
In Situ Soft X-ray Spectromicroscopy of Early Tricalcium Silicate Hydration

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
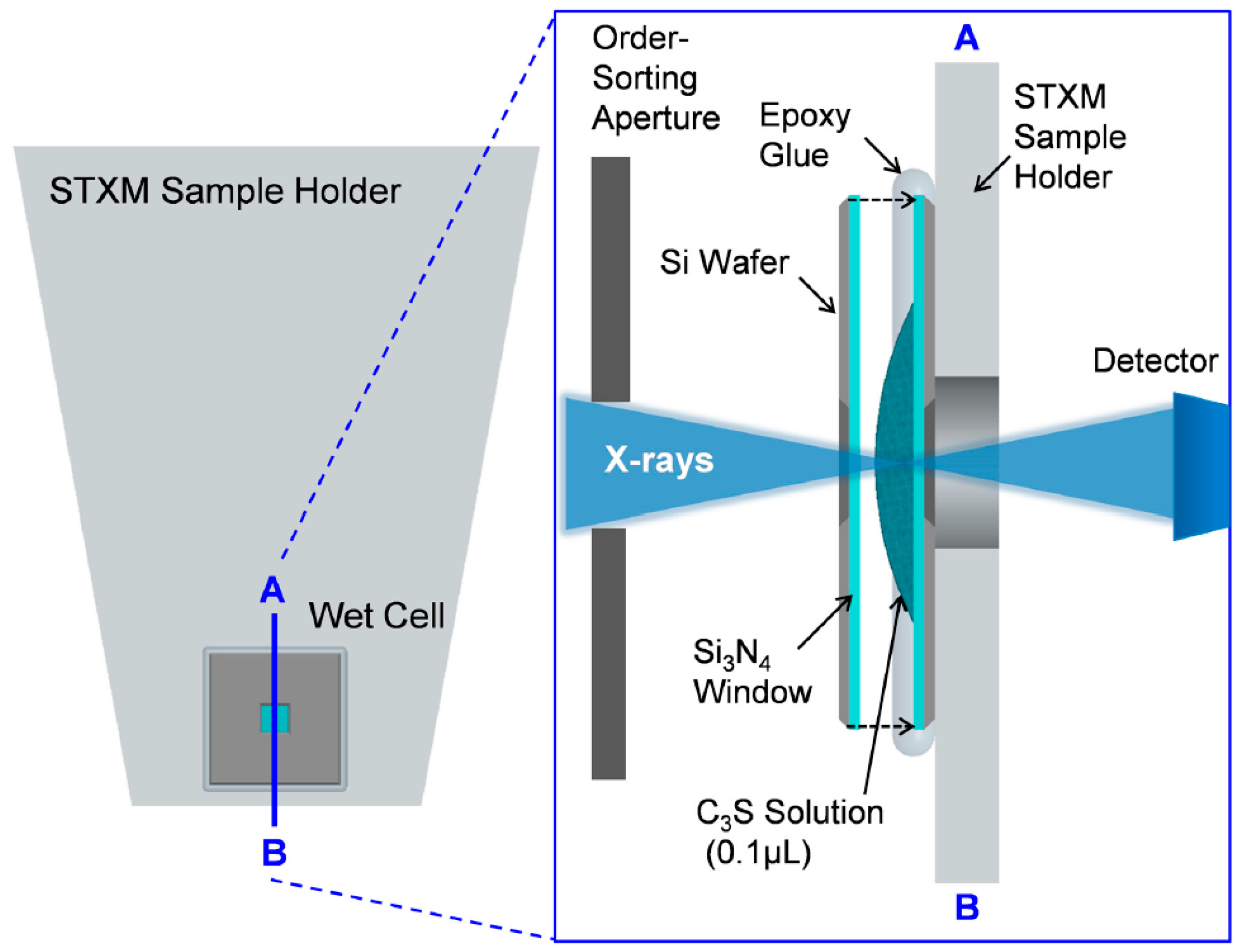
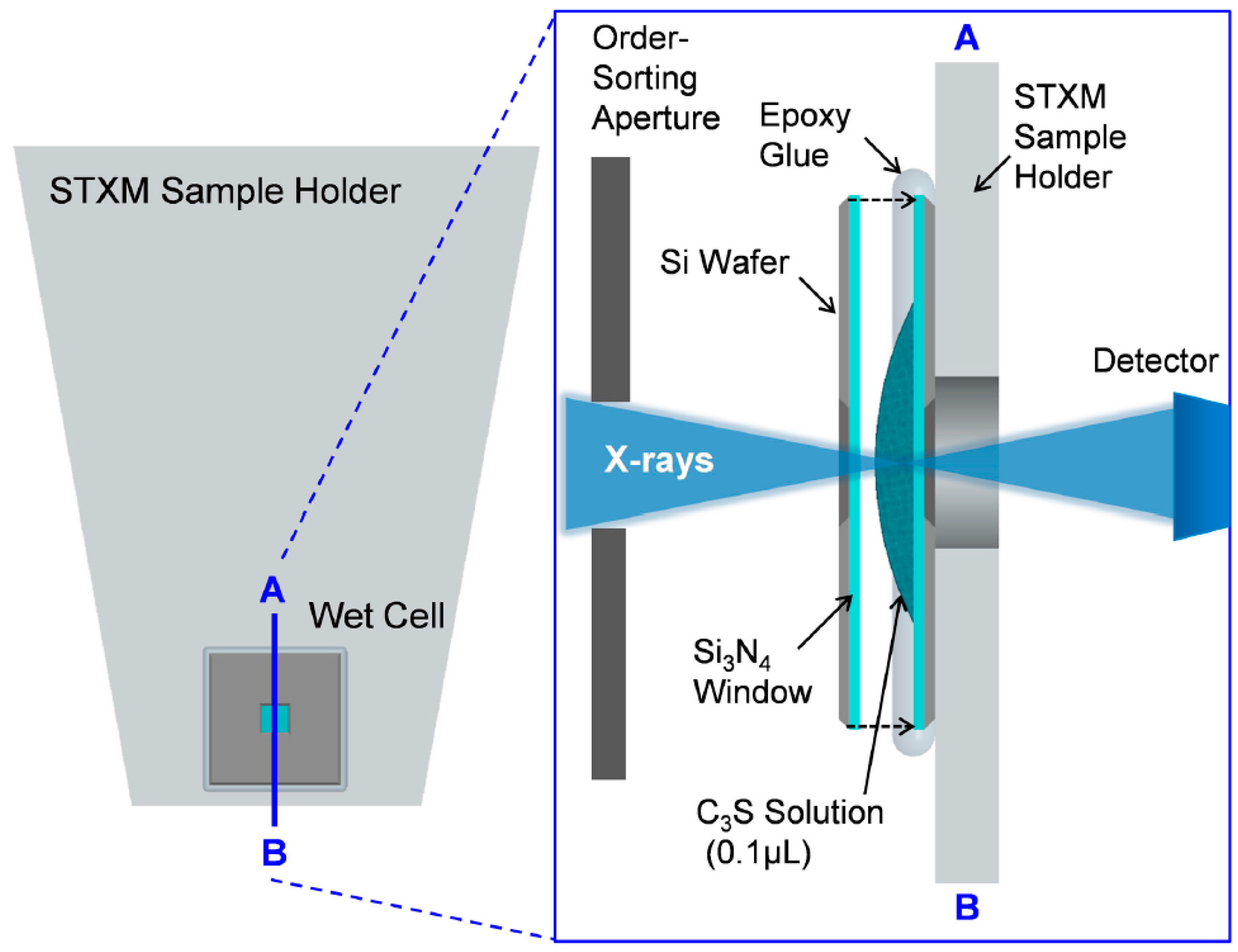
2.1. Wet Cell Production for In Situ STXM Measurement
2.2. Scanning Transmission X-ray Microscopy (STXM)
2.3. Isothermal Calorimetry
3. Results and Discussion
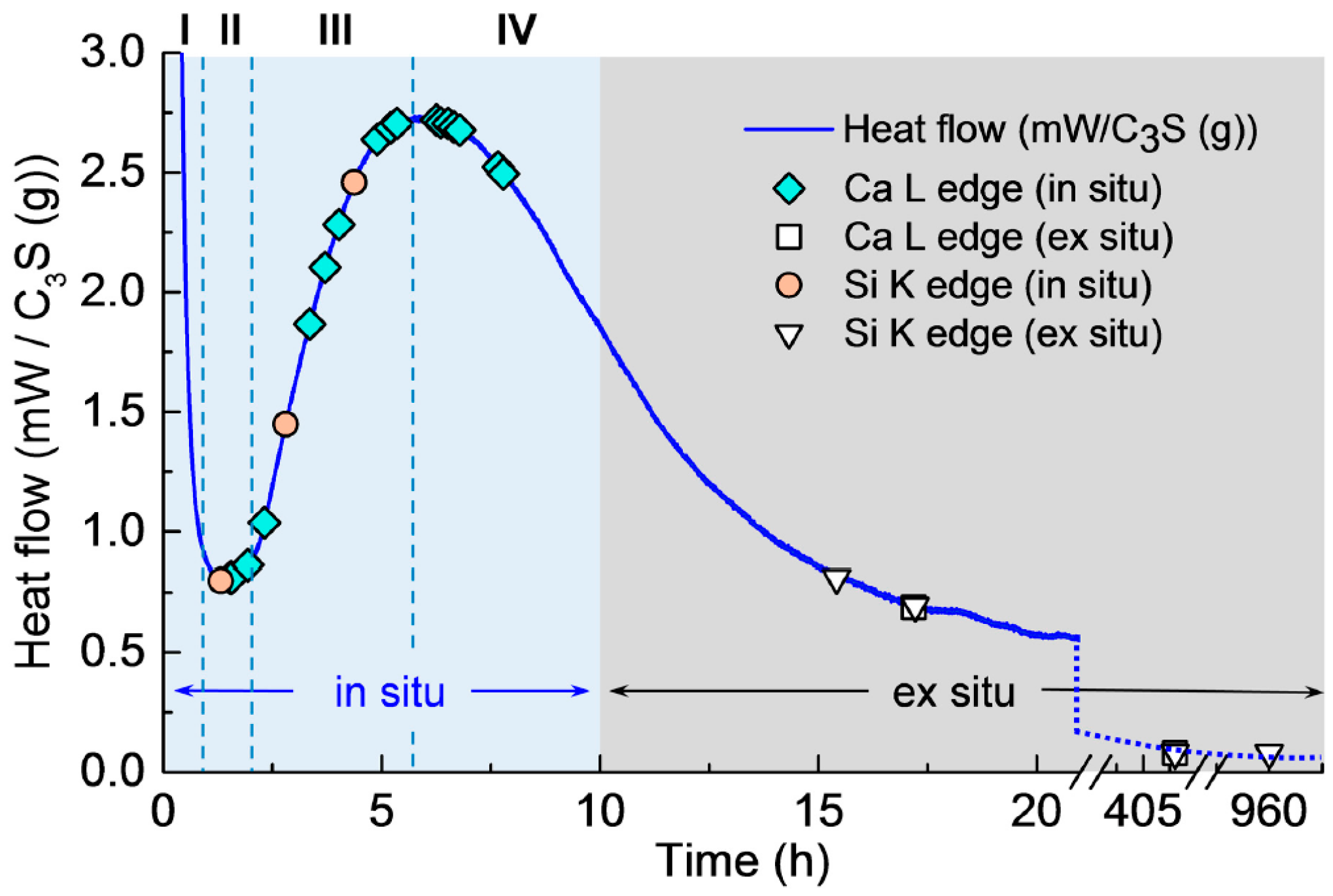
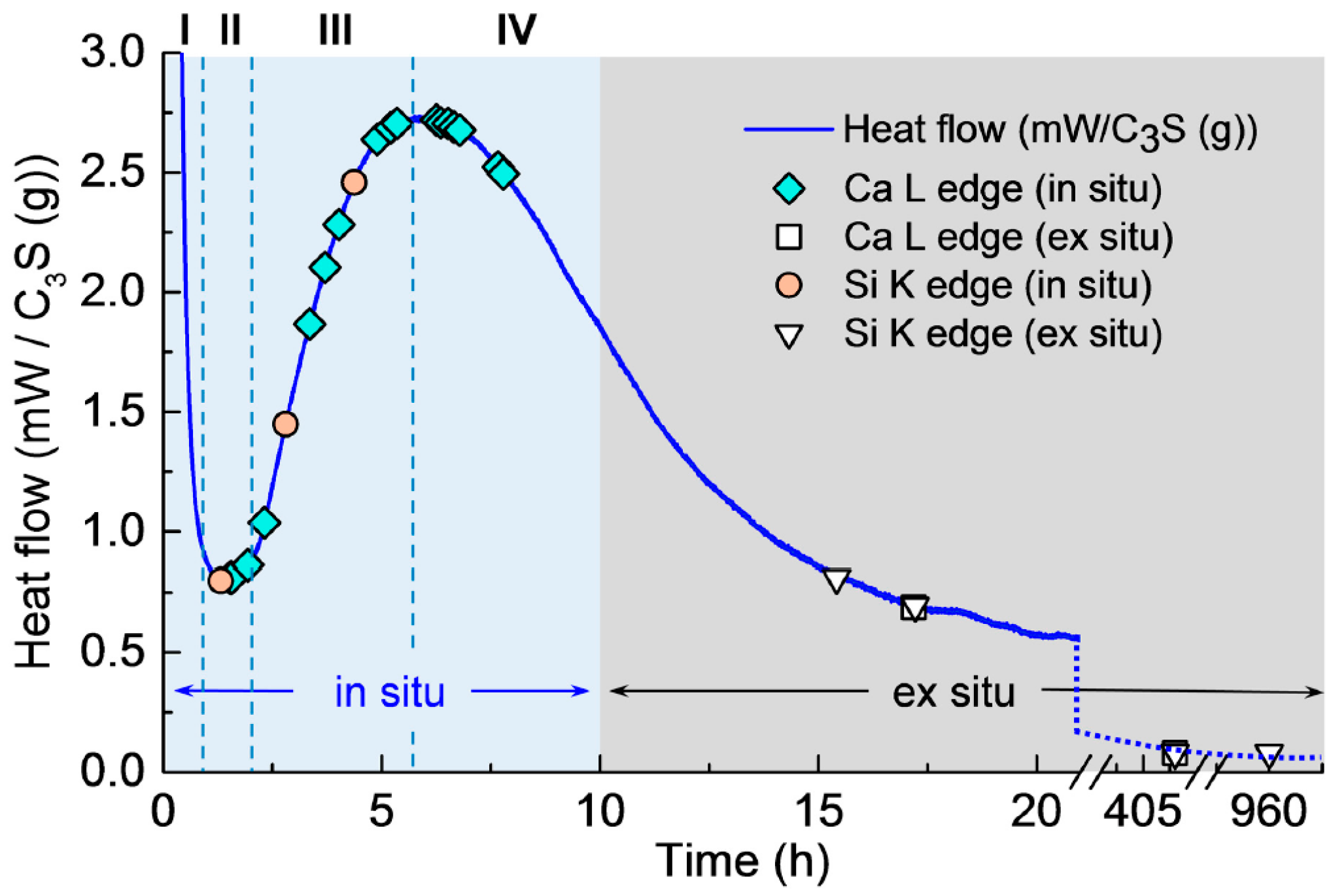
3.1. Isothermal Calorimetry
3.2. In Situ STXM at an Early Age of C3S Hydration
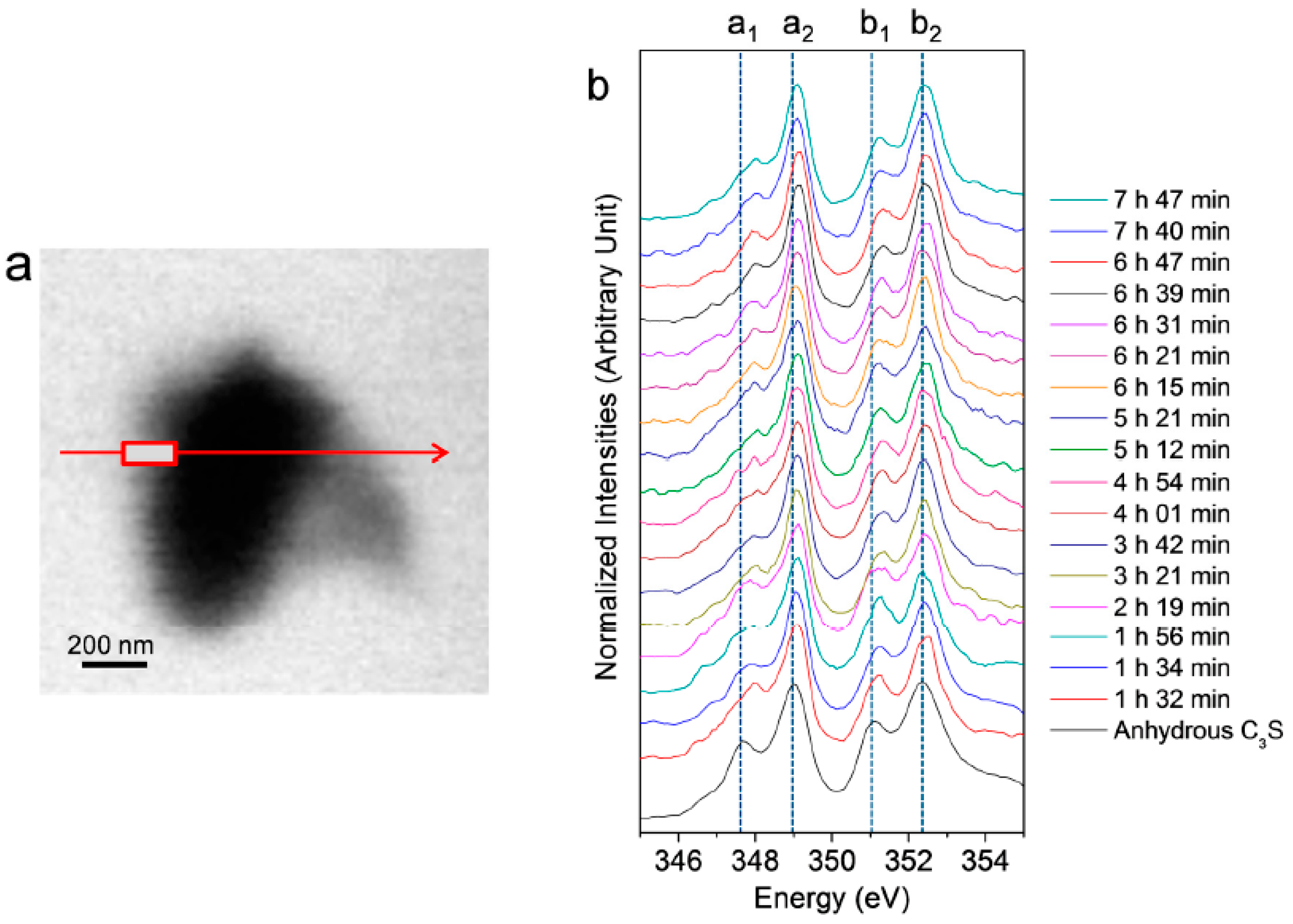
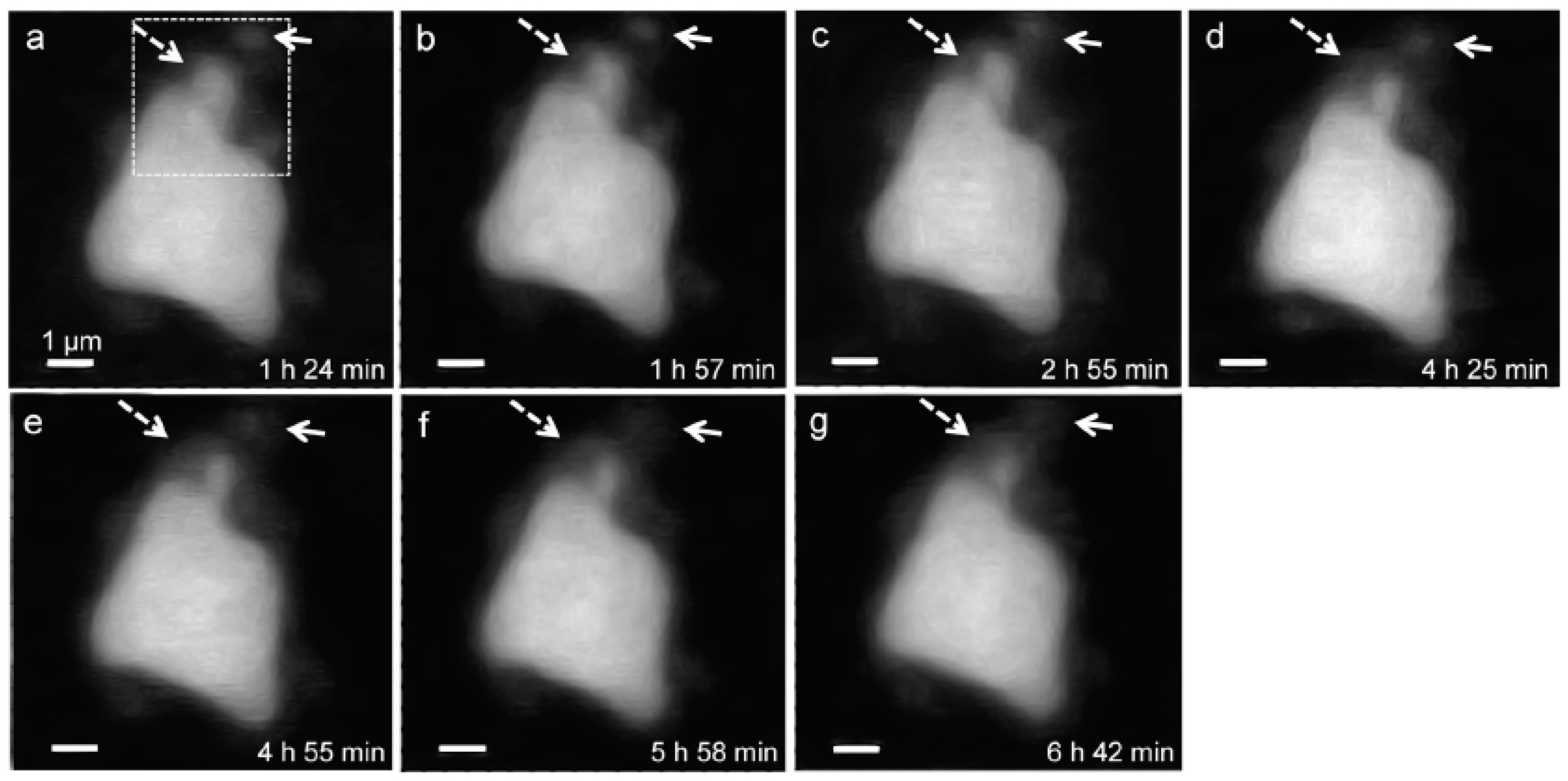
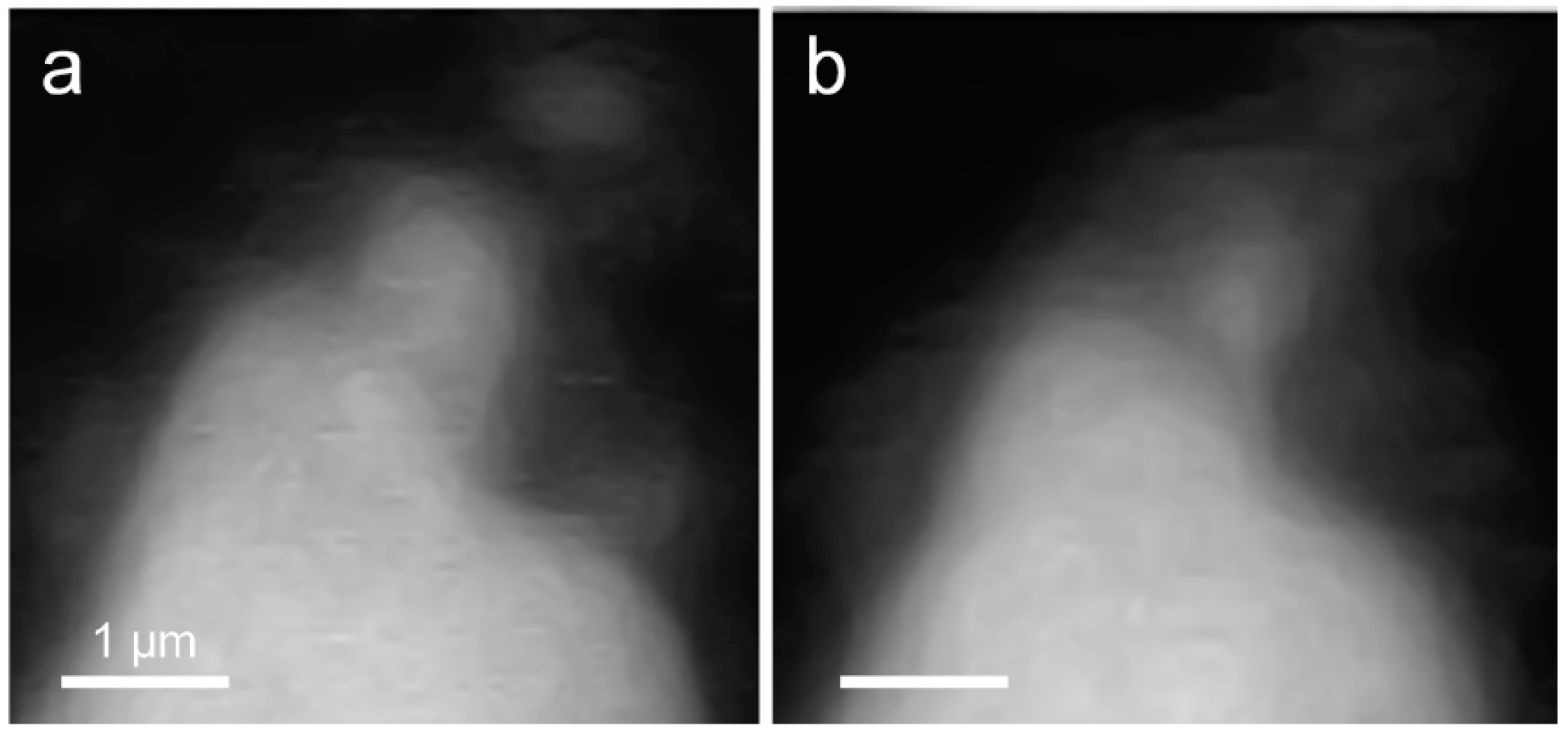
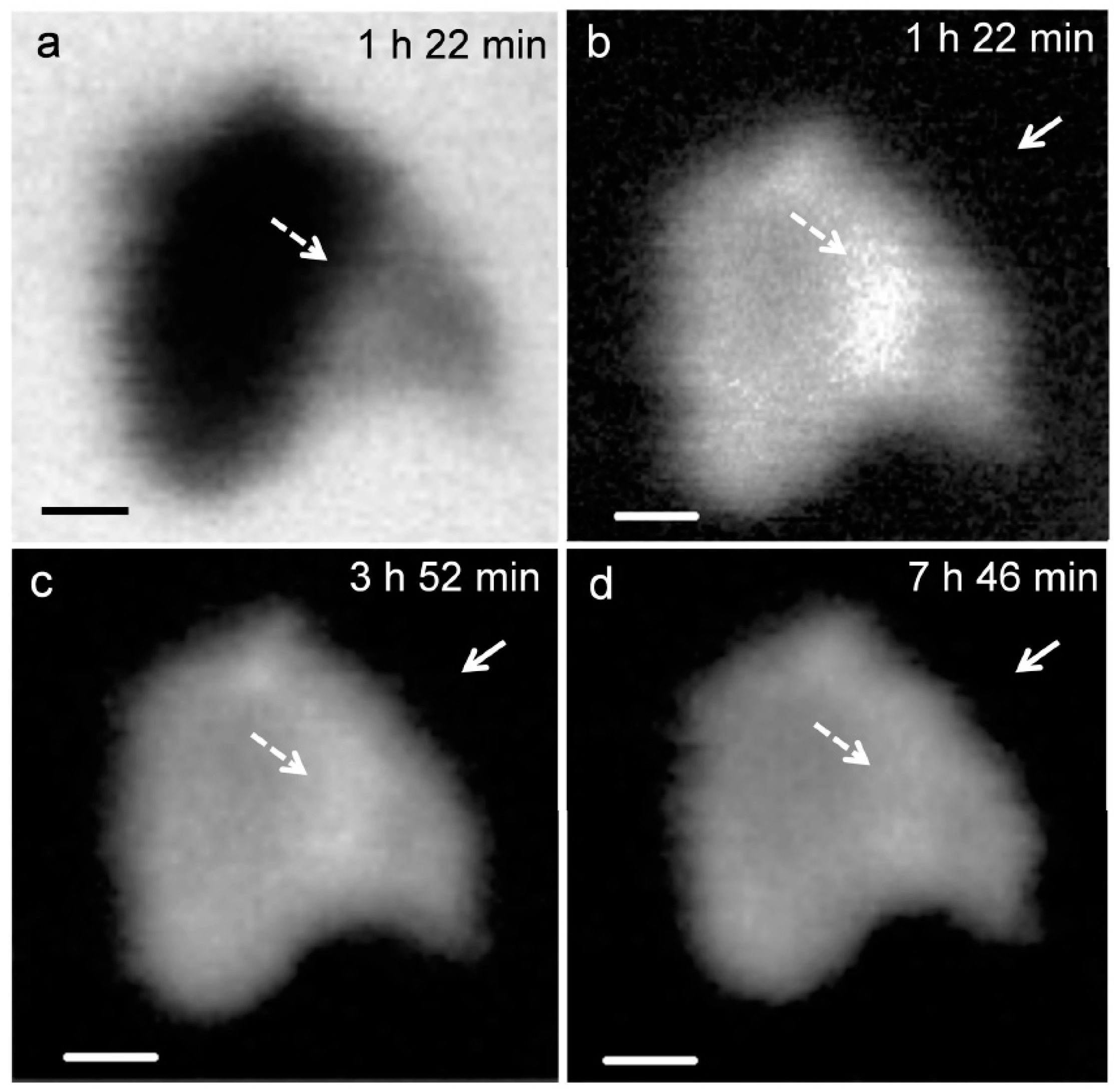
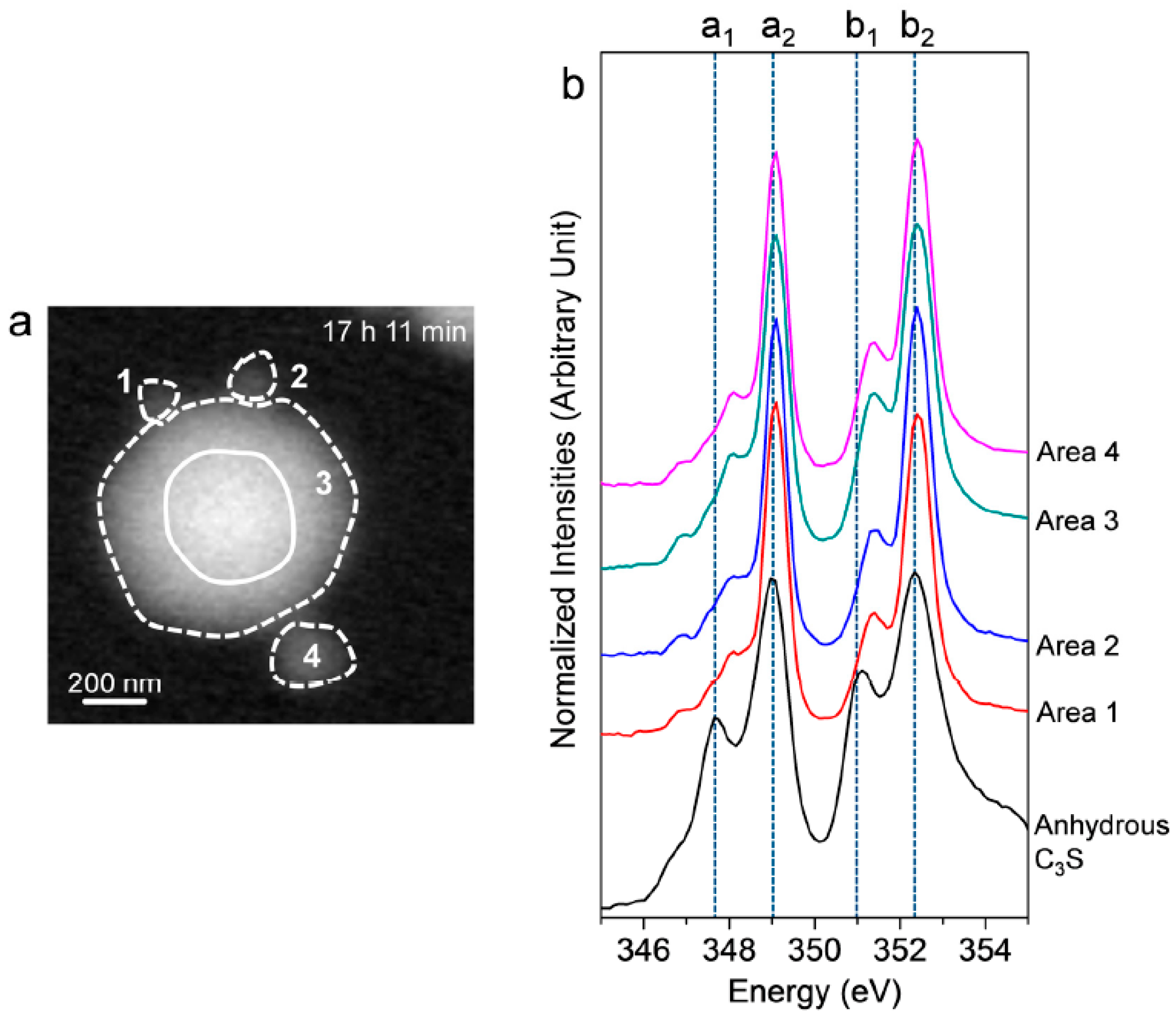
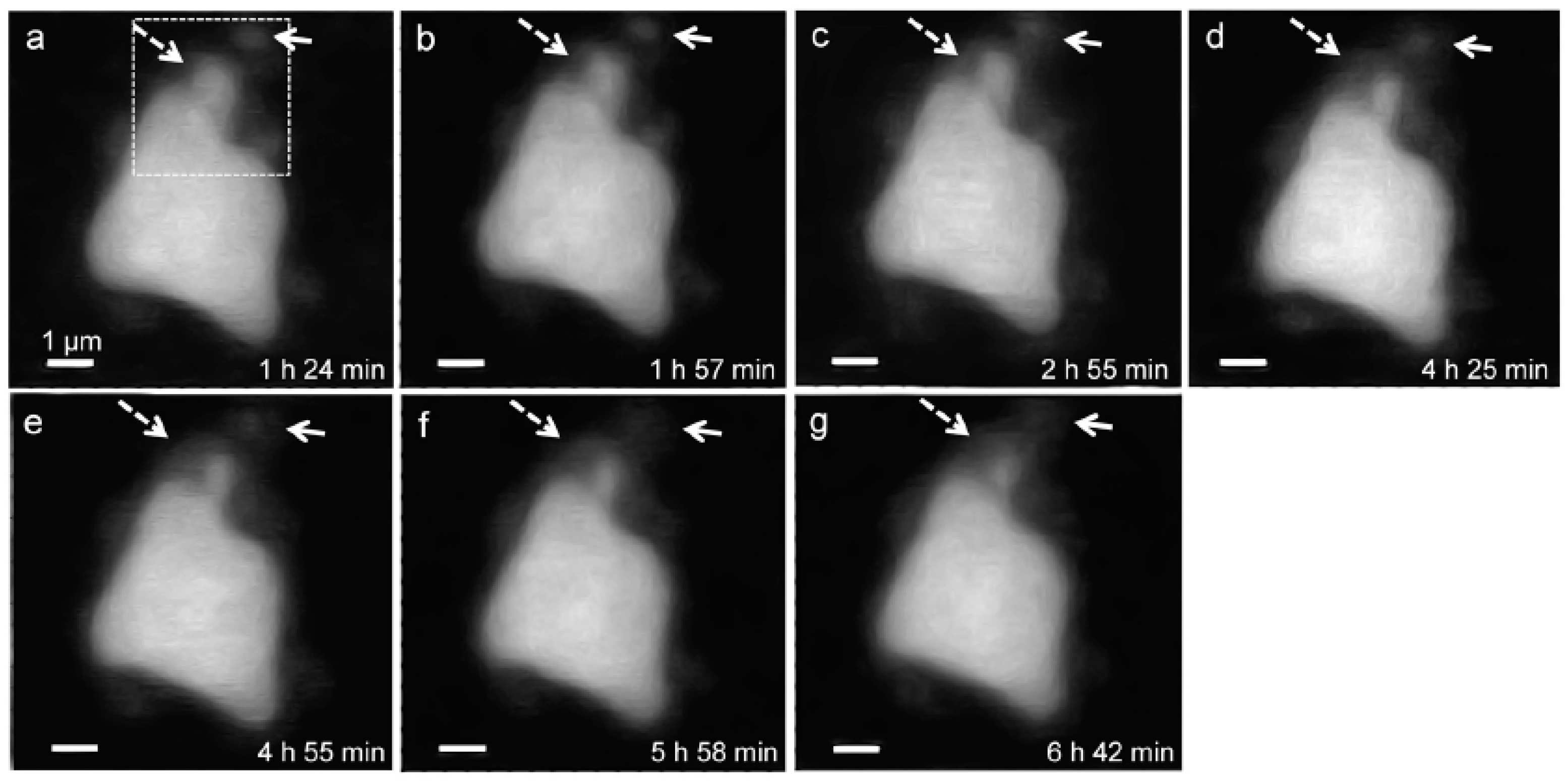
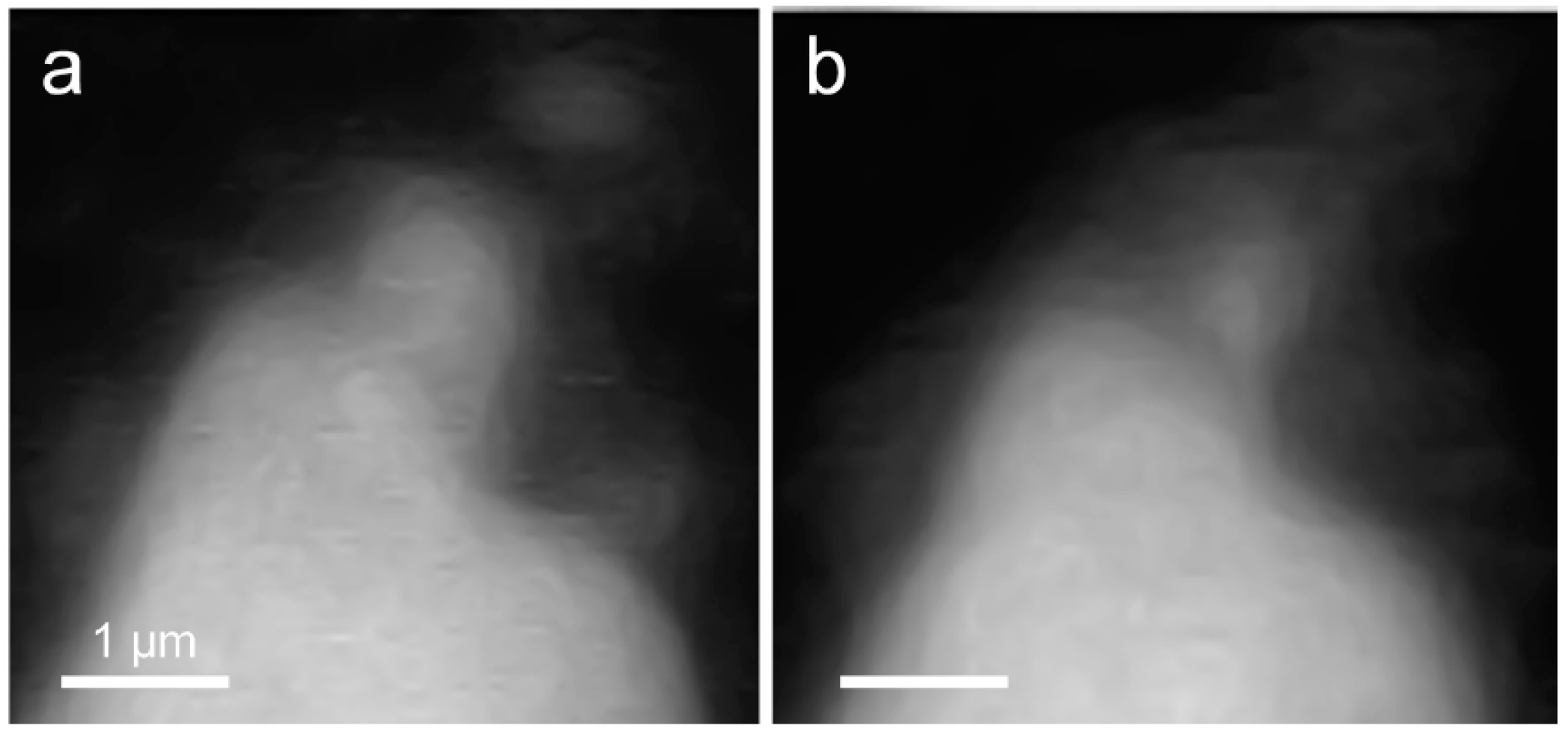
3.2.1. Morphological Observation of the C3S Hydration Process (Ca LIII,II Edge)
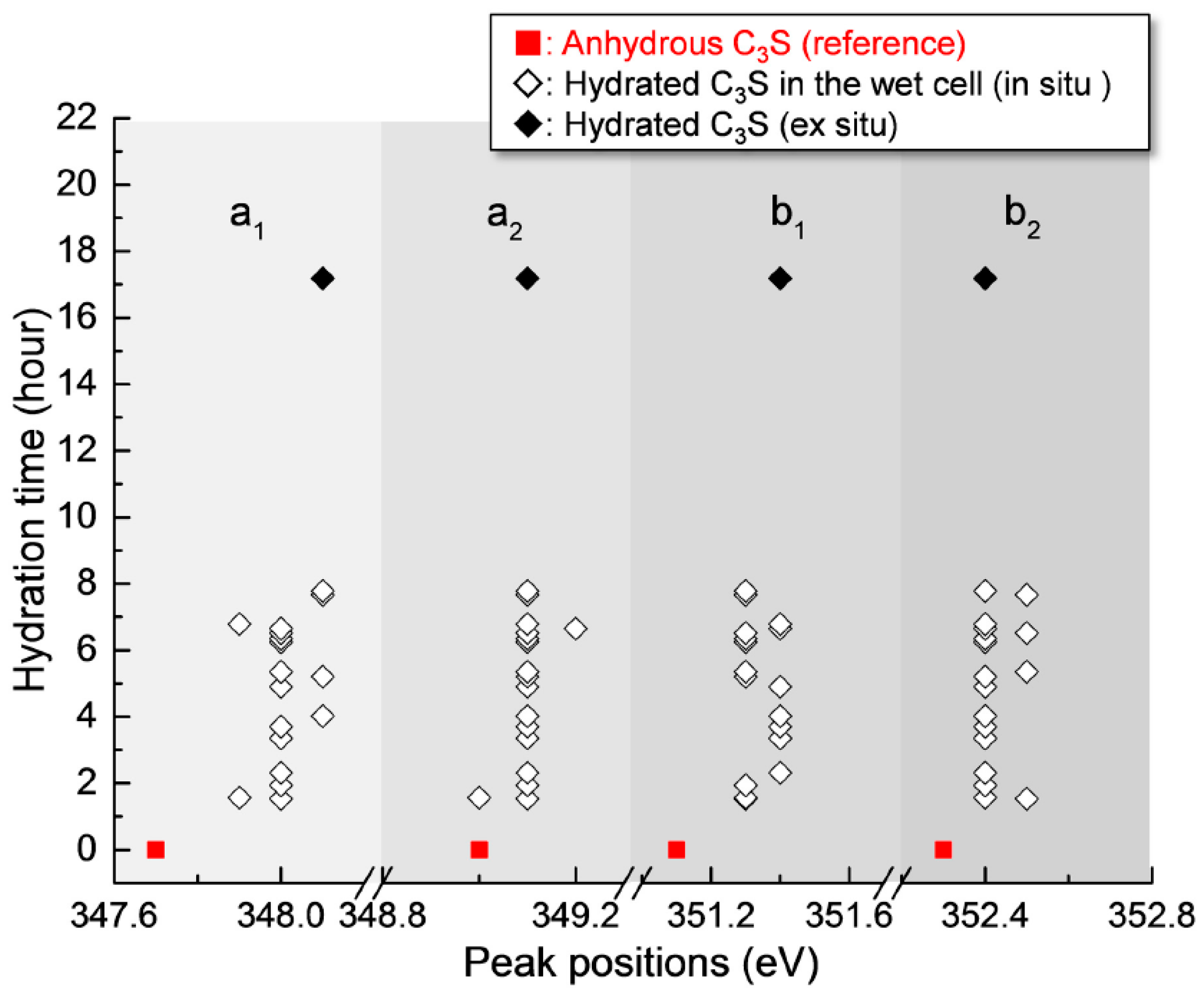
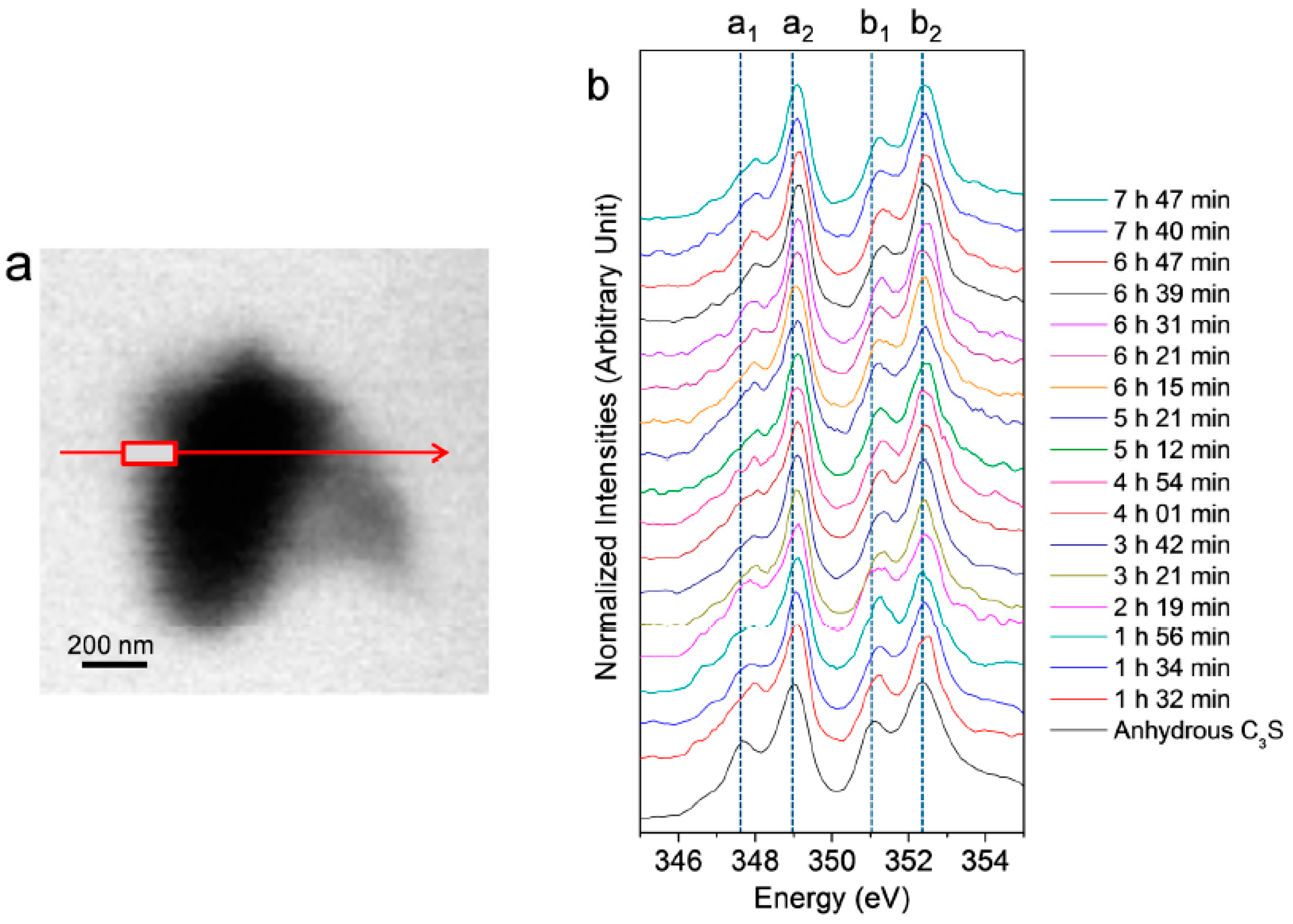
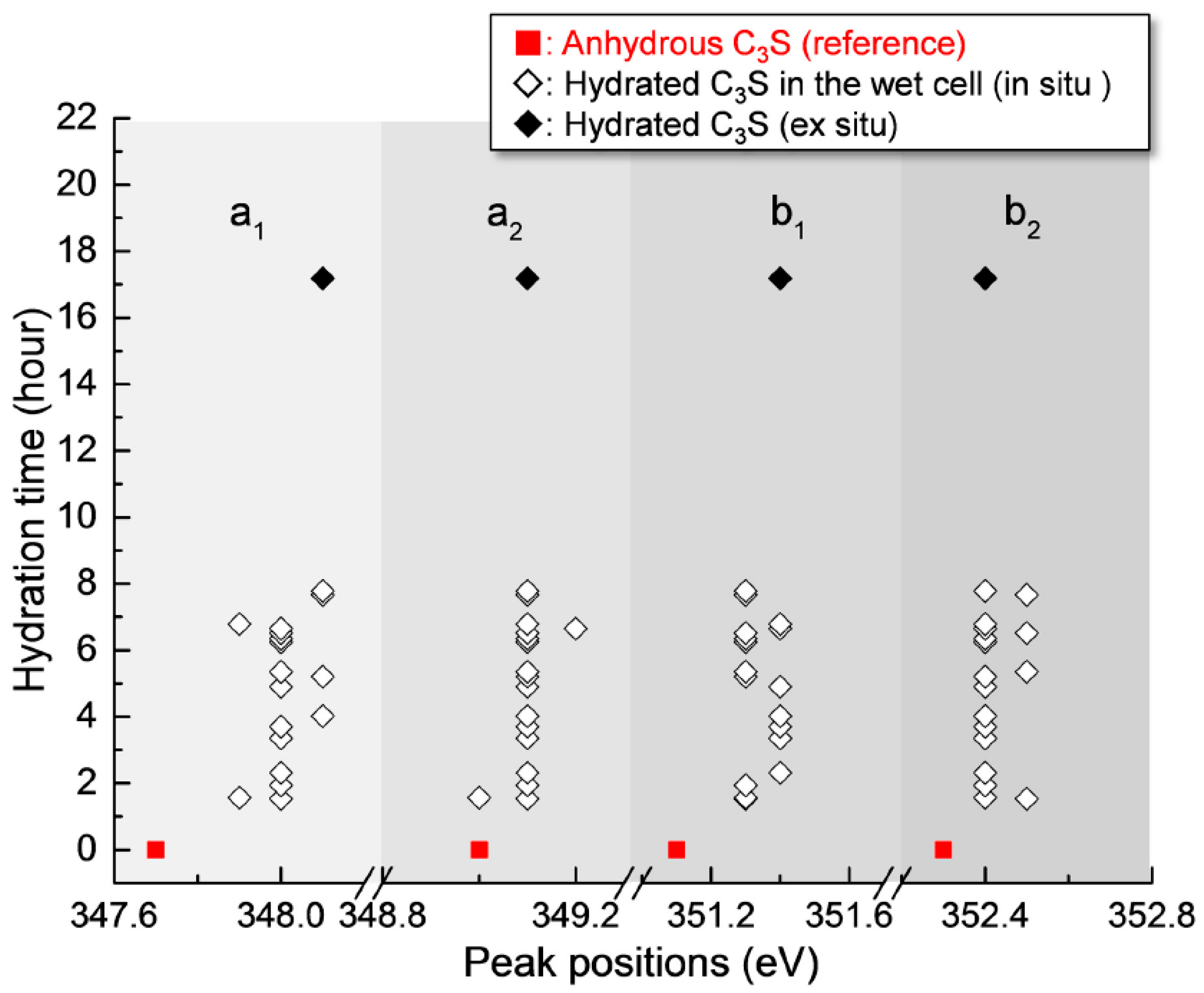
3.2.2. Ca LIII,II Edge NEXAFS Analysis
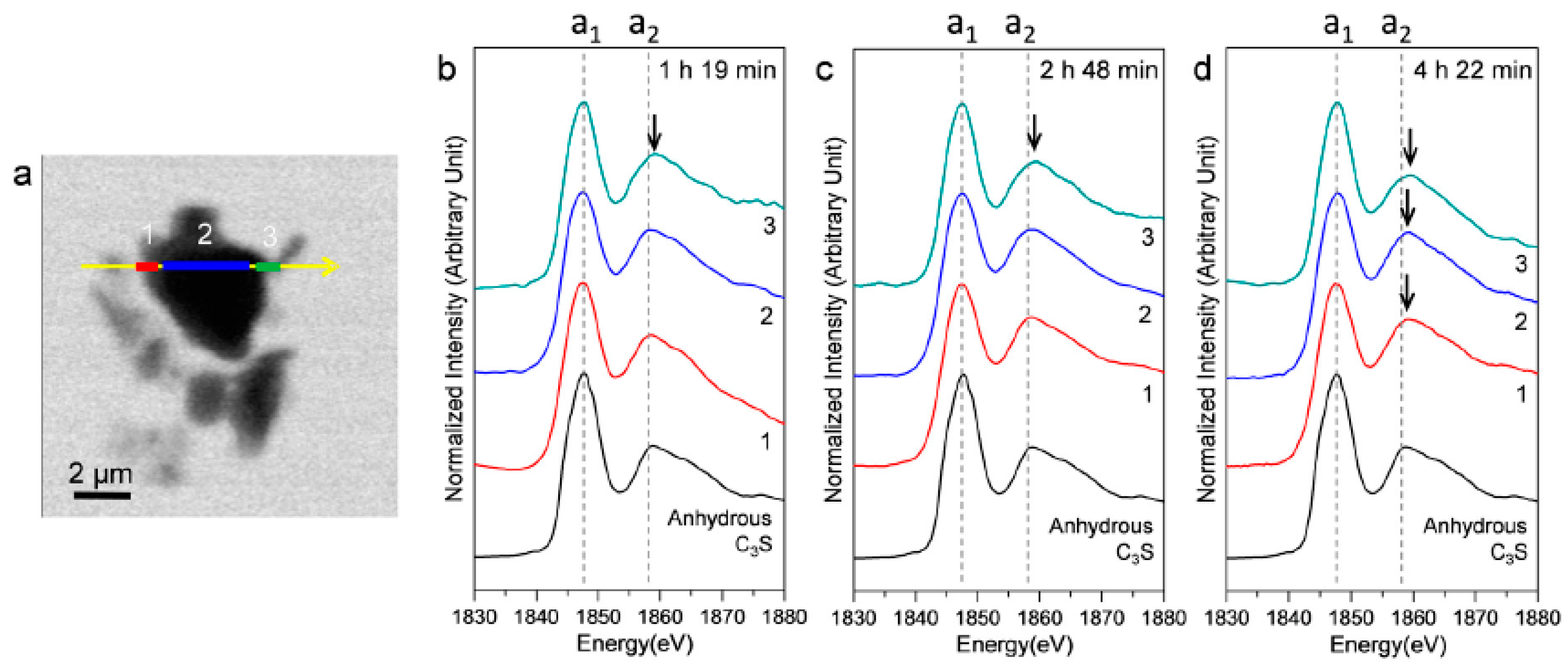
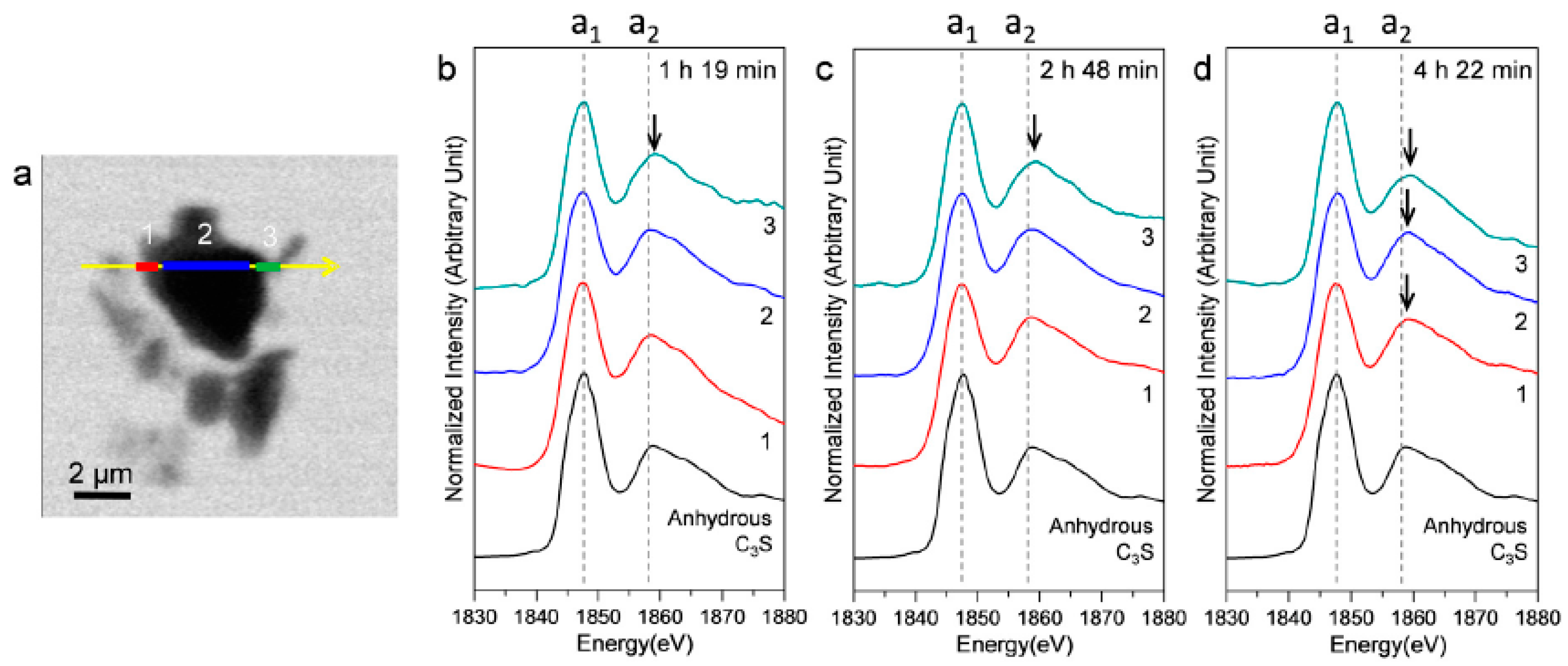
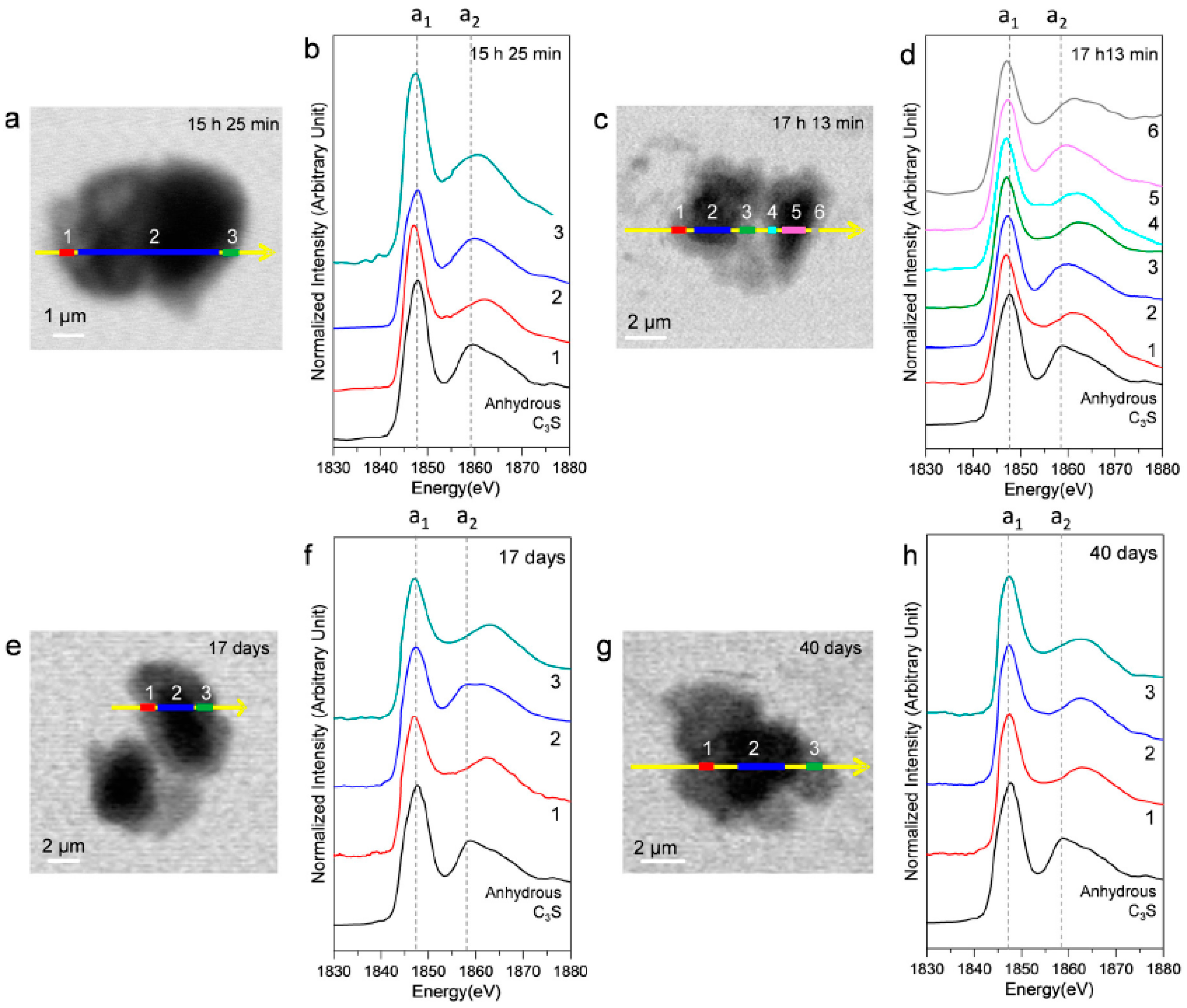
3.2.3. Morphological Observations of the C3S Hydration Process (Si K Edge)
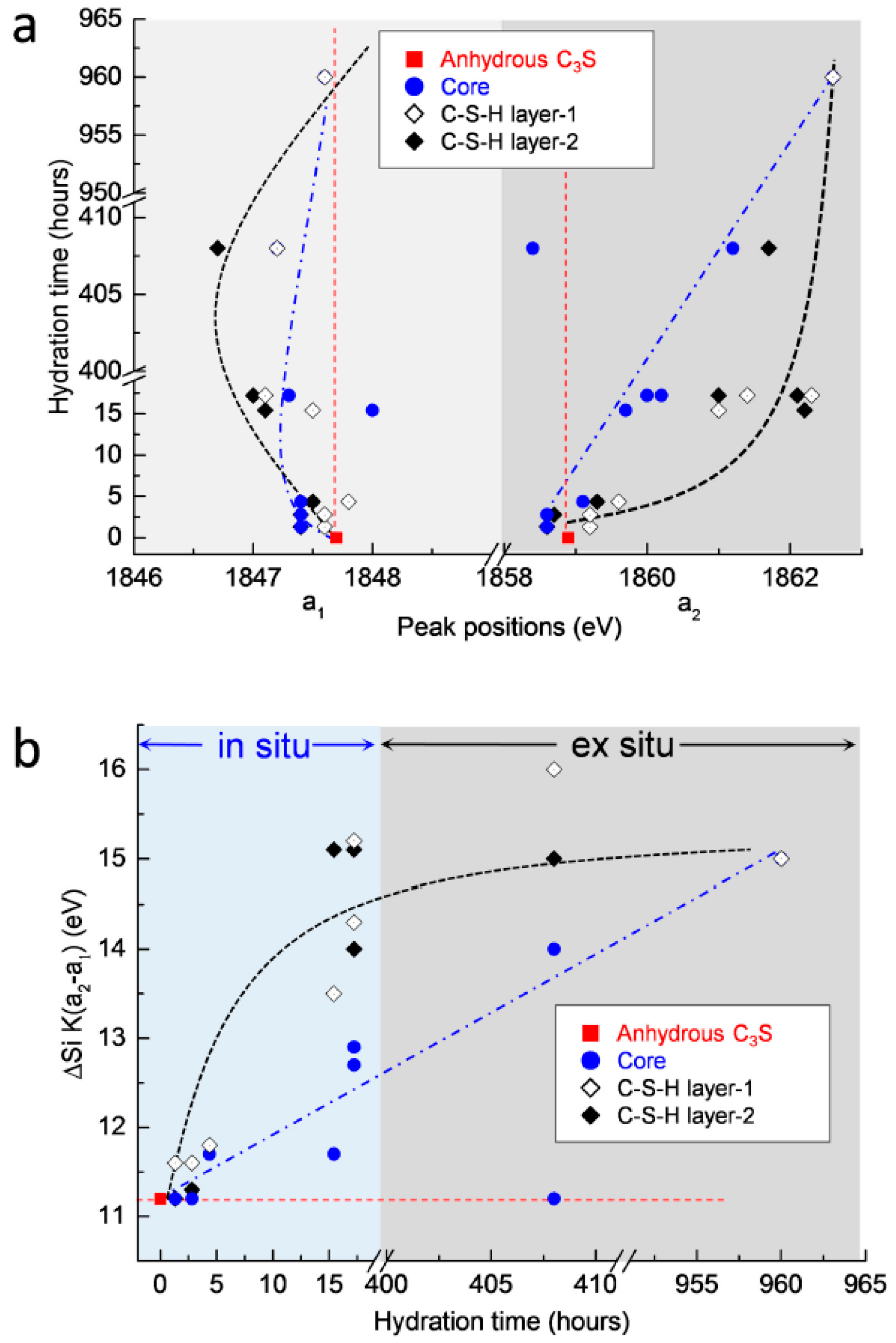
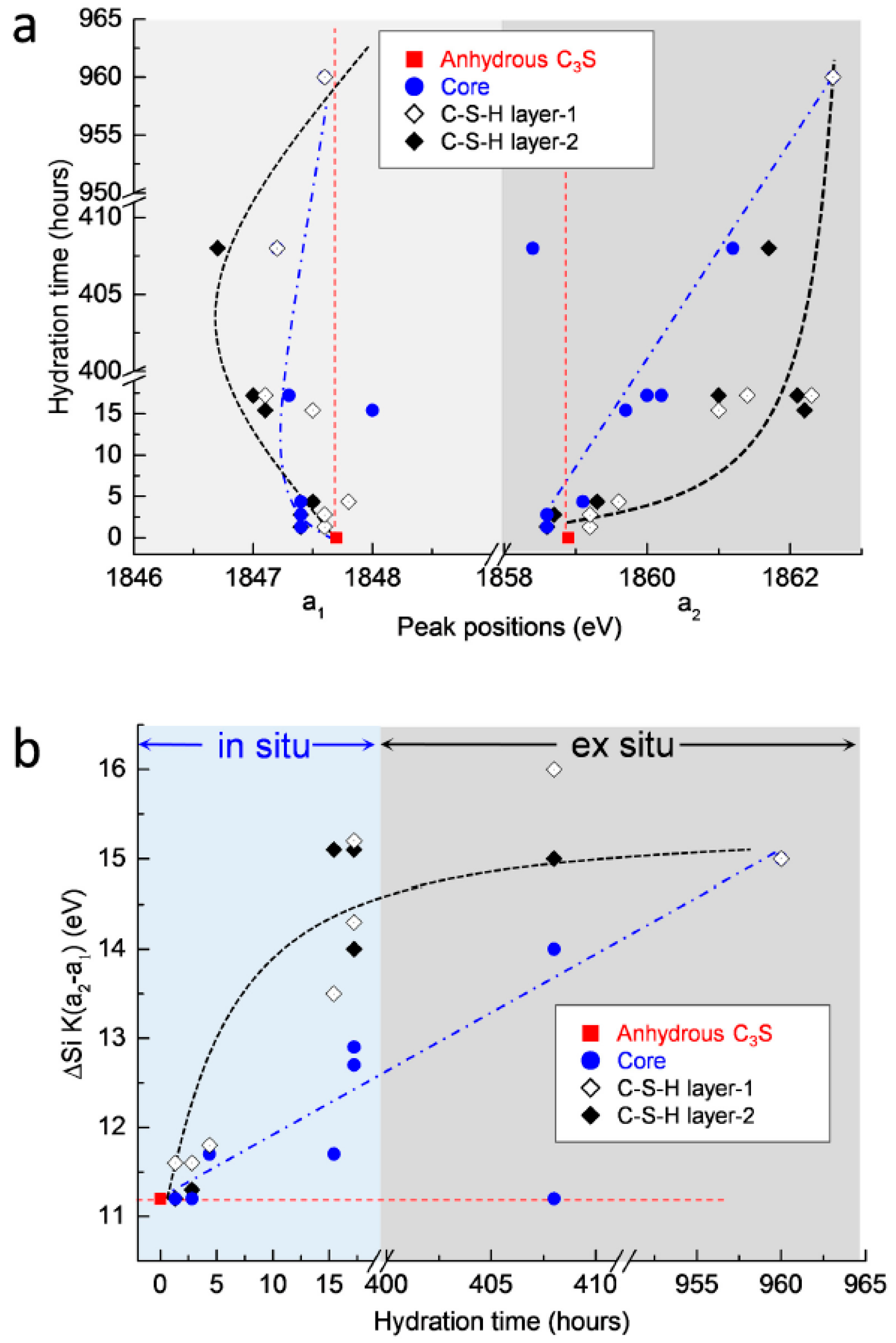
3.2.4. Si K Edge NEXAFS Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Alizadeh, R.; Raki, L.; Makar, J.M.; Beaudoin, J.J.; Moudrakovski, I. Hydration of tricalcium silicate in the presence of synthetic calcium-silicate-hydrate. J. Mater. Chem. 2009, 19, 7937–7946. [Google Scholar] [CrossRef]
- Thomas, J.J.; Jennings, H.M.; Chen, J.J. Influence of nucleation seeding on the hydration mechanisms of tricalcium silicate and cement. J. Phys. Chem. C 2009, 113, 4327–4334. [Google Scholar] [CrossRef]
- Richardson, I.G. The calcium silicate hydrates. Cem. Concr. Res. 2008, 38, 137–158. [Google Scholar] [CrossRef]
- Richardson, I.G. The nature of C–S–H in hardened cements. Cem. Concr. Res. 1999, 29, 1131–1147. [Google Scholar] [CrossRef]
- Bullard, J.W.; Jennings, H.M.; Livingston, R.A.; Nonat, A.; Scherer, G.W.; Schweitzer, J.S.; Scrivener, K.L.; Thomas, J.J. Mechanisms of cement hydration. Cem. Concr. Res. 2011, 41, 1208–1223. [Google Scholar] [CrossRef]
- FitzGerald, S.A.; Neumann, D.A.; Rush, J.J.; Bentz, D.P.; Livingston, R.A. In situ quasi-elastic neutron scattering study of the hydration of tricalcium silicate. Chem. Mater. 1998, 10, 397–402. [Google Scholar] [CrossRef]
- Scrivener, K.L.; Juilland, P.; Monteiro, P.J.M. Advances in understanding hydration of Portland cement. Cem. Concr. Res. 2015, 78, 38–56. [Google Scholar] [CrossRef]
- Nicoleau, L.; Nonat, A. A new view on the kinetics of tricalcium silicate hydration. Cem. Concr. Res. 2016, 86, 1–11. [Google Scholar] [CrossRef]
- Rodger, S.A.; Groves, G.W.; Clayden, N.J.; Dobson, C.M. Hydration of tricalcium silicate followed by Si-29 NMR with cross-polarization. J. Am. Ceram. Soc. 1988, 71, 91–96. [Google Scholar] [CrossRef]
- Rheinheimer, V.; Casanova, I. Hydration of C3S thin films. Cem. Concr. Res. 2012, 42, 593–597. [Google Scholar] [CrossRef]
- Juilland, P.; Gallucci, E.; Flatt, R.; Scrivener, K. Dissolution theory applied to the induction period in alite hydration. Cem. Concr. Res. 2010, 40, 831–844. [Google Scholar] [CrossRef]
- Bazzoni, A.; Ma, S.H.; Wang, Q.Q.; Shen, X.D.; Cantoni, M.; Scrivener, K.L. Effect of magnesium and zinc ions on the hydration kinetics of C3S. J. Am. Ceram. Soc. 2014, 97, 3684–3693. [Google Scholar] [CrossRef]
- Masoero, E.; Thomas, J.J.; Jennings, H.M. A reaction zone hypothesis for the effects of particle size and water-tocement ratio on the early hydration kinetics of C3S. J. Am. Ceram. Soc. 2014, 97, 967–975. [Google Scholar] [CrossRef]
- Hesse, C.; Goetz-Neunhoeffer, F.; Neubauer, J.; Braeu, M.; Gaeberlein, P. Quantitative in situ X-ray diffraction analysis of early hydration of portland cement at defined temperatures. Powder Diffr. 2009, 24, 112–115. [Google Scholar] [CrossRef]
- Goni, S.; Puertas, F.; Hernandez, M.S.; Palacios, M.; Guerrero, A.; Dolado, J.S.; Zanga, B.; Baroni, F. Quantitative study of hydration of C3S and C2S by thermal analysis. J. Therm. Anal. Calorim. 2010, 102, 965–973. [Google Scholar] [CrossRef] [Green Version]
- Garrault, S.; Finot, E.; Lesniewska, E.; Nonat, A. Study of C–S–H growth on C3S surface during its early hydration. Mater. Struct. 2005, 38, 435–442. [Google Scholar] [CrossRef]
- Sarkar, S.L.; Xu, A.M. Preliminary-study of very early hydration of superplasticized C3A+ gypsum by environmental SEM. Cem. Concr. Res. 1992, 22, 605–608. [Google Scholar] [CrossRef]
- Lange, D.A.; Sujata, K.; Jennings, H.M. Observations of wet cement using electron-microscopy. Ultramicroscopy 1991, 37, 234–238. [Google Scholar] [CrossRef]
- Bae, S.; Taylor, R.; Shapiro, D.; Denes, P.; Joseph, J.; Celestre, R.; Marchesini, S.; Padmore, H.; Tyliszczak, T.; Warwick, T.; et al. Soft X-ray ptychographic imaging and morphological quantification of calcium silicate hydrates (C–S–H). J. Am. Ceram. Soc. 2015, 98, 4090–4095. [Google Scholar] [CrossRef]
- Cosmidis, J.; Benzerara, K.; Nassif, N.; Tyliszczak, T.; Bourdelle, F. Characterization of ca-phosphate biological materials by scanning transmission x-ray microscopy (STXM) at the Ca L2,3−, PL2,3− and C K-edges. Acta Biomater. 2015, 12, 260–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrua, R.D.; Hitchcock, A.P.; Hon, W.B.; West, M.; Hilder, E.F. Characterization of polymer monoliths containing embedded nanoparticles by scanning transmission X-ray microscopy (STXM). Anal. Chem. 2014, 86, 2876–2881. [Google Scholar] [CrossRef] [PubMed]
- Pecher, K.; Kneedler, E.; McCubbery, D.; Meigs, G.; Tonner, B. Characterization of reactive transition metal precipitates using scanning transmission X-ray microscopy (STXM). Abstr. Pap. Am. Chem. Soc. 2000, 220, U327. [Google Scholar]
- Ha, J.; Chae, S.; Chou, K.; Tyliszczak, T.; Monteiro, P. Effect of polymers on the nanostructure and on the carbonation of calcium silicate hydrates: A scanning transmission X-ray microscopy study. J. Mater. Sci. 2012, 47, 976–989. [Google Scholar] [CrossRef]
- Ha, J.; Chae, S.; Chou, K.W.; Tyliszczak, T.; Monteiro, P.J.M. Scanning transmission X-ray microscopic study of carbonated calcium silicate hydrate. Transp. Res. Rec. 2010, 83–88. [Google Scholar] [CrossRef]
- Bae, S.; Taylor, R.; Hernández-Cruz, D.; Yoon, S.; Kilcoyne, D.; Monteiro, P.J.M. Soft X-ray spectromicroscopic investigation of synthetic C–S–H and C3S hydration products. J. Am. Ceram. Soc. 2015, 98, 2914–2920. [Google Scholar] [CrossRef]
- Kirz, J.; Jacobsen, C.; Howells, M. Soft-X-ray microscopes and their biological applications. Q. Rev. Biophys. 1995, 28, 33–130. [Google Scholar] [CrossRef] [PubMed]
- Tzvetkov, G.; Fernandes, P.; Wenzel, S.; Fery, A.; Paradossi, G.; Fink, R.H. Soft X-ray induced modifications of PVA-based microbubbles in aqueous environment: A microspectroscopy study. Phys. Chem. Chem. Phys. 2009, 11, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- Stollberg, H.; Pokorny, M.; Hertz, H.M. A vacuum-compatible wet-specimen chamber for compact X-ray microscopy. J. Microsc. Oxf. 2007, 226, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Bishnoi, S.; Scrivener, K.L. Modelling early age hydration kinetics of alite. Cem. Concr. Res. 2012, 42, 903–918. [Google Scholar] [CrossRef]
- Hu, Q.; Aboustait, M.; Kim, T.; Ley, M.T.; Hanan, J.C.; Bullard, J.; Winarski, R.; Rose, V. Direct three-dimensional observation of the microstructure and chemistry of C3S hydration. Cem. Concr. Res. 2016, 88, 157–169. [Google Scholar] [CrossRef]
- De Groot, F.M.F.; Fuggle, J.C.; Thole, B.T.; Sawatzky, G.A. 2p X-ray absorption of 3D transition-metal compounds: An atomic multiplet description including the crystal field. Phys. Rev. B 1990, 42, 5459–5468. [Google Scholar] [CrossRef]
- Ko, J.Y.P.; Zhou, X.T.; Heigl, F.; Regier, T.; Blyth, R.; Sham, T.K. X-ray absorption near-edge structure (XANES) of calcium l-3,l-2 edges of various calcium compounds and X-ray excited optical luminescence (XEOL) studies of luminescent calcium compounds. AIP Conf. Proc. 2007, 882, 538–540. [Google Scholar]
- Politi, Y.; Metzler, R.A.; Abrecht, M.; Gilbert, B.; Wilt, F.H.; Sagi, I.; Addadi, L.; Weiner, S.; Gilbert, P.U.P.A. Transformation mechanism of amorphous calcium carbonate into calcite in the sea urchin larval spicule. Proc. Natl. Acad. Sci. USA 2008, 105, 17362–17366. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, R.J.; Brown, G.E.; Xu, N.; Cong, X.D. Ca X-ray absorption spectroscopy of C–S–H and some model compounds. Adv. Cem. Res. 1997, 9, 31–36. [Google Scholar] [CrossRef]
- Li, D.; Bancroft, G.M.; Fleet, M.E.; Feng, X.H. Silicon k-edge xanes spectra of silicate minerals. Phys. Chem. Miner. 1995, 22, 115–122. [Google Scholar] [CrossRef]
- Okada, K.; Kameshima, Y.; Yasumori, A. Chemical shifts of silicon X-ray photoelectron spectra by polymerization structures of silicates. J. Am. Ceram. Soc. 1998, 81, 1970–1972. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Peak Positions (eV) | Splitting of LIII and LII (eV) | ||||
|---|---|---|---|---|---|---|
| a1 | a2 | b1 | b2 | ∆LIII (a2−a1) | ∆LII (b2−b1) | |
| Anhydrous C3S [25] | 347.7 | 349 | 351.1 | 352.3 | 1.3 | 1.2 |
| Ca(OH)2 [25] | 347.6 | 349.1 | 351 | 352.4 | 1.5 | 1.4 |
| 1 h 32 min | 348 | 349.1 | 351.3 | 352.5 | 1.1 | 1.2 |
| 1 h 34 min | 347.9 | 349 | 351.3 | 352.4 | 1.1 | 1.1 |
| 1 h 56 min | 348 | 349.1 | 351.3 | 352.4 | 1.1 | 1.1 |
| 2 h 19 min | 348 | 349.1 | 351.4 | 352.4 | 1.1 | 1 |
| 3 h 21 min | 348 | 349.1 | 351.4 | 352.4 | 1 | 1 |
| 3 h 42 min | 348 | 349.1 | 351.4 | 352.4 | 1.1 | 1 |
| 4 h 01 min | 348.1 | 349.1 | 351.4 | 352.4 | 1 | 1 |
| 4 h 54 min | 348 | 349.1 | 351.4 | 352.4 | 1.1 | 1 |
| 5 h 12 min | 348.1 | 349.1 | 351.3 | 352.4 | 1 | 1.1 |
| 5 h 21 min | 348 | 349.1 | 351.3 | 352.5 | 1.1 | 1.2 |
| 6 h 15 min | 348 | 349.1 | 351.3 | 352.4 | 1 | 1.1 |
| 6 h 21 min | 348 | 349.1 | 351.3 | 352.4 | 1 | 1.1 |
| 6 h 31 min | 348 | 349.1 | 351.3 | 352.5 | 1.1 | 1.2 |
| 6 h 39 min | 348 | 349.2 | 351.4 | 352.4 | 1.2 | 1 |
| 6 h 47 min | 347.9 | 349.1 | 351.4 | 352.4 | 1.2 | 1 |
| 7 h 40 min | 348.1 | 349.1 | 351.3 | 352.5 | 1 | 1.2 |
| 7 h 47 min | 348.1 | 349.1 | 351.3 | 352.4 | 1 | 1.1 |
| 17 h 11 min (Ex Situ) | 348.1 | 349.1 | 351.4 | 352.4 | 1 | 1 |
| Sample | Scan Areas | Peak a1 (eV) | Peak a2 (eV) | Δa2−a1 (eV) |
|---|---|---|---|---|
| Anhydrous C3S [25] | - | 1847.7 | 1858.9 | 11.2 |
| 1 h 19 min (In Situ) | 1 | 1847.4 | 1858.6 | 11.2 |
| 2 | 1847.4 | 1858.6 | 11.2 | |
| 3 | 1847.6 | 1859.2 | 11.6 | |
| 2 h 48 min (In Situ) | 1 | 1847.4 | 1858.7 | 11.3 |
| 2 | 1847.4 | 1858.6 | 11.2 | |
| 3 | 1847.6 | 1859.2 | 11.6 | |
| 4 h 22 min (In Situ) | 1 | 1847.5 | 1859.3 | 11.8 |
| 2 | 1847.4 | 1859.1 | 11.7 | |
| 3 | 1847.8 | 1859.6 | 11.8 |
| Sample | Scan Areas | Peak a1 (eV) | Peak a2 (eV) | Δa2−a1 (eV) |
|---|---|---|---|---|
| Anhydrous C3S [25] | - | 1847.7 | 1858.9 | 11.2 |
| 15 h 25 min (Ex Situ) | 1 | 1847.1 | 1862.2 | 15.1 |
| 2 | 1848 | 1859.7 | 11.7 | |
| 3 | 1847.5 | 1861 | 13.5 | |
| 17 h 13 min (Ex Situ) | 1 | 1847 | 1861 | 14 |
| 2 | 1847.3 | 1860.2 | 12.9 | |
| 3 | 1847.1 | 1862.3 | 15.2 | |
| 4 | 1847 | 1862.1 | 15.1 | |
| 5 | 1847.3 | 1860 | 12.7 | |
| 6 | 1847.1 | 1861.4 | 14.3 | |
| 17 days (Ex Situ) | 1 | 1847.2 | 1862.2 | 15 |
| 2 | 1847.2 | 1858.4, 1861.2 | 11.2, 14 | |
| 3 | 1847.2 | 1863.2 | 16 | |
| 1 | 1847.6 | 1862.6 | 15 | |
| 40 days (Ex Situ) | 2 | 1847.6 | 1862.6 | 15 |
| 3 | 1847.6 | 1862.6 | 15 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, S.; Kanematsu, M.; Hernández-Cruz, D.; Moon, J.; Kilcoyne, D.; Monteiro, P.J.M. In Situ Soft X-ray Spectromicroscopy of Early Tricalcium Silicate Hydration. Materials 2016, 9, 976. https://doi.org/10.3390/ma9120976
Bae S, Kanematsu M, Hernández-Cruz D, Moon J, Kilcoyne D, Monteiro PJM. In Situ Soft X-ray Spectromicroscopy of Early Tricalcium Silicate Hydration. Materials. 2016; 9(12):976. https://doi.org/10.3390/ma9120976
Chicago/Turabian StyleBae, Sungchul, Manabu Kanematsu, Daniel Hernández-Cruz, Juhyuk Moon, David Kilcoyne, and Paulo J. M. Monteiro. 2016. "In Situ Soft X-ray Spectromicroscopy of Early Tricalcium Silicate Hydration" Materials 9, no. 12: 976. https://doi.org/10.3390/ma9120976