
First-Principles Study of Mo Segregation in MoNi(111): Effects of Chemisorbed Atomic Oxygen

Abstract
:1. Introduction
2. Computational Methods
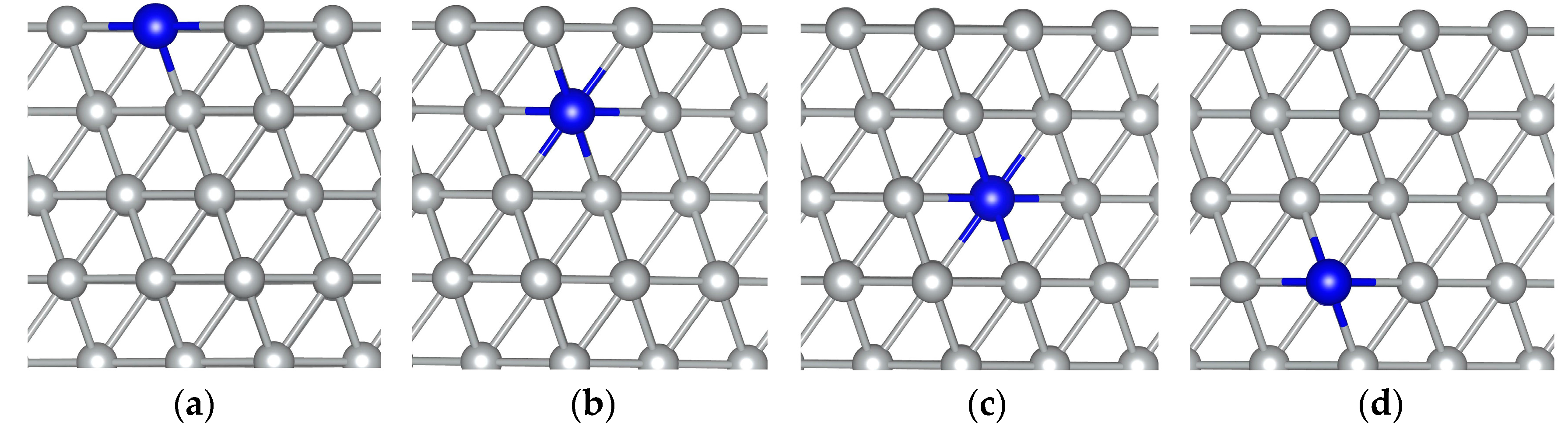
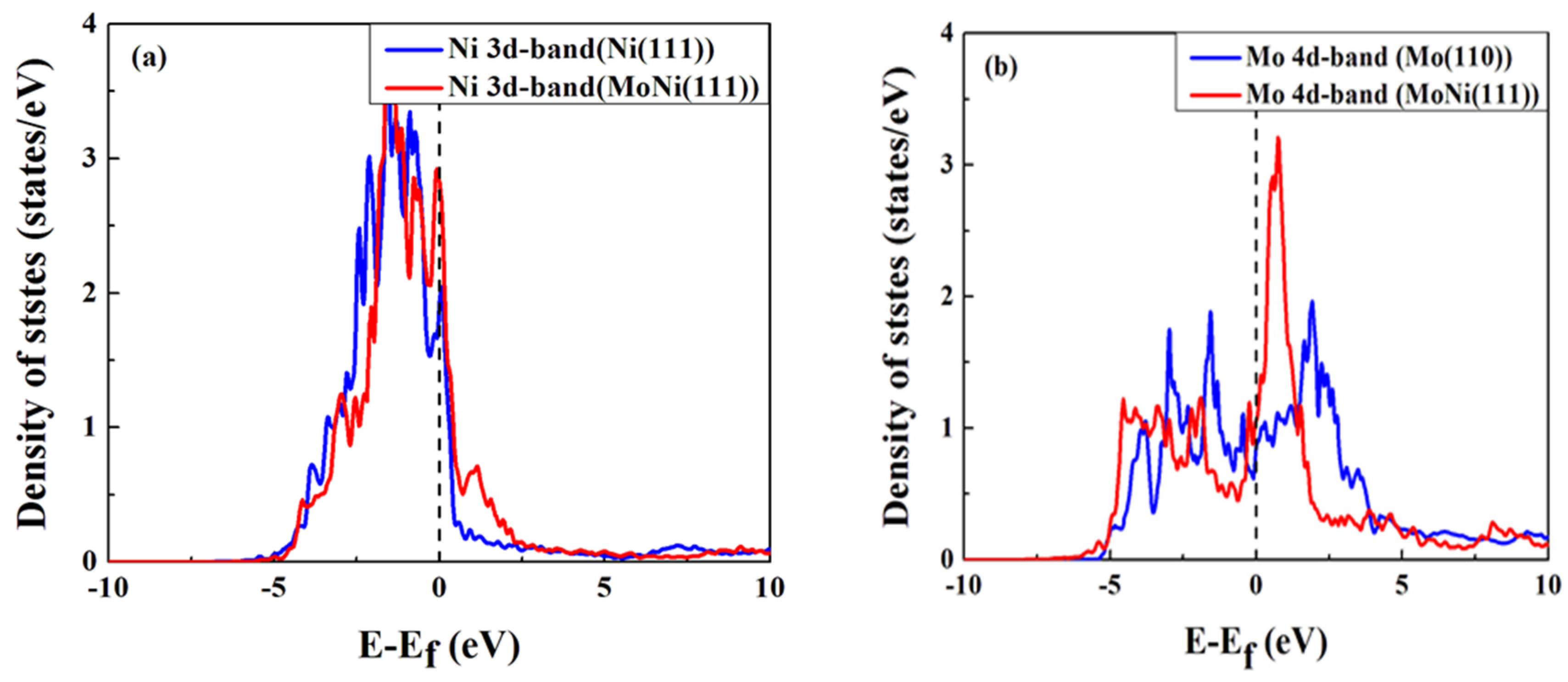
3. Results and Discussion
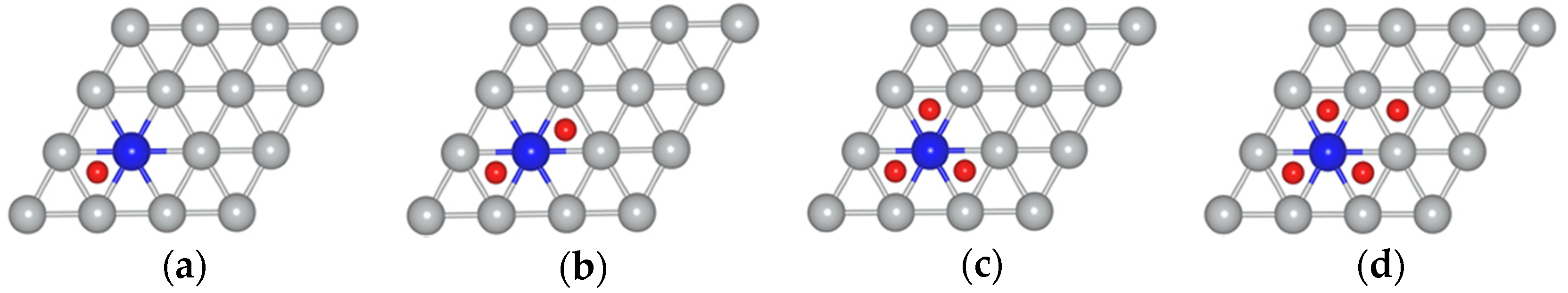

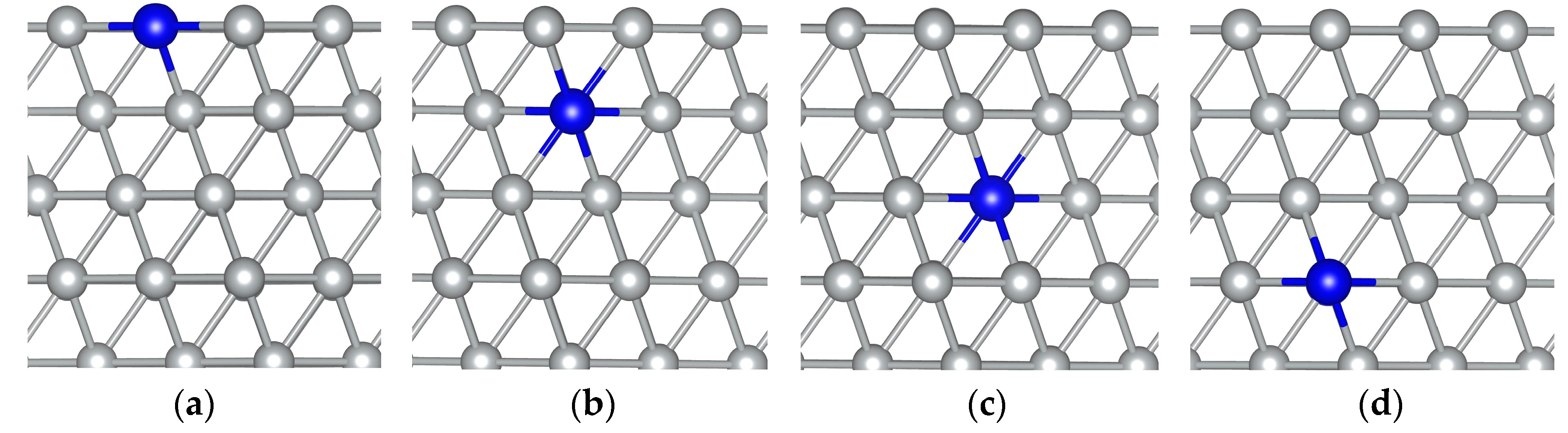{kind=link}
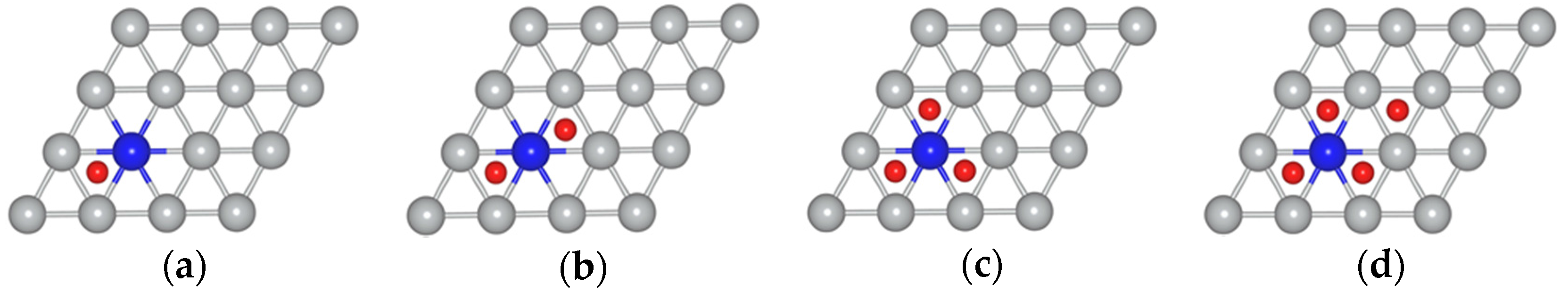{kind=link}
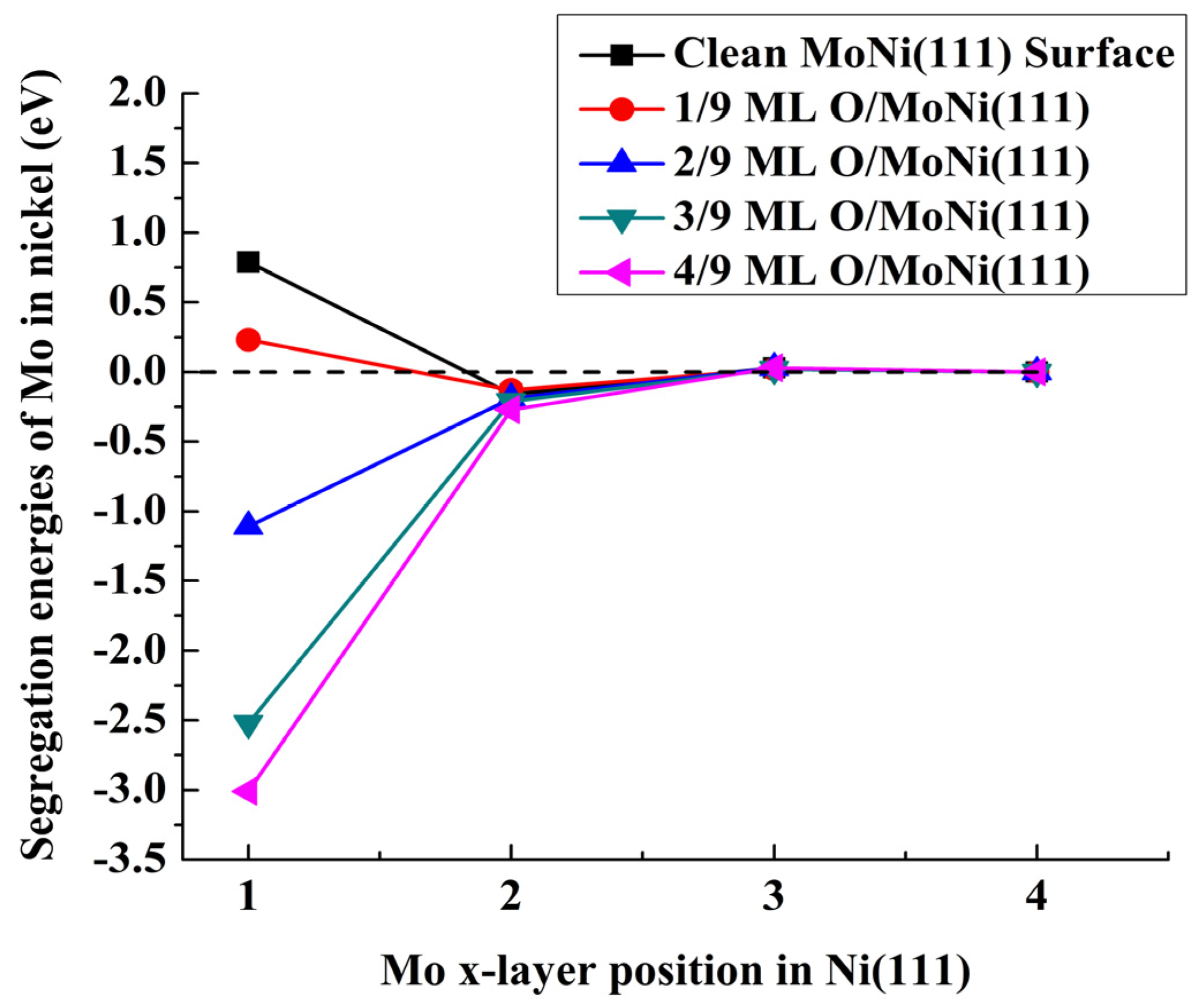{kind=link}
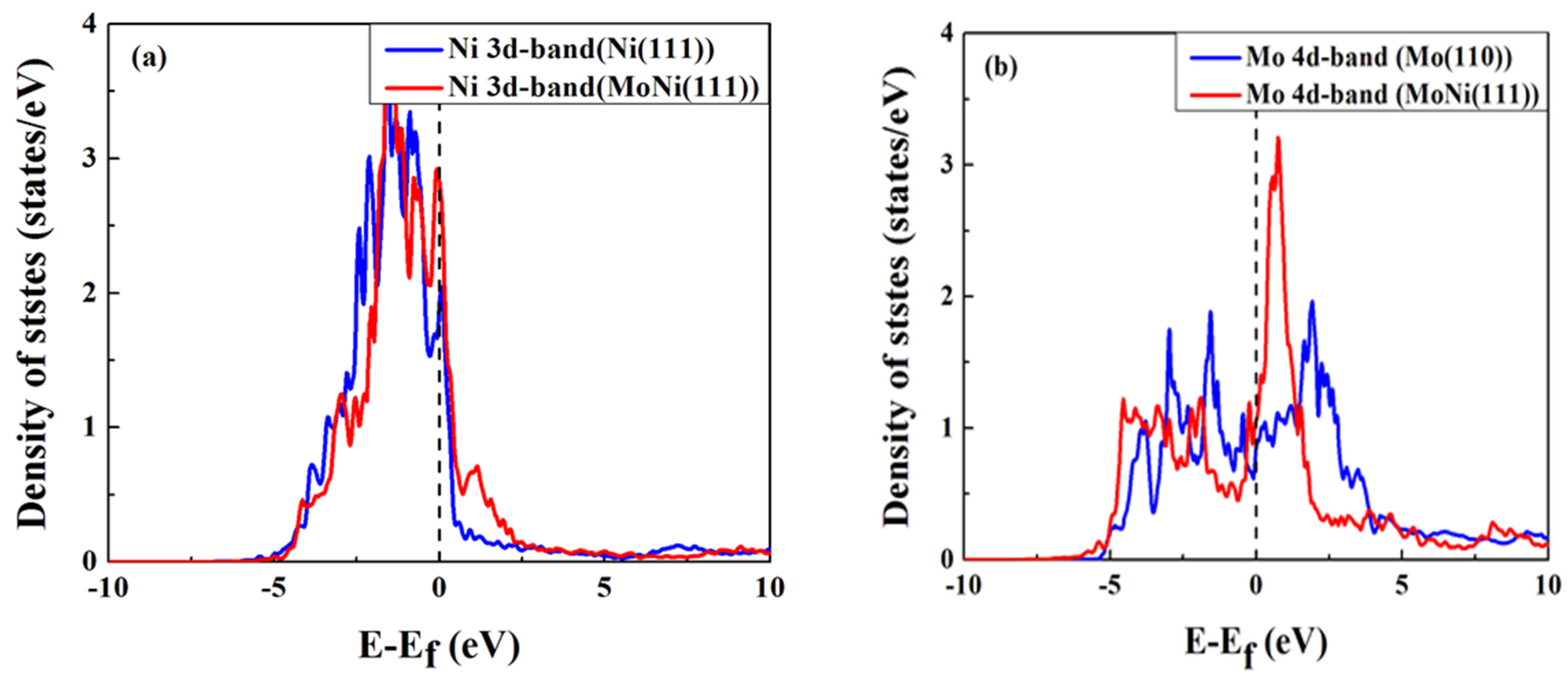{kind=link}
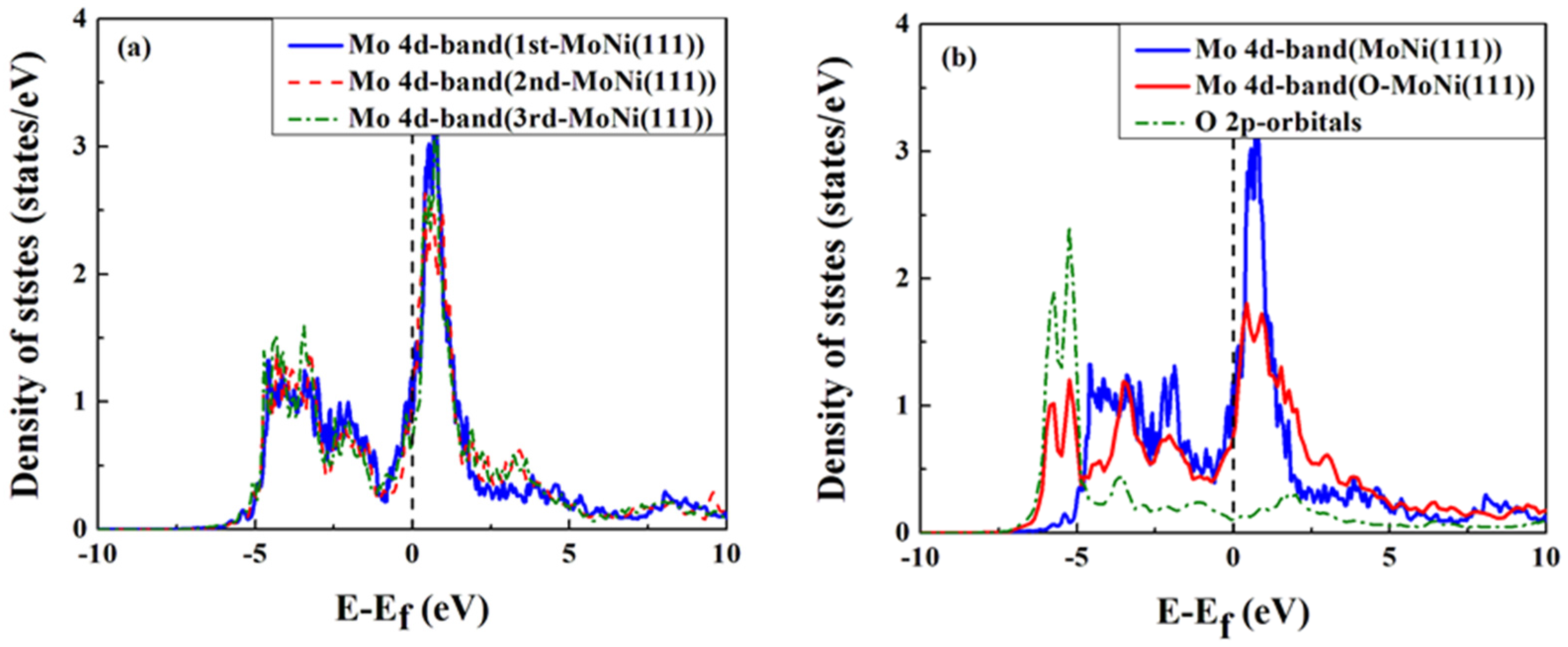{kind=link}
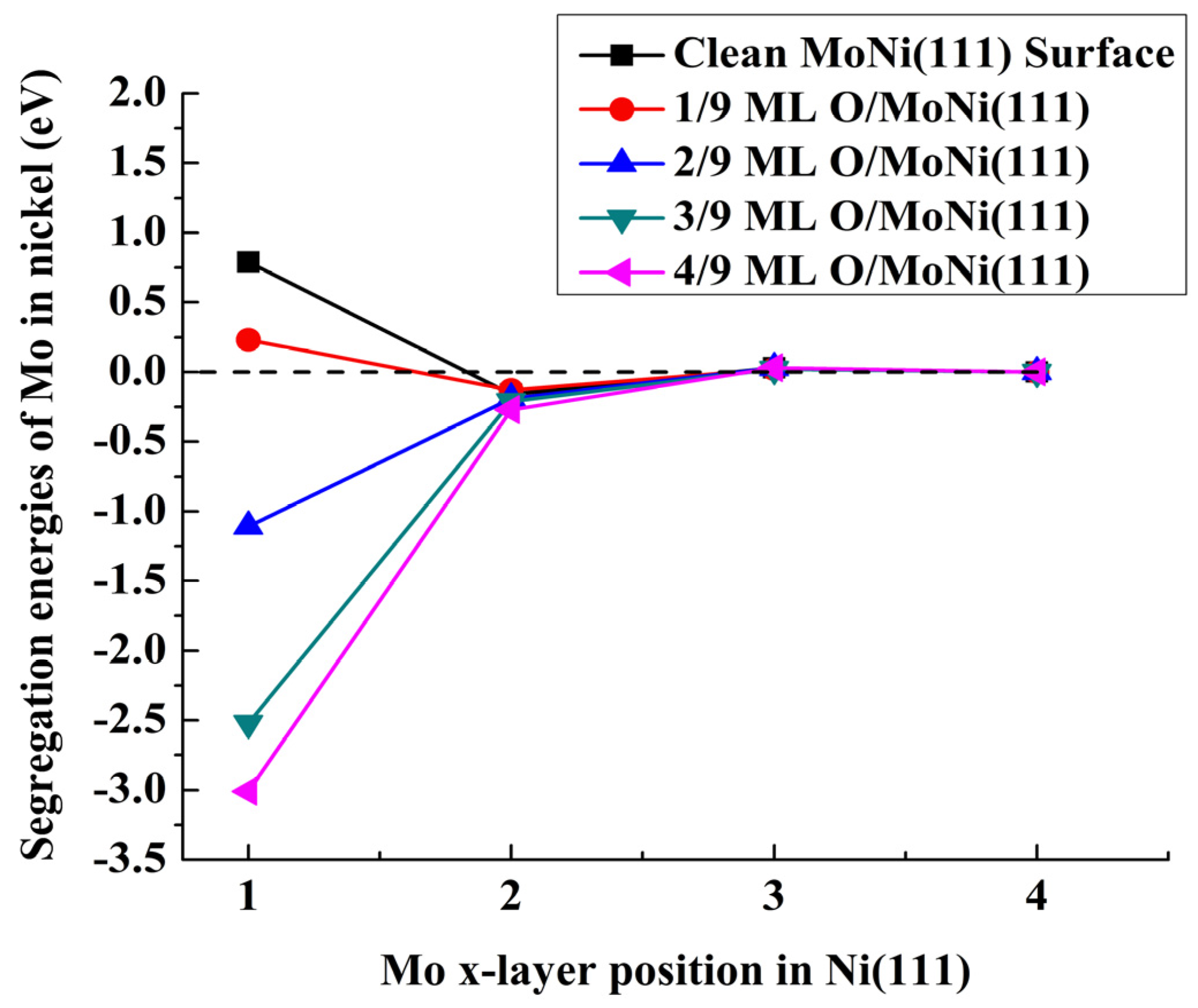
| Position of the Mo Atom | Atomic Oxygen Sub-Monolayer on MoNi(111) Alloy | ||||
|---|---|---|---|---|---|
| 0 | 1/9 ML | 2/9 ML | 3/9 ML | 4/9 ML | |
| Etot (eV) | |||||
| First layer | 0.79 | 0.23 | −1.11 | −2.52 | −3.01 |
| Second layer | −0.16 | −0.13 | −0.19 | −0.21 | −0.27 |
| Third layer | 0.03 | 0.02 | 0.03 | 0.02 | 0.03 |
| Fourth layer | 0 | 0 | 0 | 0 | 0 |
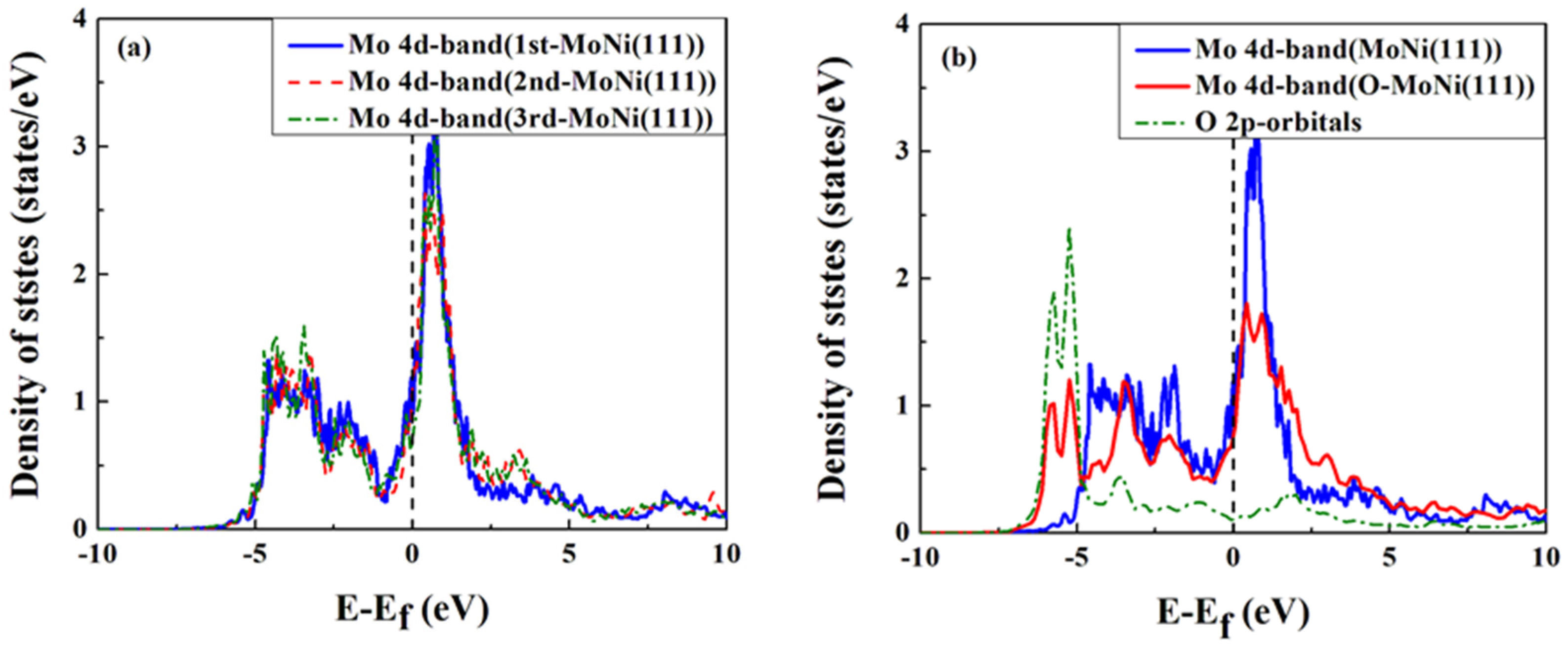
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lendzion-Bielun, Z.; Narkiewicz, U.; Arabczyk, W. Cobalt-based catalysts for ammonia decomposition. Materials 2013, 6, 2400–2409. [Google Scholar] [CrossRef]
- Liu, J.; Xu, C.; Liu, C.; Feng, W.; Liu, H.; Jing, J.; Li, Z. Impact of Cu-Pt nanotubes with a high degree of alloying on electro-catalytic activity toward oxygen reduction reaction. Electrochim. Acta 2015, 152, 425–432. [Google Scholar] [CrossRef]
- Lloyd, A.C.; Noël, J.J.; Mcintyre, S.; Shoesmith, D.W. Cr, Mo and W alloying additions in Ni and their effect on passivity. Electrochim. Acta 2004, 49, 3015–3027. [Google Scholar] [CrossRef]
- Yarulin, A.; Berguerand, C.; Alonso, A.O.; Yuranov, I.; Kiwi-Minsker, L. Increasing Pt selectivity to vinylaniline by alloying with Zn via reactive metal-support interaction. Catal. Today 2015, 256, 241–249. [Google Scholar] [CrossRef]
- Bowker, R.H.; Ilic, B.; Bo, A.C.; Reynolds, M.A.; Murray, B.D.; Bussell, M.E. Carbazole hydrodenitrogenation over nickel phosphide and Ni-rich bimetallic phosphide catalysts. Appl. Catal. A 2014, 482, 221–230. [Google Scholar] [CrossRef]
- Iizuka, Y.; Inoue, R.; Miura, T.; Morita, N.; Toshima, N.; Honma, T.; Oji, H. Chemical environment of Ag atoms contained in AuAg bimetallic catalysts and the generation of the catalytic activity for CO oxidation. Appl. Catal. A 2014, 483, 63–75. [Google Scholar] [CrossRef]
- Kang, J.; Wang, R.; Wang, H.; Liao, S.; Key, J.; Linkov, V.; Ji, S. Effect of Ni Core structure on the electrocatalytic activity of Pt-Ni/C in methanol oxidation. Materials 2013, 6, 2689–2700. [Google Scholar] [CrossRef]
- Zhu, H.; Anjum, D.H.; Wang, Q.; Abou-Hamad, E.; Emsley, L.; Dong, H.; Laveille, P.; Li, L.; Samal, A.K.; Basset, J.M. Sn surface-enriched Pt-Sn bimetallic nanoparticles as a selective and stable catalyst for propane dehydrogenation. J. Catal. 2014, 320, 52–62. [Google Scholar] [CrossRef]
- Andersson, K.J.; Federico, C.V.; Jan, R.; Ib, C. Adsorption-driven surface segregation of the less reactive alloy component. J. Am. Chem. Soc. 2009, 131, 2404–2407. [Google Scholar] [CrossRef] [PubMed]
- Menning, C.A.; Chen, J.G. General trend for adsorbate-induced segregation of subsurface metal atoms in bimetallic surfaces. J. Chem. Phys. 2009, 130, 363–366. [Google Scholar] [CrossRef]
- Dhouib, A.; Guesmi, H. DFT study of the M segregation on MAu alloys (M = Ni, Pd, Pt) in presence of adsorbed oxygen O and O2. Chem. Phys. Lett. 2012, 521, 98–103. [Google Scholar] [CrossRef]
- Guesmi, H.; Louis, C.; Delannoy, L. Chemisorbed atomic oxygen inducing Pd segregation in PdAu(111) alloy: Energetic and electronic DFT analysis. Chem. Phys. Lett. 2011, 503, 97–100. [Google Scholar] [CrossRef]
- Kitchin, J.R.; Reuter, K.; Scheffler, M. Alloy surface segregation in reactive environments: First-principles atomistic thermodynamics study of Ag3Pd(111) in oxygen atmospheres. Phys. Rev. B Condens. Matter Mater. Phys. 2008, 77, 439–446. [Google Scholar] [CrossRef]
- Mainardi, D.S.; Balbuena, P.B. Monte Carlo simulation of Cu-Ni nanoclusters: Surface segregation studies. Langmuir 2001, 17, 2047–2050. [Google Scholar] [CrossRef]
- Oetelaar, D.; Nooij, W.; Oerlemans, S.; Gon, D. Surface segregation in supported Pd-Pt nanoclusters and alloys. J. Phys. Chem. B 1998, 102, 3445–3455. [Google Scholar] [CrossRef]
- Mainardi, D.S.; Balbuena, P.B. Surface segregation in bimetallic nanoclusters: Geometric and thermodynamic effects. Int. J. Quantum Chem 2001, 85, 580–591. [Google Scholar] [CrossRef]
- Wang, J.W.; Wang, Y.F.; Wang, L.G.; Yu, Y.L.; Zhang, J.G.; Wang, L.S. Optimization of the electrocatalytic properties of the NiMoCo foam electrode for water electrolysis by post-treatment processing. Rare Met. 2015, 34, 802–807. [Google Scholar] [CrossRef]
- Zhang, X.M.; Liu, J.C.; Tang, J.G.; Li, L.; Chen, M.A.; Liu, S.D.; Zhu, B. Element segregation on the surfaces of pure aluminum foils. Appl. Surf. Sci. 2010, 256, 7300–7304. [Google Scholar] [CrossRef]
- Zafeiratos, S.; Piccinin, S.; Teschner, D. Alloys in catalysis: Phase separation and surface segregation phenomena in response to the reactive environment. Catal. Sci. Technol. 2012, 2, 1787–1801. [Google Scholar] [CrossRef]
- Guesmi, H. Theoretical insights on the effect of reactive gas on the chemical ordering of gold-based alloys. Gold Bull. 2013, 46, 213–219. [Google Scholar] [CrossRef]
- Juárez, M.F.; Soldano, G.; Guesmi, H.; Tielens, F.; Santos, E. Catalytic properties of Au electrodes modified by an underlayer of Pd. Surf. Sci. 2015, 631, 235–247. [Google Scholar] [CrossRef]
- Sansa, M.; Dhouib, A.; Guesmi, H. Density functional theory study of CO-induced segregation in gold-based alloys. J. Chem. Phys. 2014, 141. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Thrimurthulu, G.; Delannoy, L.; Louis, C.; Mottet, C.; Creuze, J.; Legrand, B.; Guesmi, H. Evidence of Pd segregation and stabilization at edges of AuPd nano-clusters in the presence of CO: A combined DFT and DRIFTS study. J. Catal. 2013, 308, 272–281. [Google Scholar] [CrossRef]
- Sun, Q.; Reuter, K.; Scheffler, M. Effect of a humid environment on the surface structure of RuO2(110). Phys. Rev. B Condens. Matter Mater. Phys. 2003, 67, 920–925. [Google Scholar] [CrossRef]
- Silva, A.M.; Achete, C.A.; Capaz, R.B. First-principles study of oxygen-induced copper segregation in Cu3Au(111). Chem. Phys. 2013, 410, 99–102. [Google Scholar] [CrossRef]
- Wang, L.; Kuklja, M.M. First-principles study of small aluminum clusters: Oxygen adsorptions, oxidation and phase stability. J. Phys. Chem. Solids 2010, 71, 140–144. [Google Scholar] [CrossRef]
- Zunger, A.; Wang, L.G.; Hart, G.L.W.; Sanati, M. Obtaining Ising-like expansions for binary alloys from first principles. Model. Simul. Mater. Sci. Eng. 2002, 10, 685–706. [Google Scholar] [CrossRef]
- Chen, W.; Schmidt, D.; Schneider, W.F.; Wolverton, C. Ordering and oxygen adsorption in Au–Pt/Pt(111) surface alloys. J. Phys. Chem. C 2011, 115, 17915–17924. [Google Scholar] [CrossRef]
- Han, B.C.; Ven, A.V.D.; Ceder, G.; Hwang, B.J. Surface segregation and ordering of alloy surfaces in the presence of adsorbates. Phys. Rev. B Condens. Matter Mater. Phys. 2005, 72. [Google Scholar] [CrossRef]
- Kukushkin, R.G.; Bulavchenko, O.A.; Kaichev, V.V.; Yakovlev, V.A. Influence of Mo on catalytic activity of Ni-based catalysts in hydrodeoxygenation of esters. Appl. Catal. B 2015, 163, 531–538. [Google Scholar] [CrossRef]
- Yuan, C.; Nan, Y.; Wang, X.; Wang, J.; Lv, D.; Li, X. The SiO2 supported bimetallic Ni-Ru particles: A good sulfur-tolerant catalyst for methanation reaction. Chem. Eng. J. 2015, 260, 1–10. [Google Scholar] [CrossRef]
- Malaibari, Z.O.; Croiset, E.; Amin, A.; Epling, W. Effect of interactions between Ni and Mo on catalytic properties of a bimetallic Ni-Mo/Al2O3 propane reforming catalyst. Appl. Catal. A 2015, 490, 80–92. [Google Scholar] [CrossRef]
- Xun, T.; Li, X.; Yang, C.; Lu, J.; Lin, Z. Noble fabrication of Ni-Mo cathode for alkaline water electrolysis and alkaline polymer electrolyte water electrolysis. Int. J. Hydrog. Energy 2014, 39, 3055–3060. [Google Scholar]
- Marcus, P.; Chandler, C.; Chadli, H.; Wynblatt, P. Surface composition and structure of nickel-molybdenum single crystal alloys. Appl. Surf. Sci. 1989, 37, 33–43. [Google Scholar] [CrossRef]
- Nilekar, A.U.; Ruban, A.V.; Mavrikakis, M. Surface segregation energies in low-index open surfaces of bimetallic transition metal alloys. Surf. Sci. 2009, 603, 91–96. [Google Scholar] [CrossRef]
- Ruban, A.; Skriver, H.; Nørskov, J.K. Surface segregation energies in transition-metal alloys. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 59, 15990–16000. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B Condens. Matter Mater. Phys. 1993, 49, 14251–14269. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 1994, 50, 2665–2668. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B Condens. Matter Mater. Phys. 1989, 40, 3616–3621. [Google Scholar] [CrossRef]
- Neugebauer, J.; Scheffler, M. Adsorbate-substrate and adsorbate-adsorbate interactions of Na and K adlayers on Al(111). Phys. Rev. B Condens. Matter Mater. Phys. 1992, 46, 16067–16080. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Hendrik, J.; James, D. Special points for Brillouin-zone integrations. Phys. Rev. B Condens. Matter Mater. Phys. 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- DeBoer, F.R.; Boom, R.; Miedema, A.R. Cohesion in Metals, 2nd ed.; North-Holland Physics Publishing: Amsterdam, The Netherlands, 1989; pp. 657–660. [Google Scholar]
- Florencio, J.; Ren, D.M.; Tsong, T.T. Absolute composition depth-profiles in surface segregation of Pt-Rh alloys. Surf. Sci. 1996, 345, 29–33. [Google Scholar] [CrossRef]
- Helfensteyn, S.; Luyten, J.; Feyaerts, L.; Creemers, C. Modelling surface phenomena in Pd-Ni alloys. Appl. Surf. Sci. 2003, 75, 844–849. [Google Scholar] [CrossRef]
- Kuntze, J.; Speller, S.; Heiland, W.; Deurinck, P.; Creemers, C.; Atrei, A.; Bardi, U. Surface structure and segregation profile of the alloy Au3Pd(110): Experiment and theory. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 60, 9010–9018. [Google Scholar] [CrossRef]
- Ren, D.M.; Qin, J.H.; Wang, J.B.; Tsong, T.T. Oscillatory compositional depth profiles in surface segregation of a Pt-Rh alloy. Phys. Rev. B Condens. Matter Mater. Phys. 1993, 47, 3944–3946. [Google Scholar] [CrossRef]
- Tsong, T.T.; Ren, D.M.; Ahmad, M. Atomic-layer by atomic-layer compositional depth profiling: Surface segregation and impurity cosegregation of Pt-Rh and Pt-Ru alloys. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 38, 7428–7435. [Google Scholar] [CrossRef]
- Kerr, J.A.; Trotman-Dickenson, A.F. Strengths of chemical bonds. In CRC Handbook of Chemistry and Physics, 62nd ed.; Weast, R.C., Astle, M.J., Eds.; CRC Press, Inc.: Boca Raton, FL, USA, 1982; pp. F180–F190. [Google Scholar]
- Hammer, B.; Morikawa, Y.; Nøskov, J.K. CO chemisorption at metal surfaces and overlayers. Phys. Rev. Lett. 1996, 76, 2141–2144. [Google Scholar] [CrossRef] [PubMed]
- Allinger, N.L.; Zhou, X.; Bergsma, J. Molecular mechanics parameters. J. Mol. Struct. THEOCHEM 1994, 312, 69–83. [Google Scholar] [CrossRef]
- Wang, L.G.; Tsymbal, E.Y.; Jaswal, S.S. Structural and magnetic properties of clean and methylthiolate-adsorbed Co(0001) surfaces: A first-principles study. J. Magn. Magn. Mater. 2005, 286, 119–123. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Y.; Xiao, W.; Wang, J.; Wang, L. First-Principles Study of Mo Segregation in MoNi(111): Effects of Chemisorbed Atomic Oxygen. Materials 2016, 9, 5. https://doi.org/10.3390/ma9010005
Yu Y, Xiao W, Wang J, Wang L. First-Principles Study of Mo Segregation in MoNi(111): Effects of Chemisorbed Atomic Oxygen. Materials. 2016; 9(1):5. https://doi.org/10.3390/ma9010005
Chicago/Turabian StyleYu, Yanlin, Wei Xiao, Jianwei Wang, and Ligen Wang. 2016. "First-Principles Study of Mo Segregation in MoNi(111): Effects of Chemisorbed Atomic Oxygen" Materials 9, no. 1: 5. https://doi.org/10.3390/ma9010005