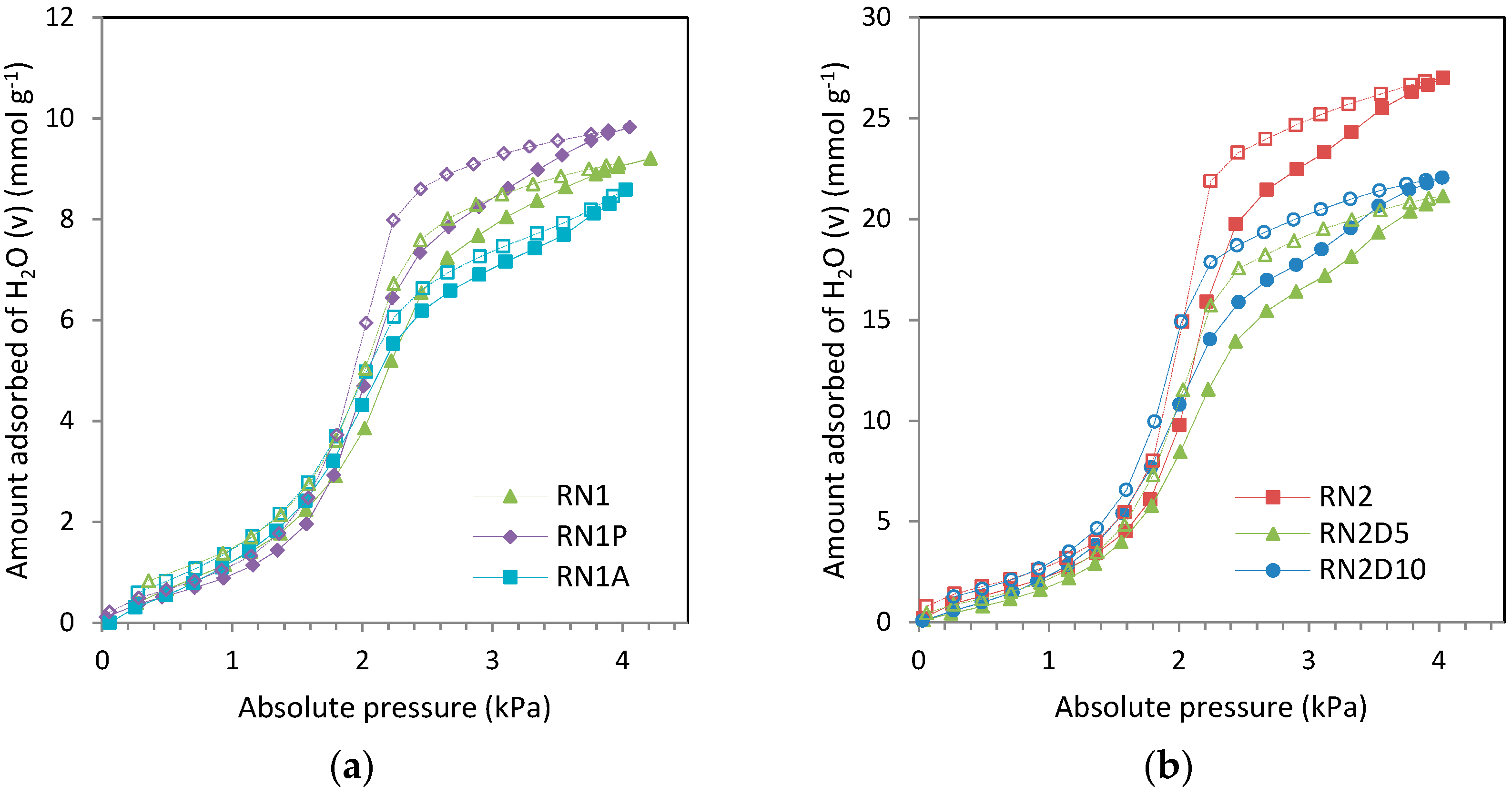
In spite of that, the micropore volume obtained for RN1P was higher (0.17 cm−3·g−1) than that calculated from nitrogen adsorption (0.11 cm−3·g−1). Depending on the pore shape and size, the packing of water molecules may not be as effective as that of gases because of the requirement of a correct orientation for hydrogen bonding. As a result, water molecules occupy only a fraction of the micropore volume while gases, such as argon, can occupy effectively the whole volume. In rare cases where micropores are very small, water might access due to its smaller size while argon or nitrogen could not.
Moreover although the estimated micropore volume of RN1 is lower than that reported in the textural characterization, the widening of the pores by means of the heat treatment leads to higher water adsorption on RN1P thereby obtaining an overestimated micropore volume.
In the case of the E parameter, the data presented in this work suggest that no direct correlation exists between E and the classical characteristic energy, E0, associated with type I isotherms. The highest value is registered for RN1P (3.6 kJ·mol−1) and may be ascribed to the volume of micropores of the sample (W0,N2 = 0.11). This could be explained by the fast filling of the micropores that leads to the cooperative adsorption of clusters in the form of multilayer on the carbon surface. These clusters of water act as secondary active centers by means of hydrogen bonding and increase the value of E due to their evident affinity. The lower values were obtained for the aminated samples (about 2–3 kJ·mol−1) owing to the presence of amine groups that diminish the number of oxygen surface functional groups besides blocking the entrance to the micropores.
Even though water adsorption on the carbons is of type V, the DA equation was insufficient to describe experimental data at low relative pressures. This is because the DA equation only describes the volume filling of micropores at medium and high pressures and does not take into account the presence of surface oxygen functional groups responsible of water vapor adsorption at low pressures.
3.3.3. Do-Junpirom-Do (DJD)
Water adsorption isotherms were fitted to the Do-Junpirom-Do (DJD) equation [
14].
The parameters
S0,
Kf,
αμ,
Cμs, and
Kμ of the DJD model were adjusted to minimize the sum of square residuals between the experimental adsorption data and the values calculated using Equation (3) at the adsorption temperature. The portion of the isotherm corresponding to the filling of the micropores (
p/p0 > 0.2) was first fitted to obtain
Cμs, and
Kμ; then, a new optimization was run departing from these preliminary adjusted parameters by fitting all the parameters simultaneously using the total sum of the square residuals of adsorption as the objective function. The value of
m was set to
m =
αμ +
1. The relaxation equilibrium constant for water desorption from the micropores,
KRμ, was fitted to minimize the sum of square residuals between the experimental desorption data and the values calculated using Equation (4). In order to fit the parent activated carbons, RN1 and RN2, previous results optimized by our research group were used and these values were applied as initial in the fitting of the treated samples [
15,
39].
Heat treatment reduces the number of oxygen surface functional groups in sample RN1P, leads to a decrease in the micropore volume of the parent carbon (see
Table 2) but increases the total water vapor uptake of RN1P. Therefore the parameters
S0 and
Cμs vary for the heat treated sample RN1P with respect to the parent RN1. Moreover
Cμs was set to be greater or equal to the physical upper limit (6.08 mmol·g
−1) which is calculated assuming that all the micropore volume is completely full of liquid water at the adsorption temperature.
In the case of the acid treated sample RN1A and the aminated samples RN2D5 and RN2D10 the values of Cμs optimized for the parent activated carbons were set as constants in the fitting. Although these samples present a reduction in microporosity with respect to the parent carbons, their performance in water vapor adsorption only seems to be affected in the pressure range assigned to the contribution of oxygen surface functional groups where they show a decrease in the total water vapor uptake capacity. However, the water vapor uptake corresponding to the filling of micropores remains unaltered with respect to the parent samples. Thus only the parameter S0 is considered to be affected by the effect of these post-treatments.
The values of the DJD model parameters optimized following the aforementioned criteria can be found in
Table 8.
It can be observed that the DJD model (Equation (3)) describes adequately the adsorption branch of the isotherms in the entire pressure range at the evaluated temperature. However, the DJD model for desorption (Equation (4)) presents greater deviations from the experimental desorption data: it underestimates the uptake at high pressures, particularly for RN2, which corresponds to the emptying of the widest micropores. This seems to be an intrinsic limitation of the model. Besides, only one parameter (KRμ) was fitted using the experimental desorption data; the rest of the model parameters (S0, Kf, αμ, Cμs, and Kμ) were fitted to minimize the sum of square residuals between the experimental adsorption data and the adsorption model (Equation (3)).
The values obtained for
S0 represent between 7% and 30% of the total oxygen content of the carbons, which was to be expected, as not all of the oxygen is available for H
2O adsorption. The critical size of the water cluster to enter the micropores,
αμ, is related to the micropore size and also varies with the concentration of functional groups; when their amount is large the water clusters are small and
vice versa. This is due to the functional groups that can contribute to stabilize smaller clusters, allowing their confinement within the micropores [
14].
Regarding the parent activated carbons, the value of
α = 6 obtained for RN1 is relatively low, which is attributed to the narrow size of the micropores of this carbon. In fact, this value is lower than that obtained for RN2 which is produced from the same precursor by activation with CO
2 and presents slightly higher oxygen content but wider micropores (see
Table 2).
For parent carbon RN1, Cμs represents 57% of the physical upper limit (11.06 mmol·g−1). This packing fraction in the micropores is also significantly lower for RN1 than for RN2 (74%), due to the narrower pore size of the carbon activated in air.
Despite that the size of the water cluster for the samples with low content of oxygen surface functional groups should be larger than for the parent carbon, in the case of RN1P the value of α remains constant. For this carbon, the value of Cμs represents 100% of the physical upper limit, due to its poor microporosity development as previously commented within the DA section.
Acid treatment led to a decrease in the size of the water cluster (carbon RN1A) due to the presence of remaining chloride groups that could contribute to stabilize smaller clusters. The number of primary centers represented by S0 also decreases because of the unavailability of these groups for water vapor adsorption after this treatment.
Likewise, impregnation with amines followed the same trend as acid treatment: decrease in the size of the cluster due to the presence of amine that acts as oxygen surface functional group and stabilizes the clusters inside the pores. Moreover S0 decreases owing to the effect of the wet impregnation.
In order to describe the water vapor adsorption performance of the studied samples, the main advantages of the DJD model are that it provides a fair description of the adsorption branch in the entire relative pressure range and that it is based on the specific mechanism of water vapor adsorption on carbon materials.









{kind=link}
{kind=link}
{kind=link}
{kind=link}