Liquid Crystalline π-Conjugated Copolymers Bearing a Pyrimidine Type Mesogenic Group


Abstract
:1. Introduction
2. Experimental Section
2.1. Materials and Methods
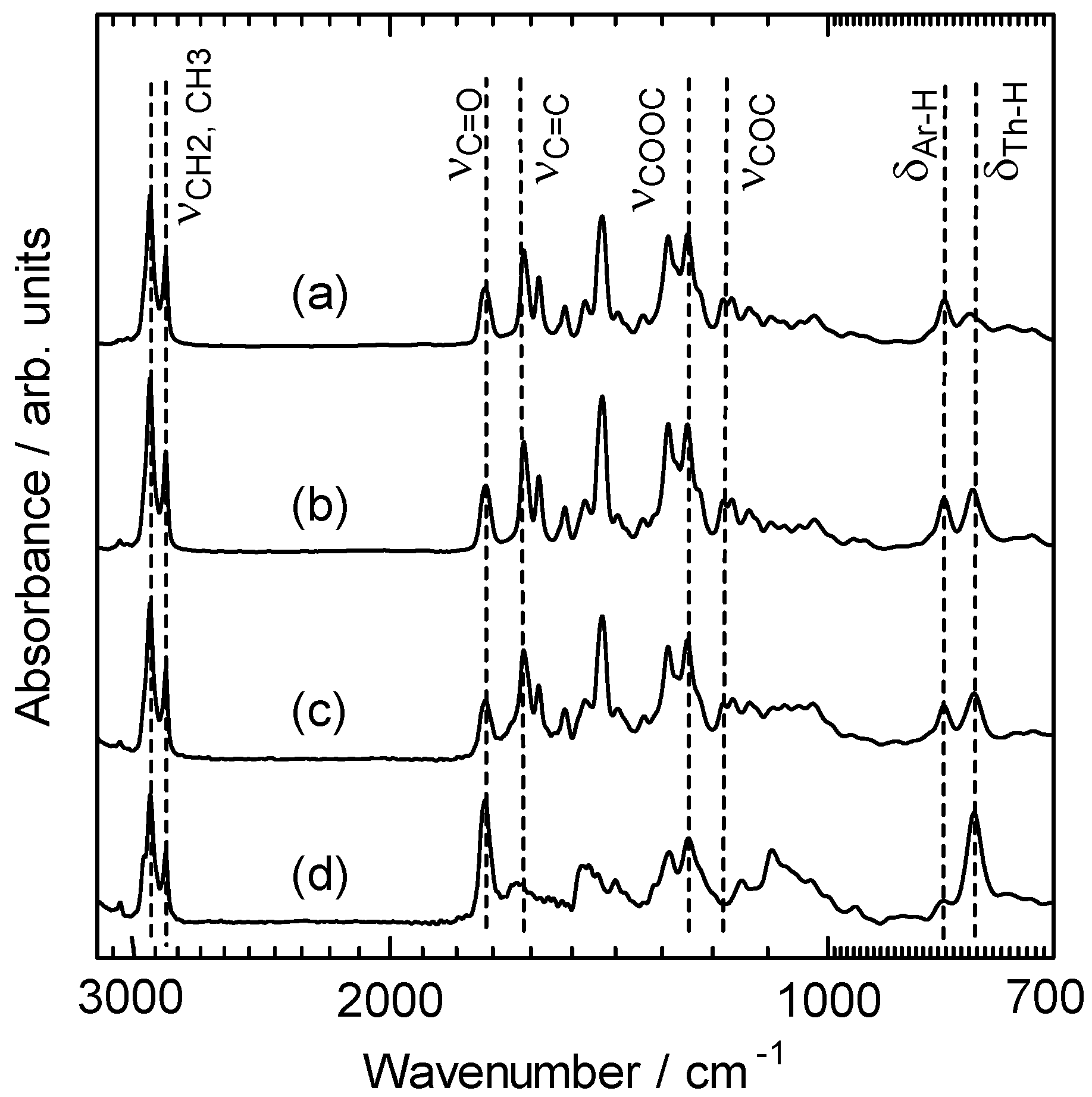
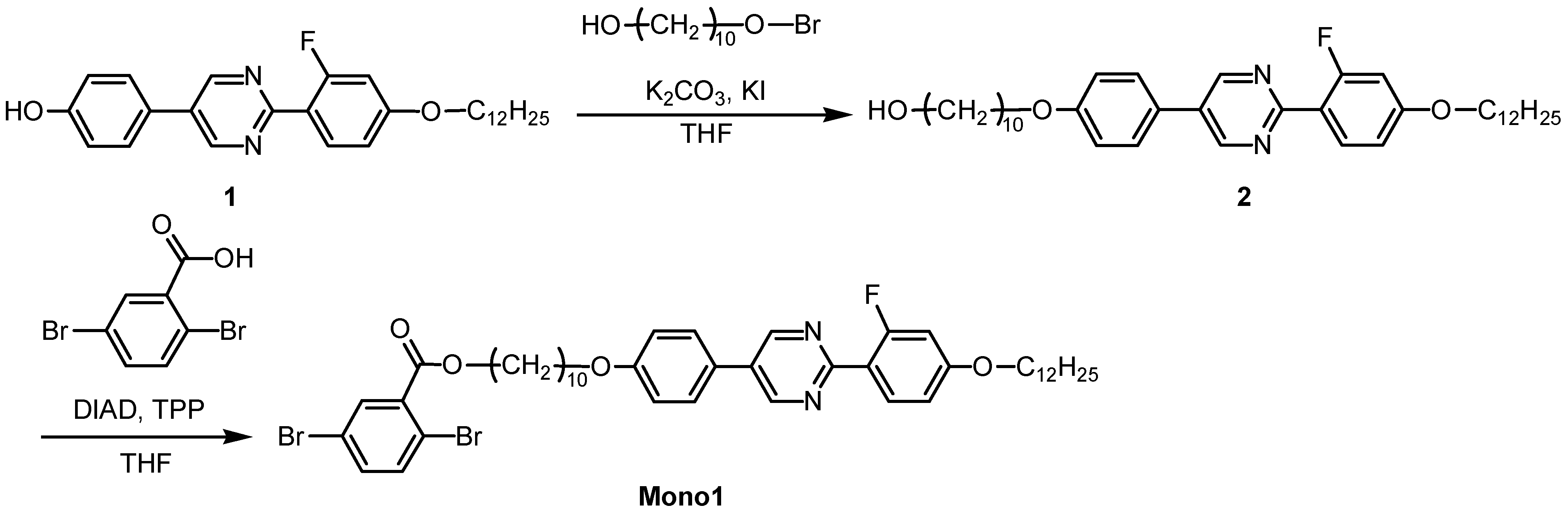
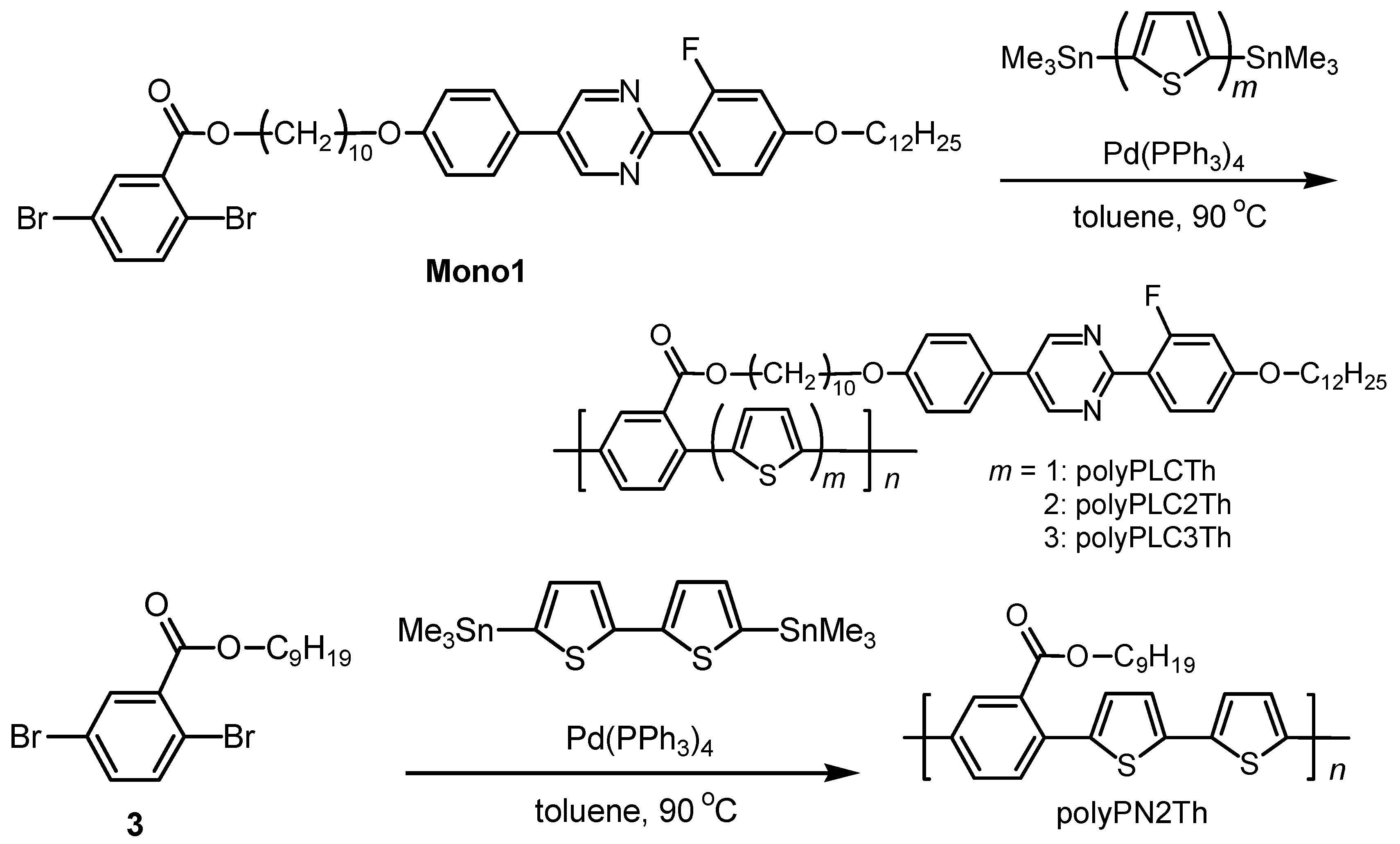
2.2. Monomer synthesis

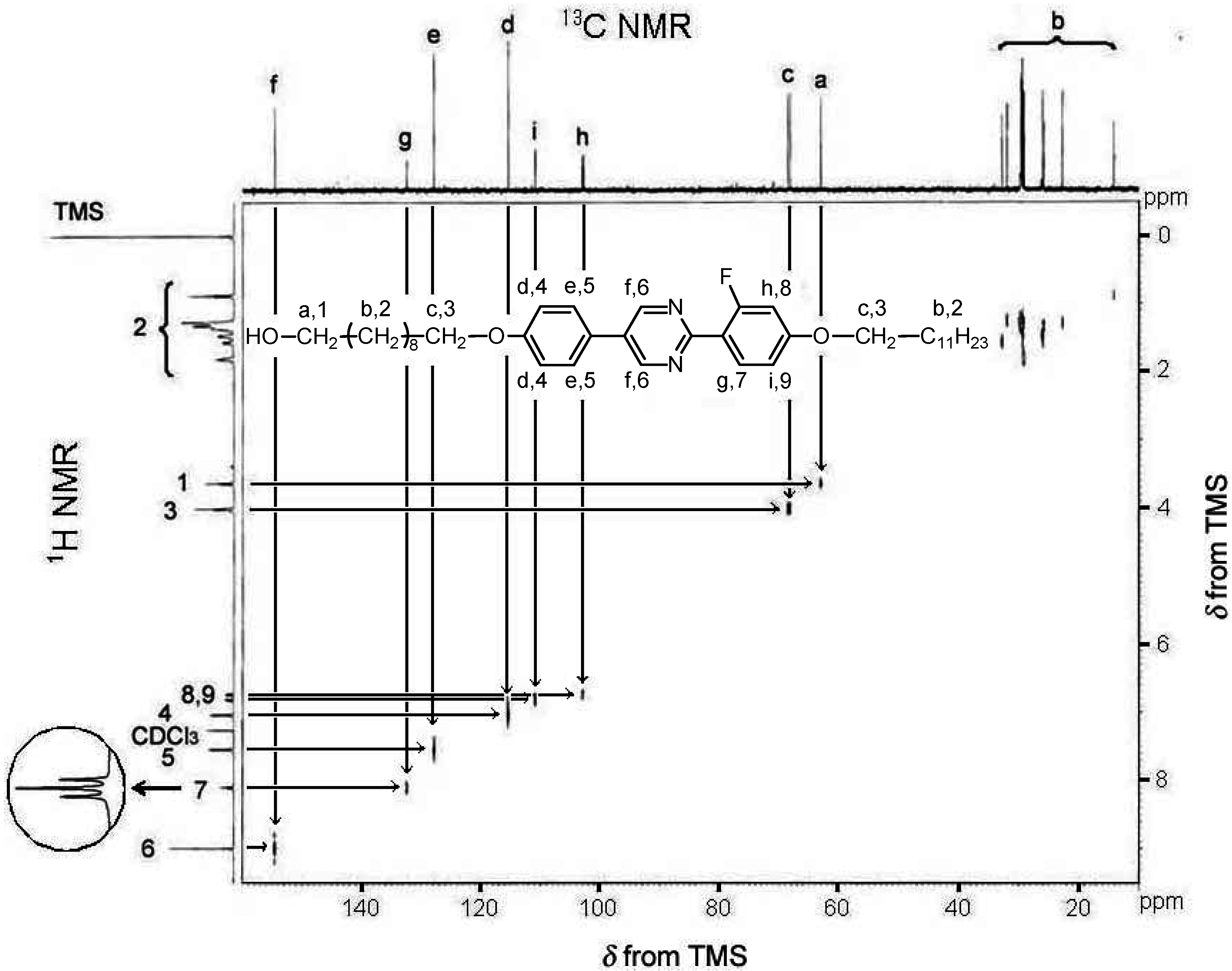
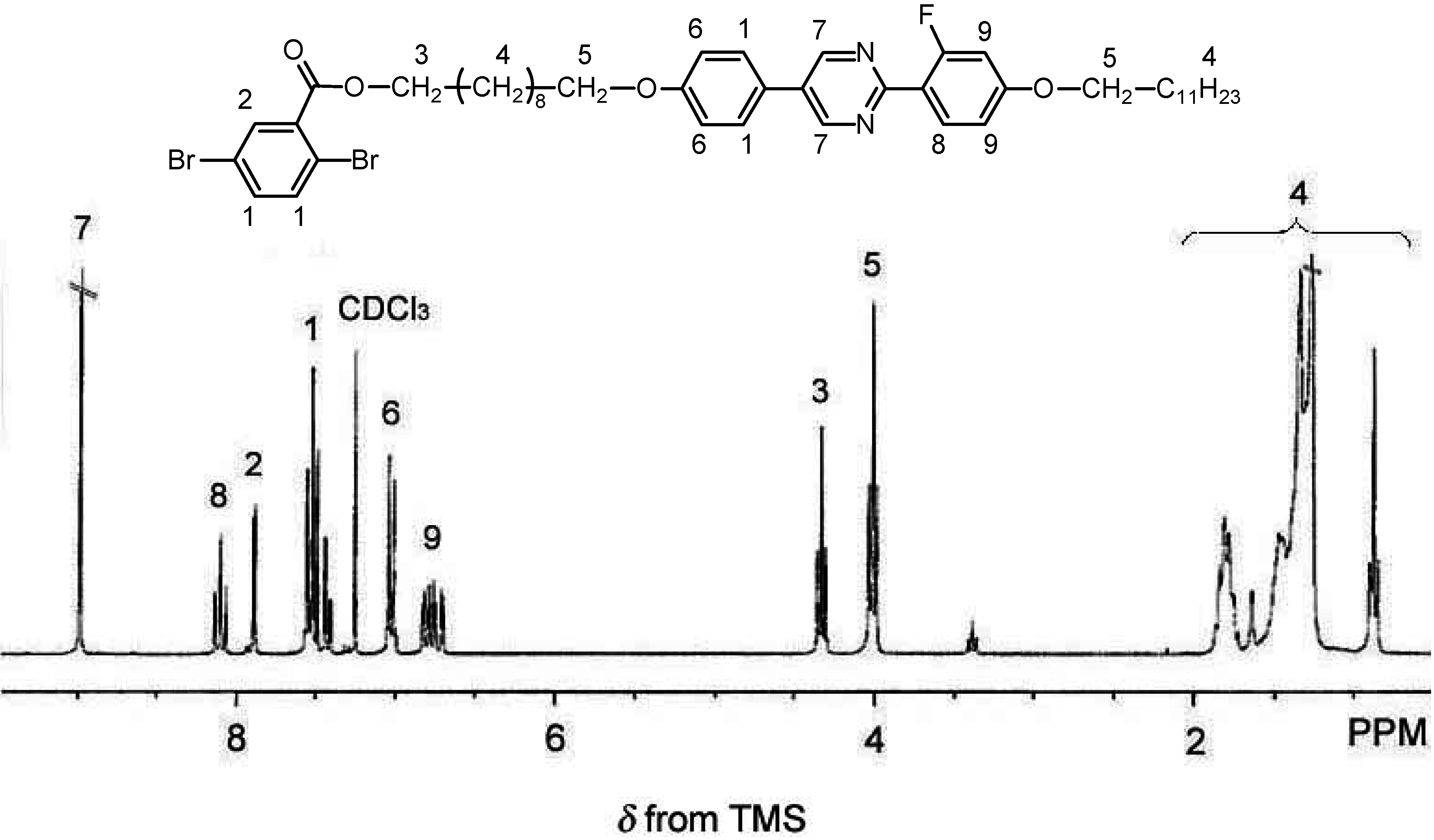
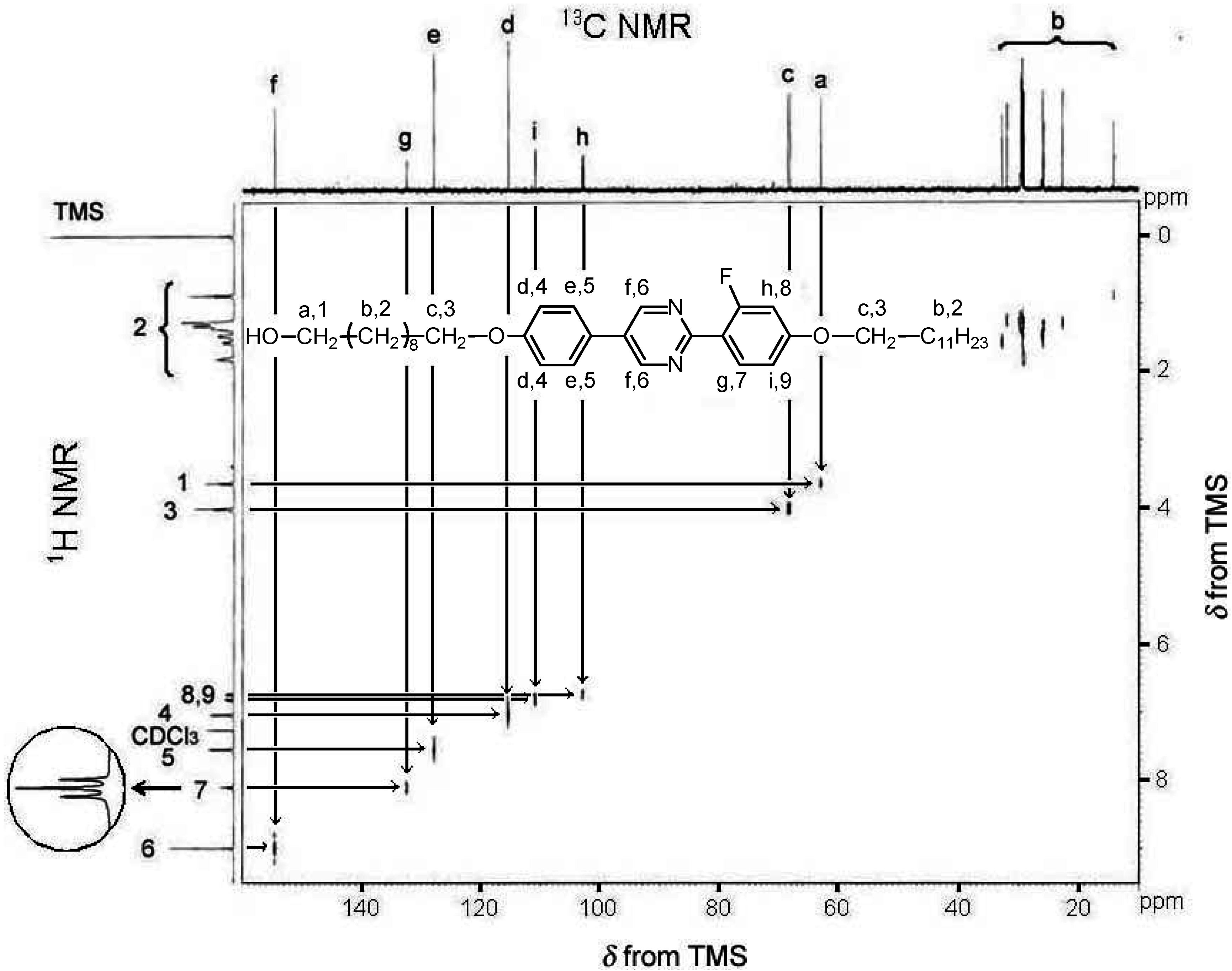
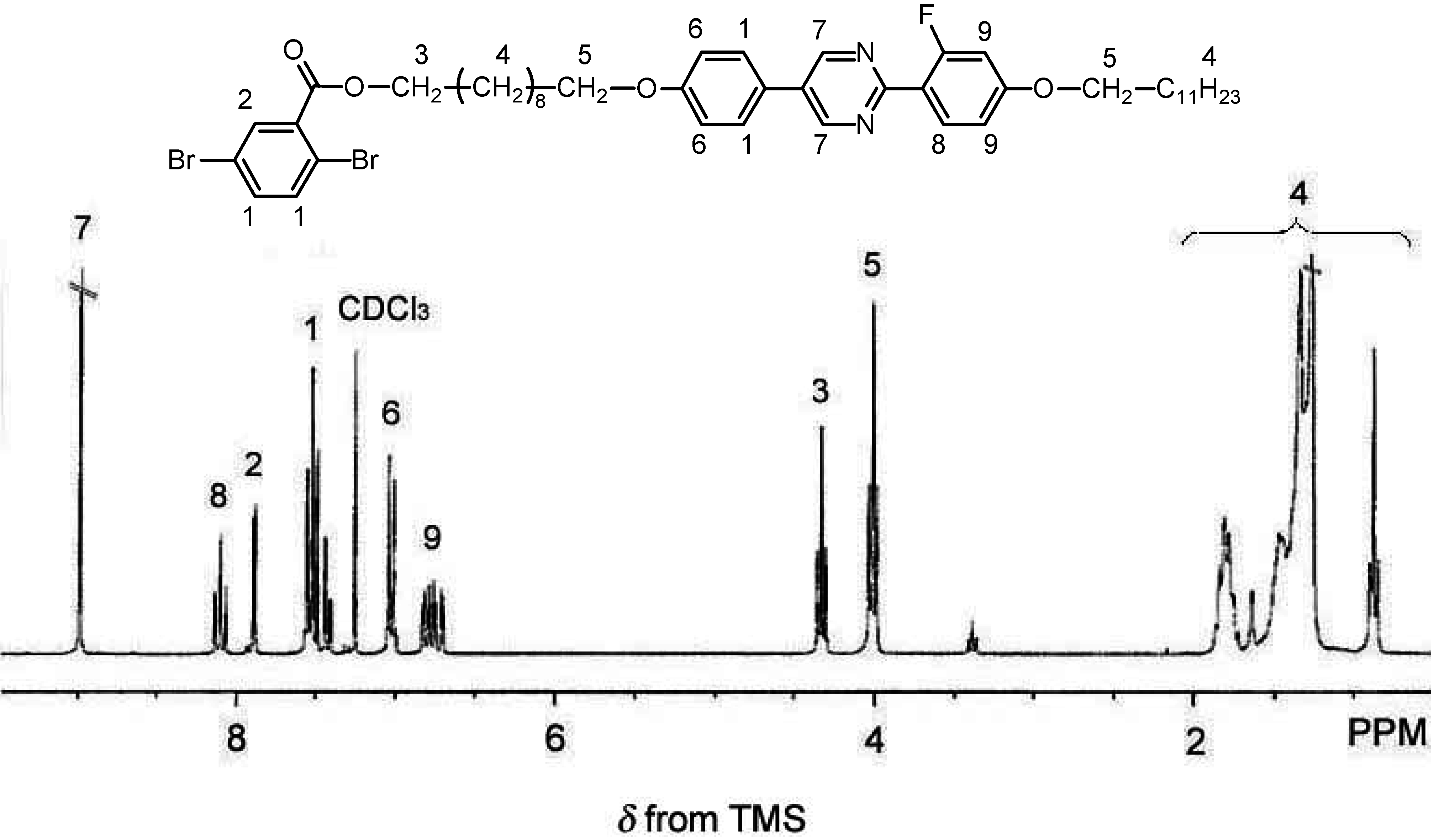
2.3. Polymerization


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Mn (×103)a, b | Mw (×103)a, c | MWDa | Yield (%)d | 19F δ (ppm)e |
|---|---|---|---|---|---|
| PolyPLCTh | 4.9 | 6.5 | 1.3 | 94 | 113 |
| PolyPLC2Th | 3.6 | 4.5 | 1.3 | 89 | 113 |
| PolyPLC3Th | 4.2 | 5.6 | 1.3 | 81 | 113 |
| PolyPN2Th | 2.1 | 3.0 | 1.4 | 50 | — |
- Polystyrene standard.
- Number average molecular weight.
- Weight average molecular weight.
- Calculated by (Ws / (Wp · Wm) ) · 100, Ws: weight of the polymer sample (g), Wp: molecular weight of mru of the polymer (g/mol), Wm: molar mass of the monomer (mol)
- δ from trifluorotoluene in 19F NMR.
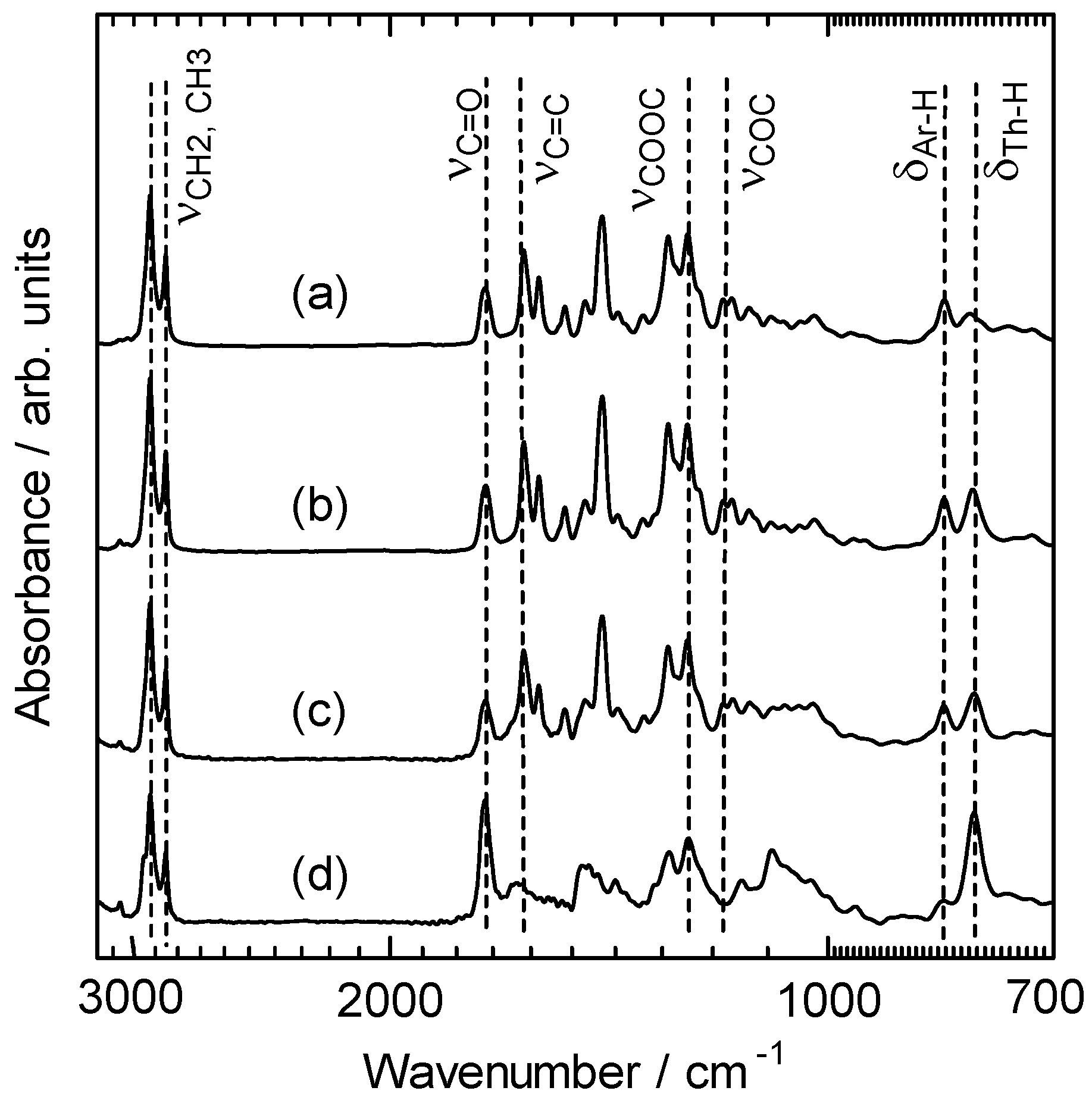
3. Results and Discussion
3.1. Ultraviolet–visible absorption spectra

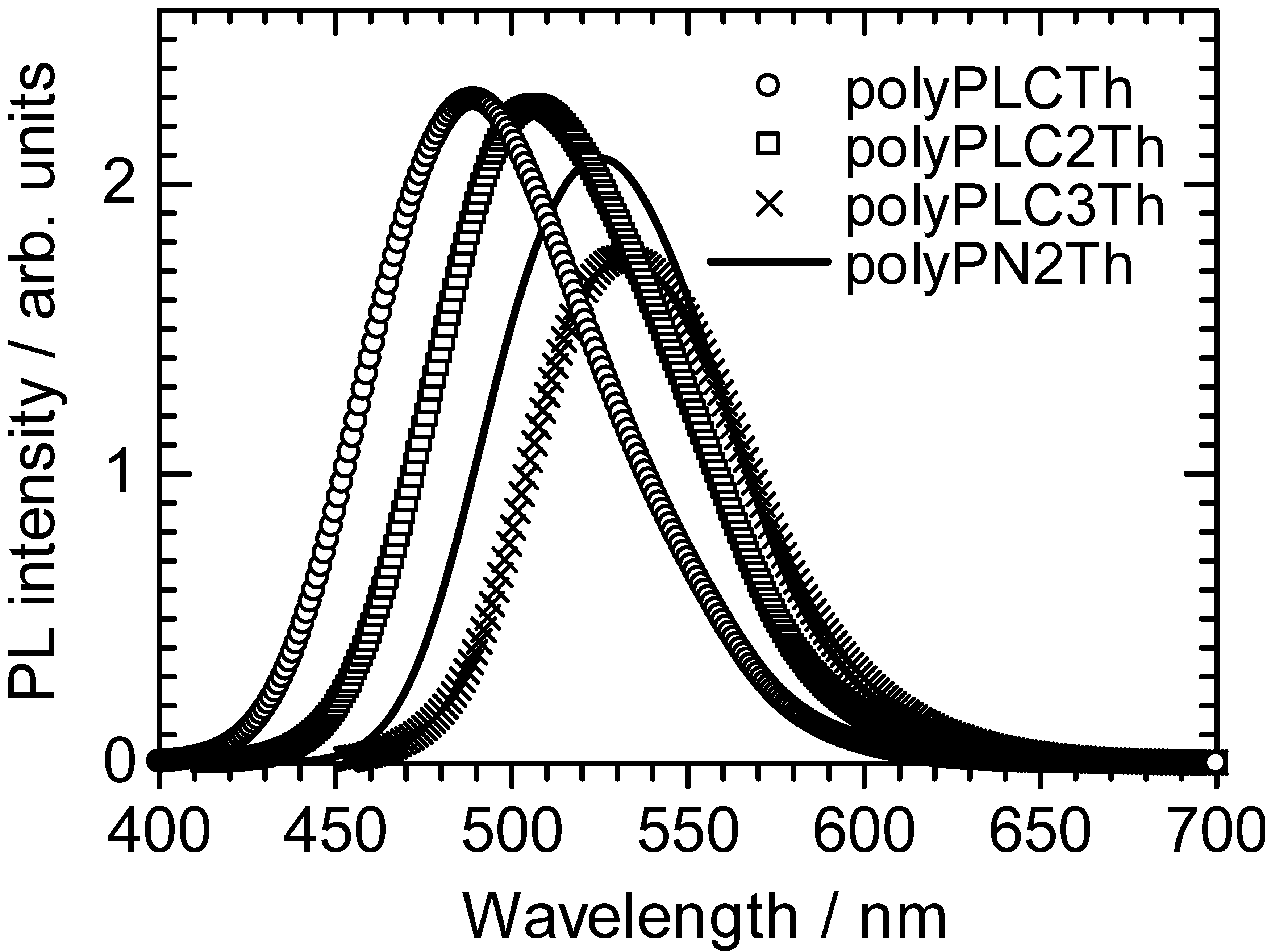
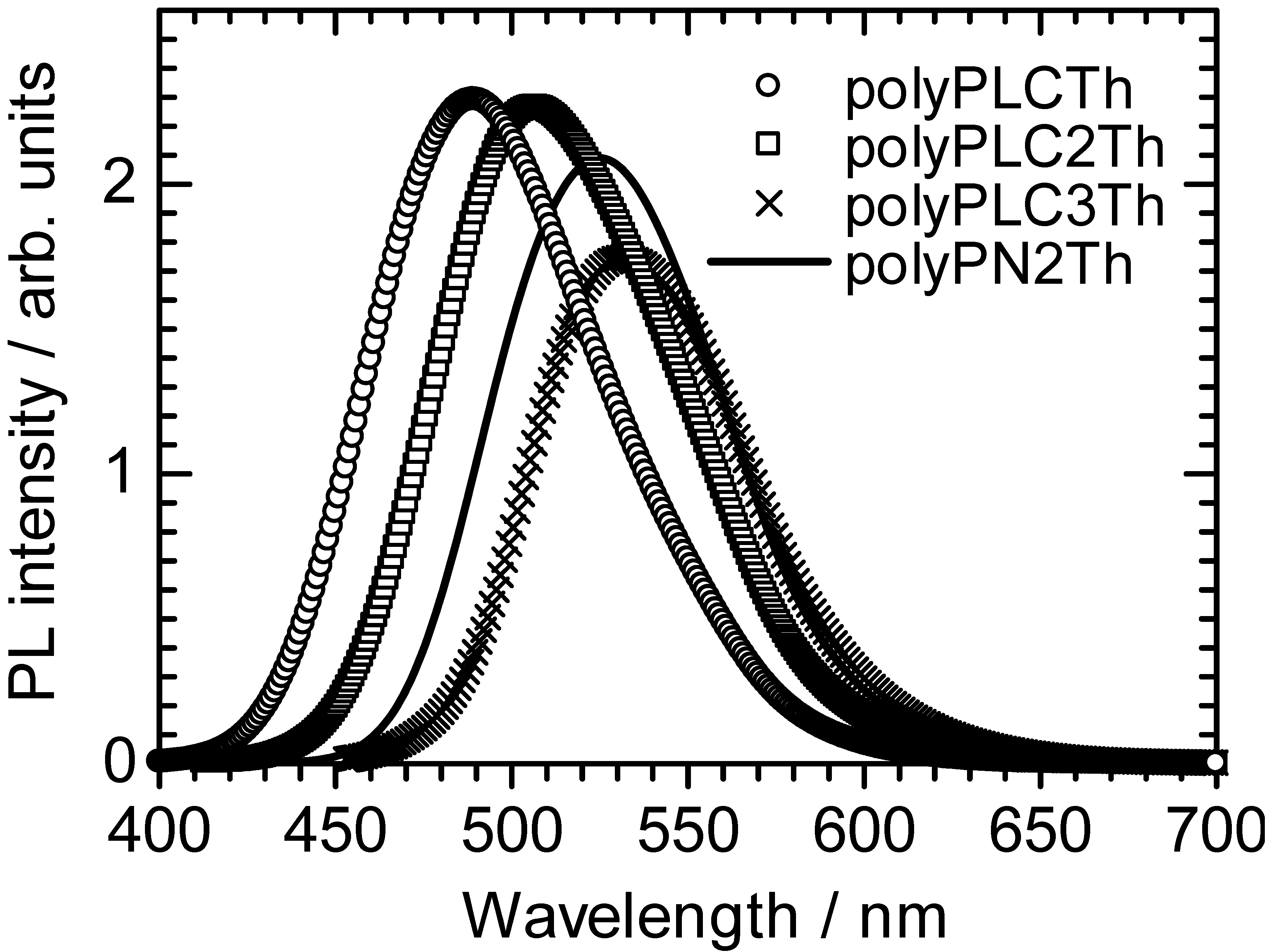
3.2. Photoluminescence
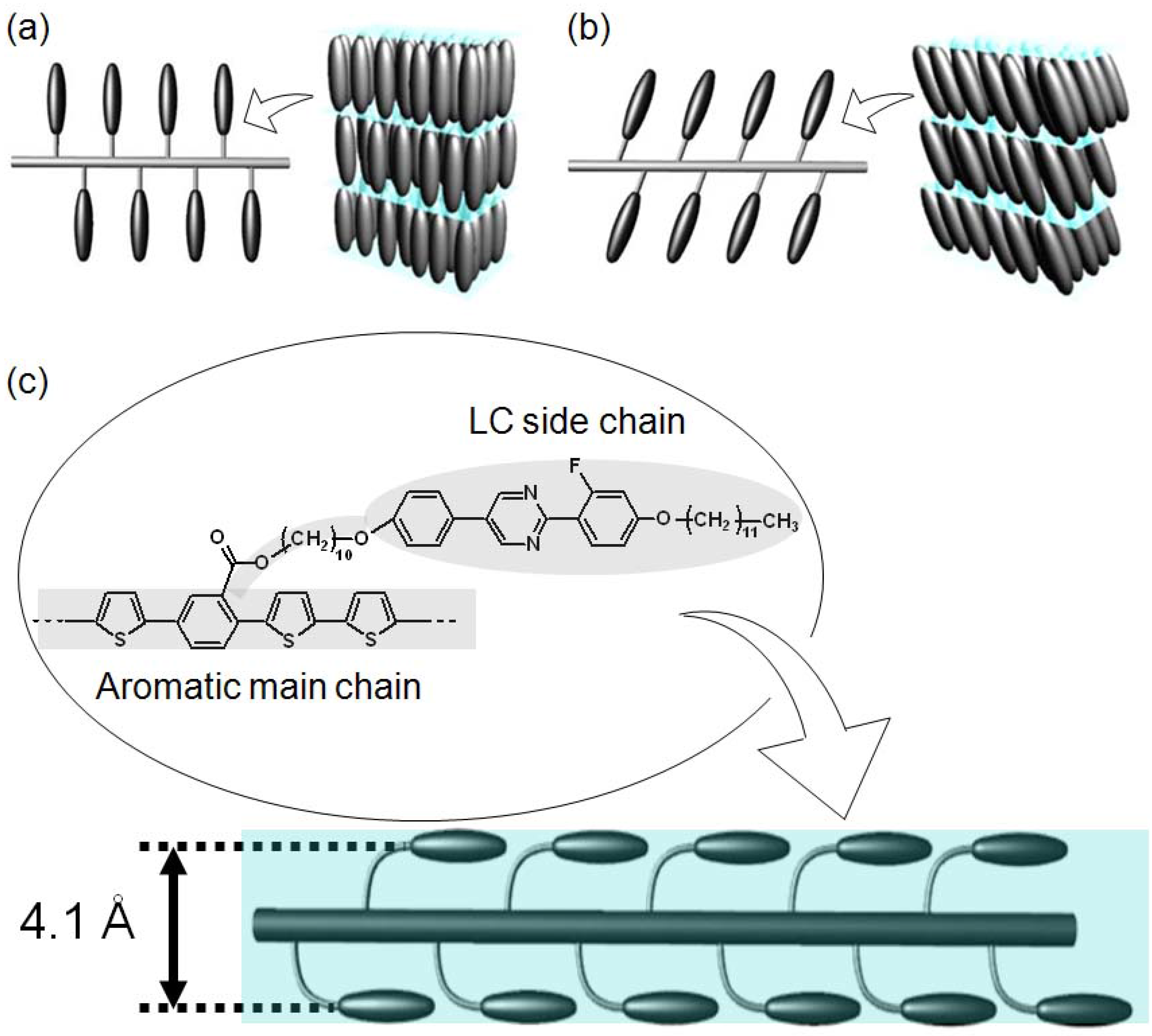
3.3. Polarizing optical microscopy observation


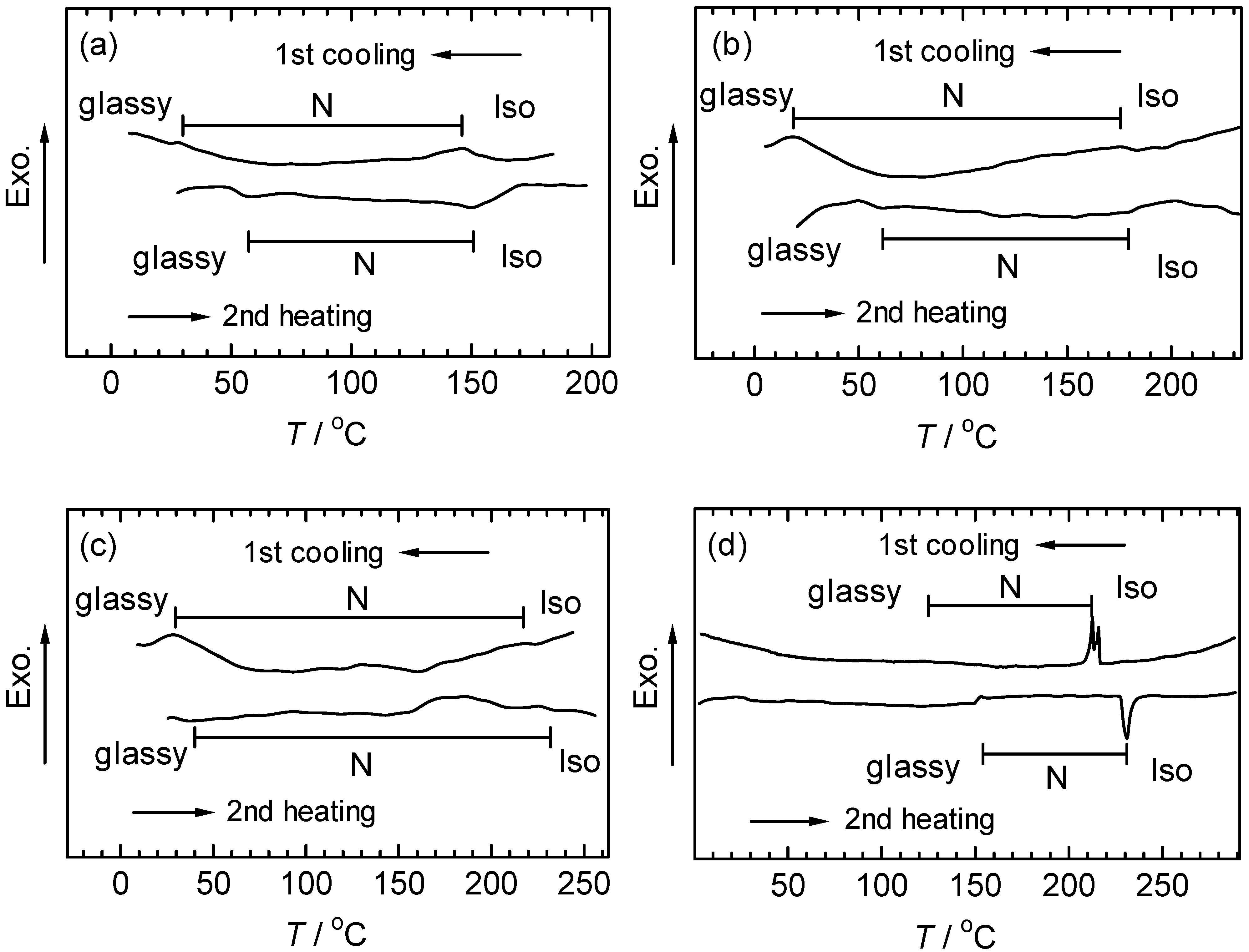
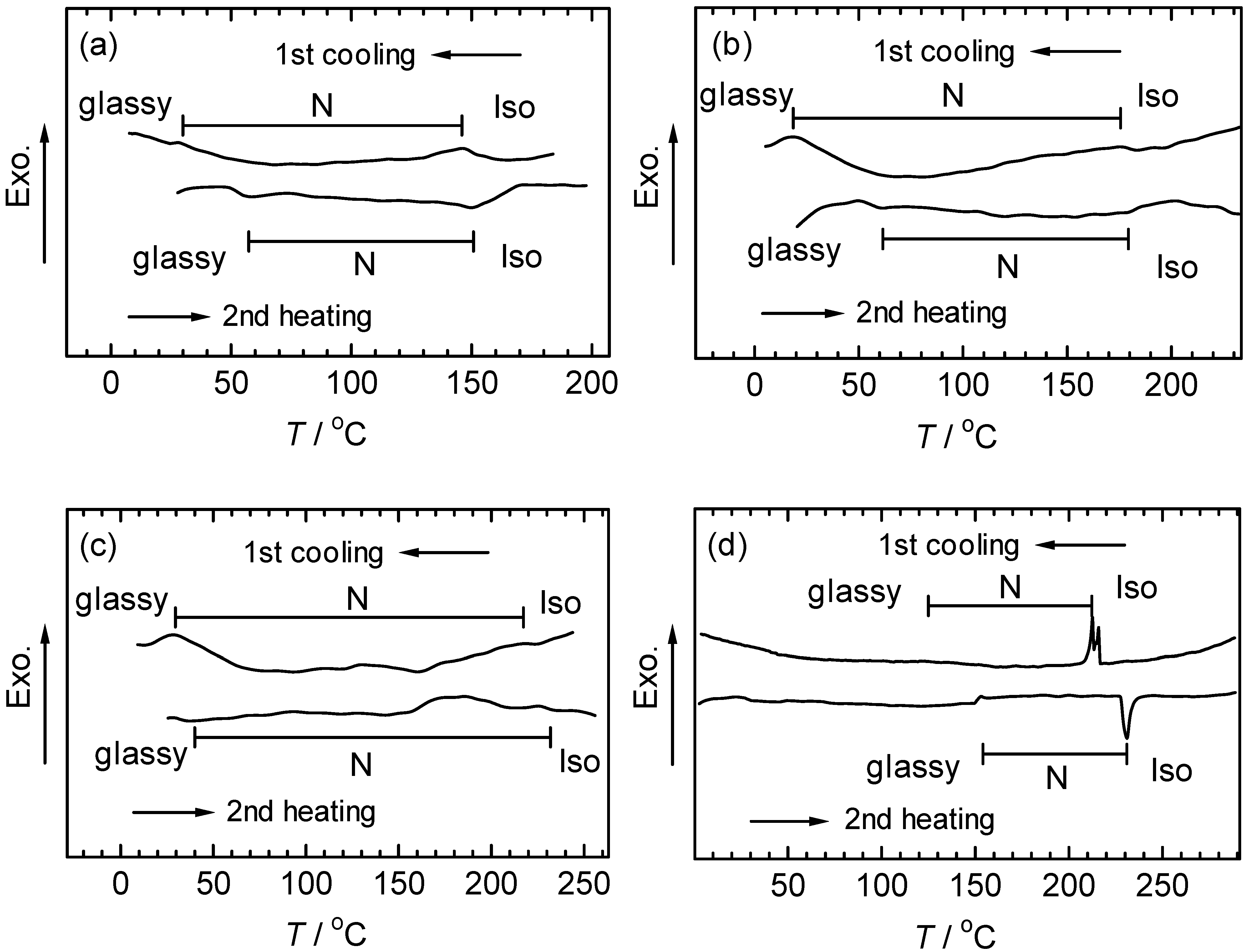
3.4. Differential scanning calorimetry
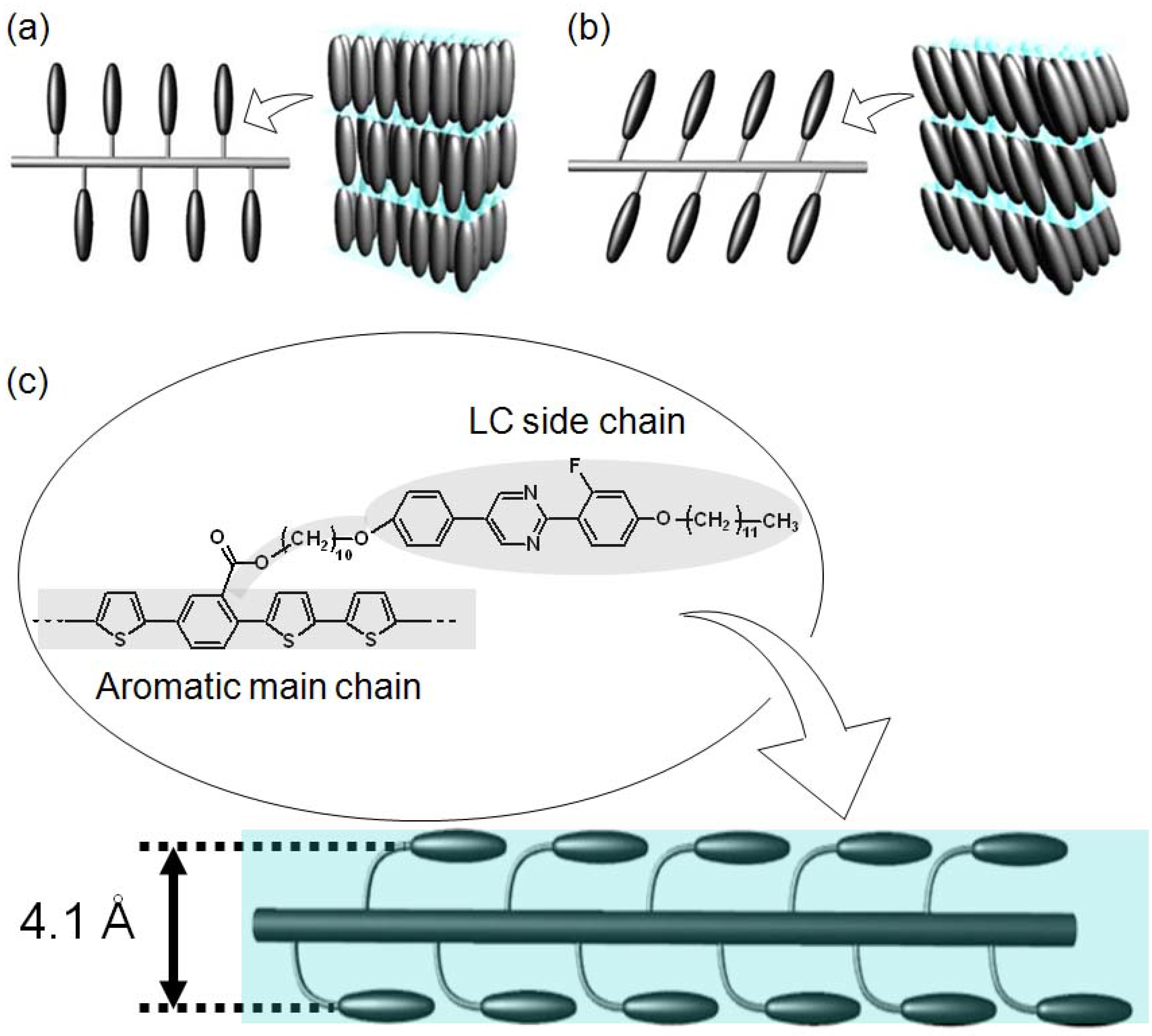
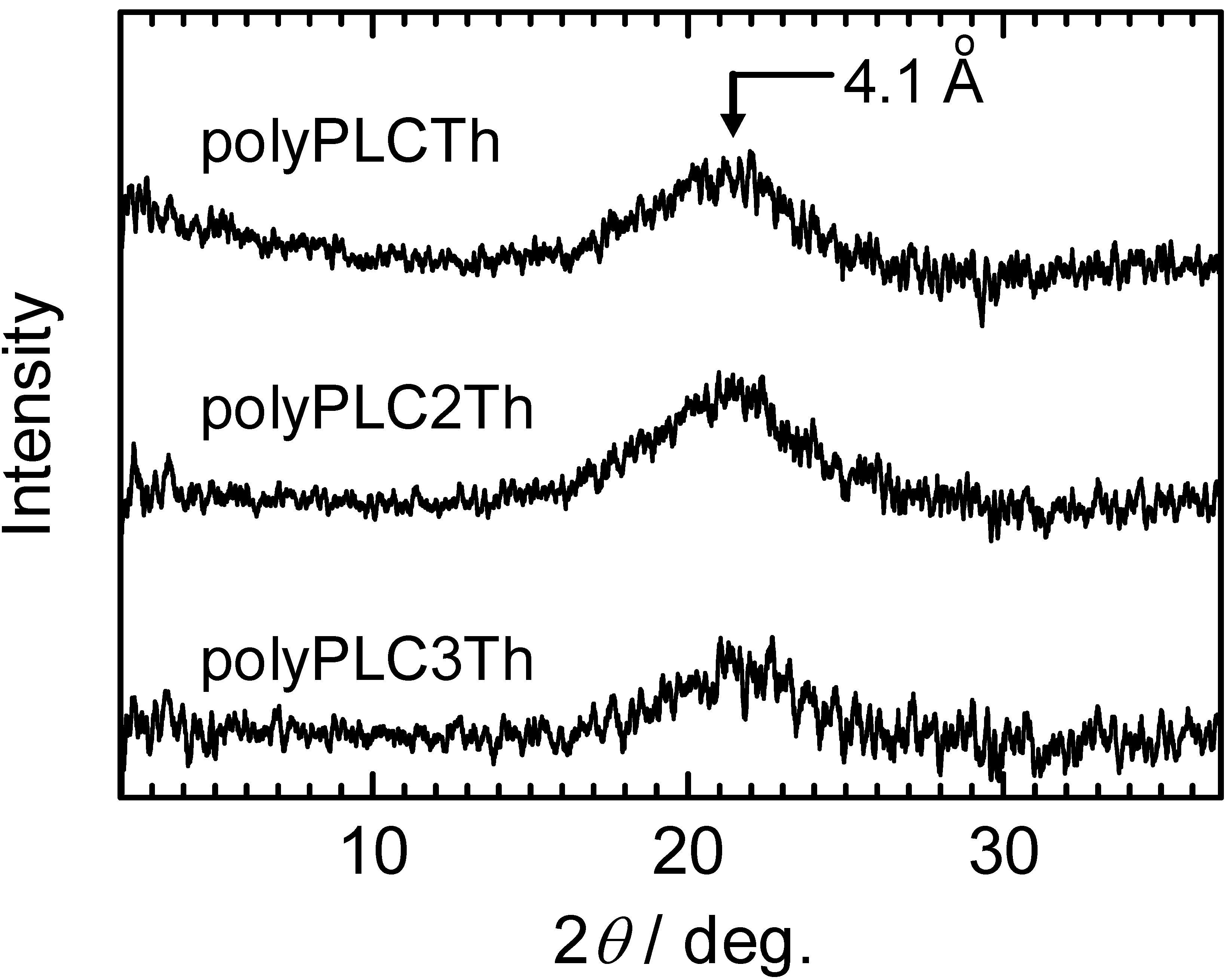
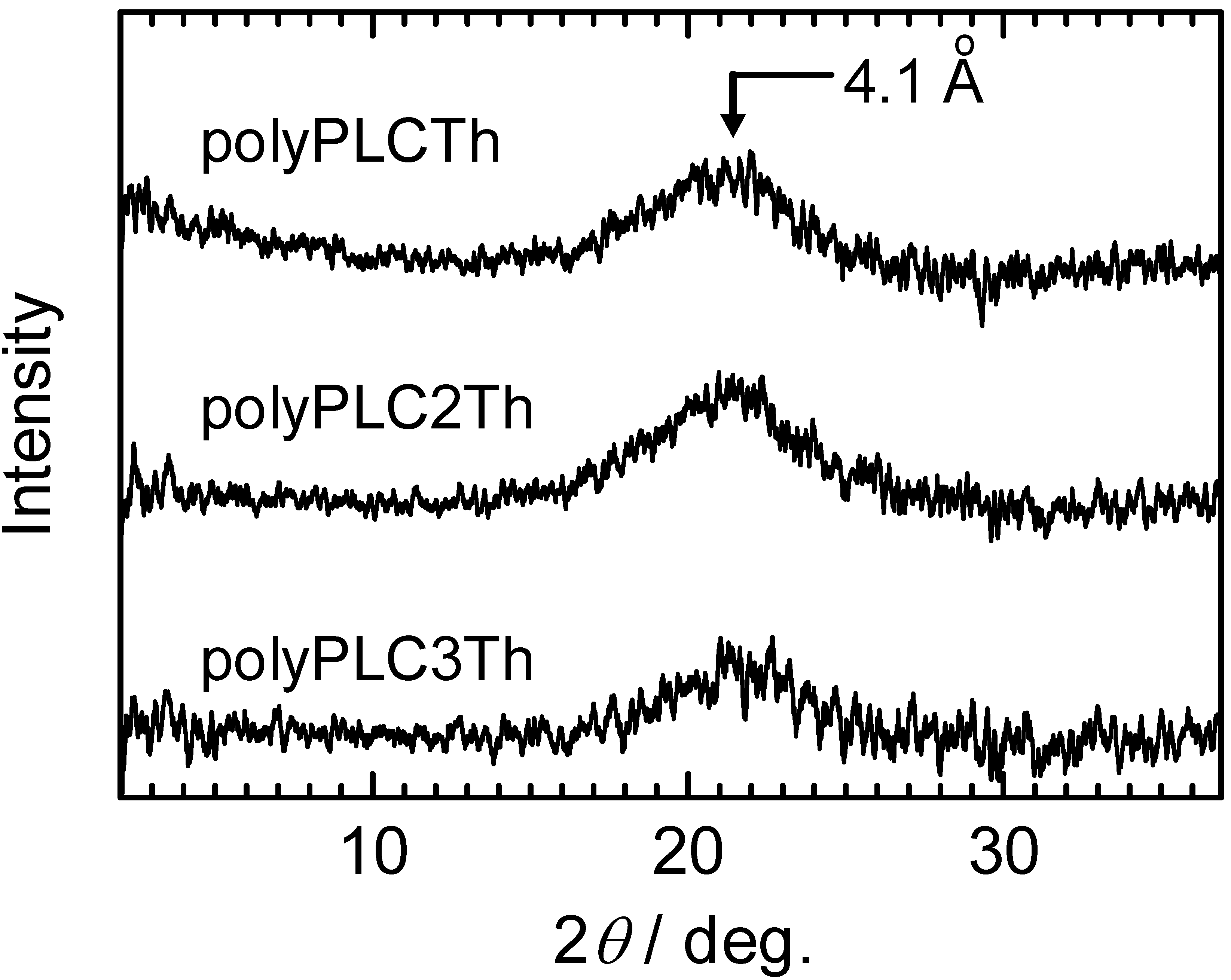
3.5. X-ray diffractometry
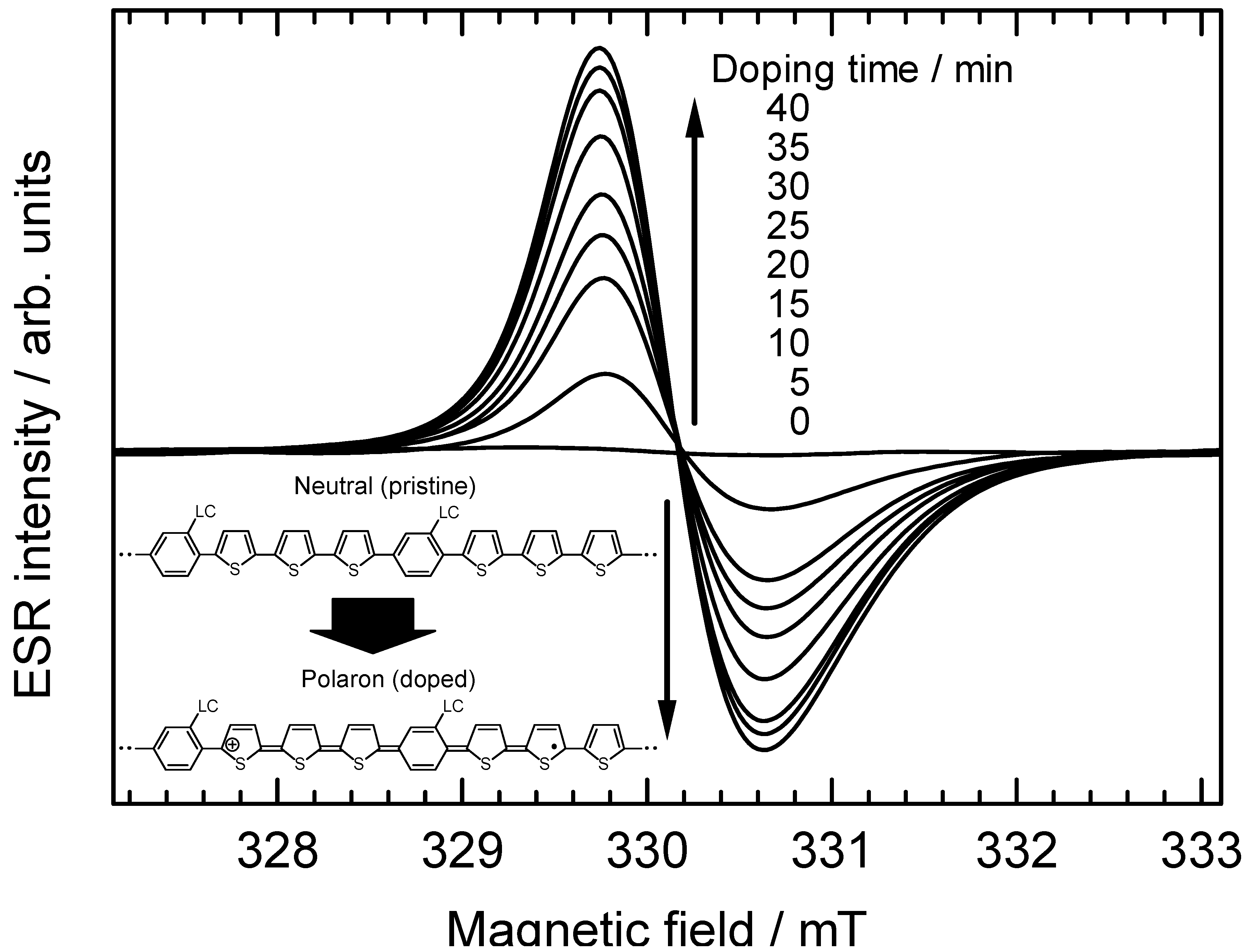
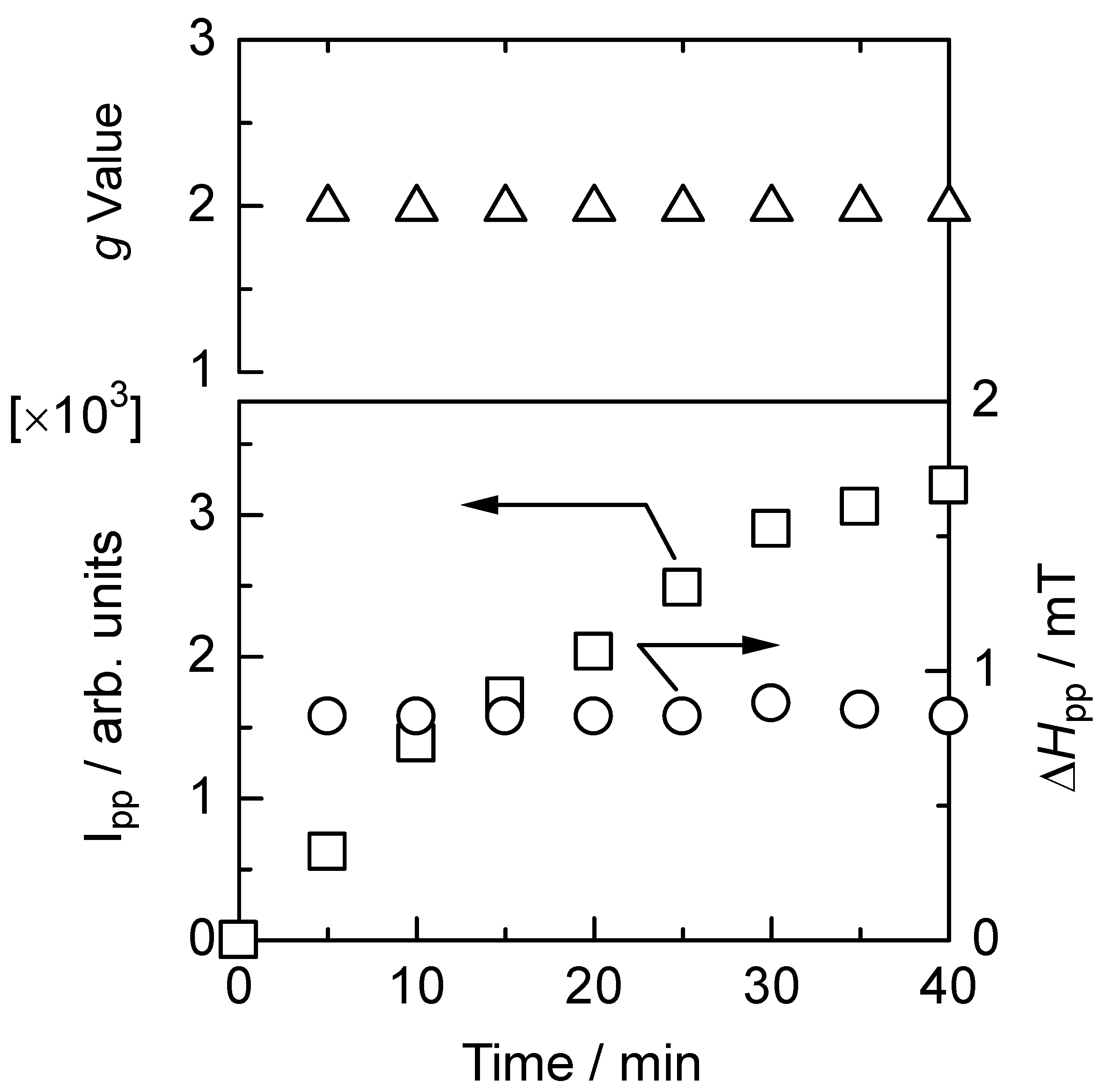
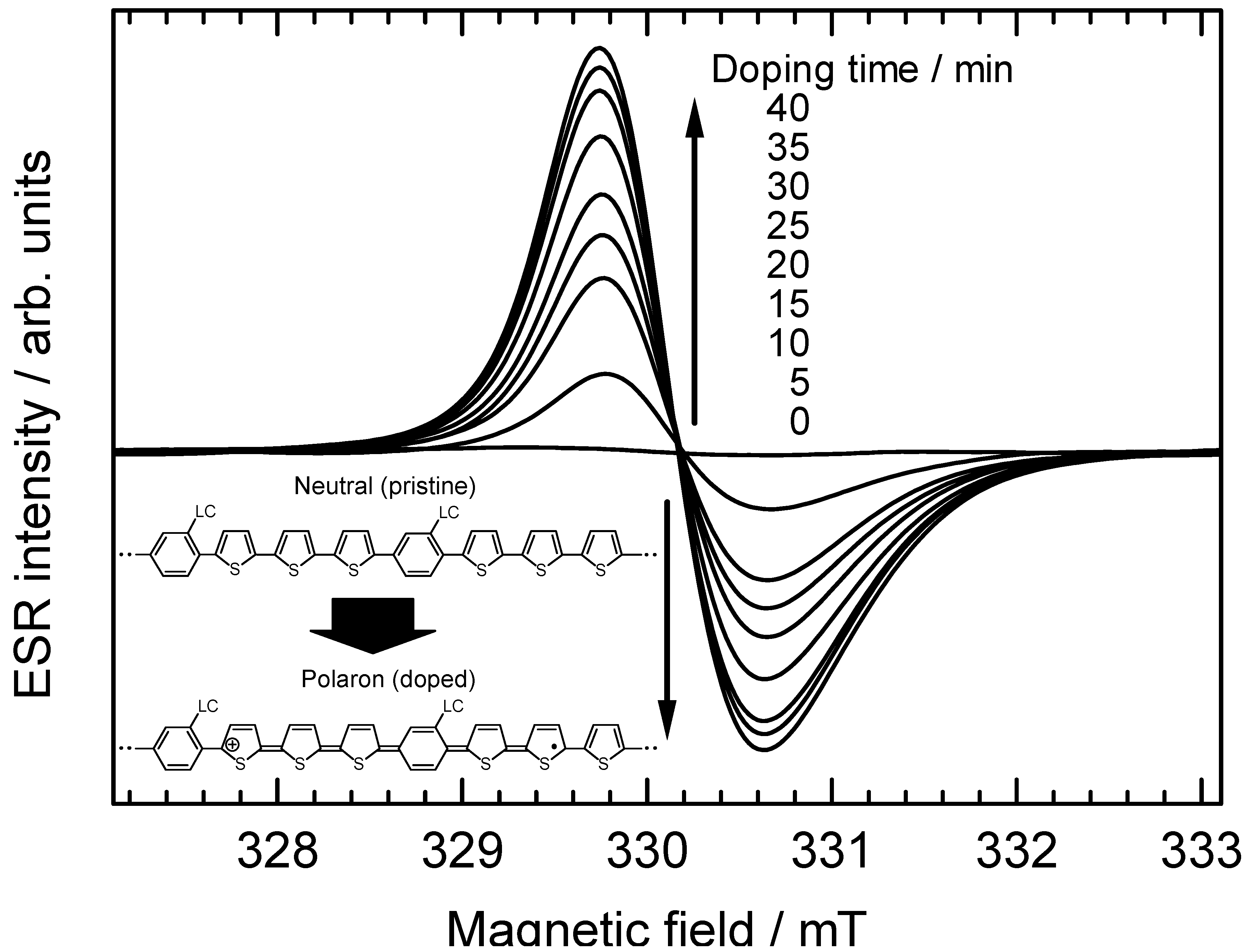
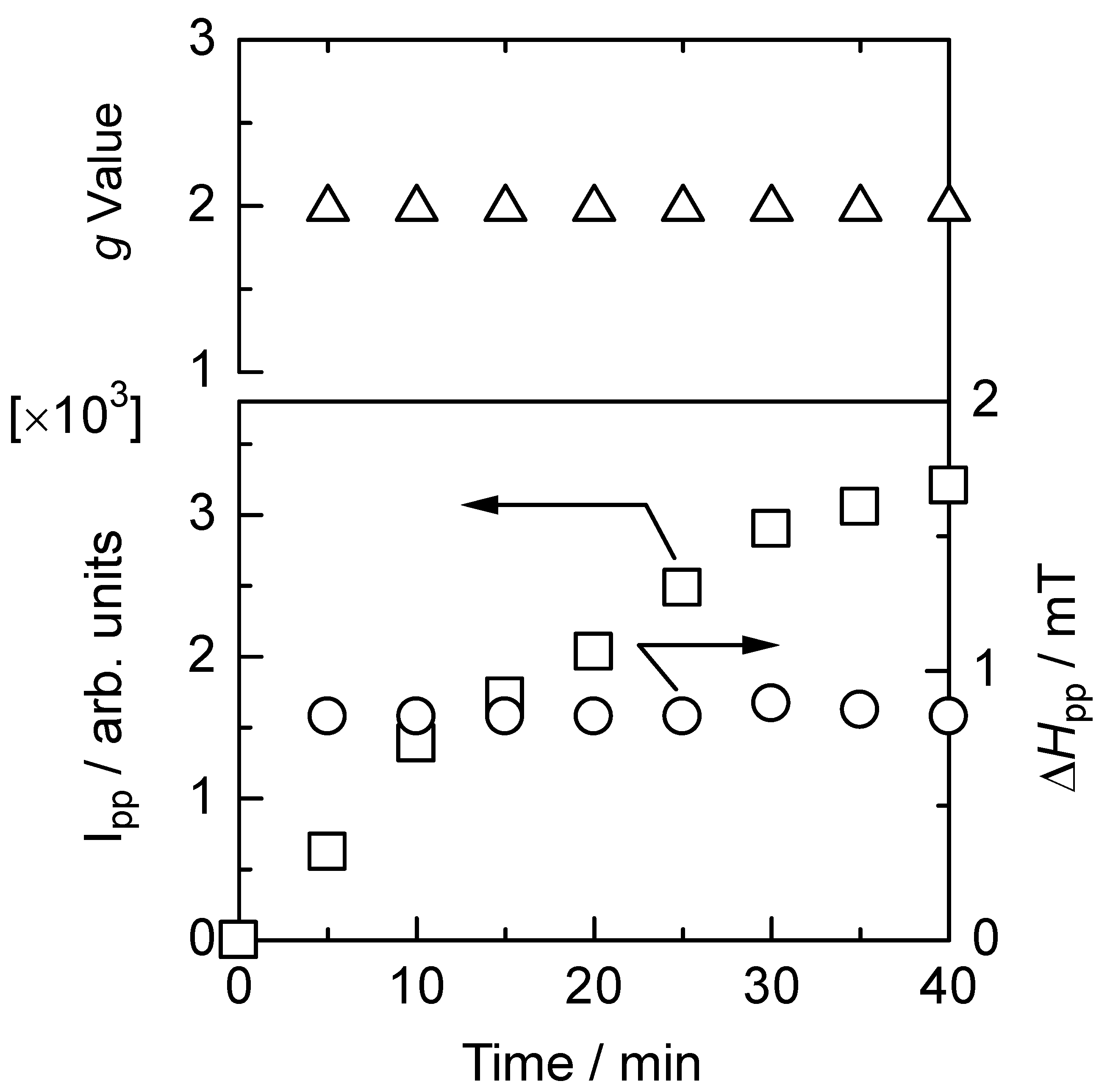
3.6. Electron spin resonance analysis
4. Conclusions
Acknowledgements
References
- Ueyama, K. Selected Issues in Liquid Crystal Elastomers and Gels. Macromolecules 2007, 40, 2277–2288. [Google Scholar] [CrossRef]
- Bergmann, F.H.G.; Finkelmann, H. Liquid-Crystalline Main-Chain Elastomers. Macromol. Rapid Commun. 1997, 18, 353–360. [Google Scholar]
- Dubois, C.J.; Barny, L.P.; Mauzac, M.; Noel, C. Behavior and Properties of Side Chain Thermotropic Liquid Crystal Polymers. Acta Polym. 1997, 48, 47–87. [Google Scholar] [CrossRef]
- Zhang, D.; Liu, Y.; Wan, X.; Zhou, Q. Synthesis of a New Side-Chain Type Liquid Crystal Polymer Poly[dicyclohexylvinylterephthalate]. Macromolecules 1999, 32, 4494–4496. [Google Scholar] [CrossRef]
- Choi, J.S.; Jin, H.S.; Park, W.J.; Cho, N.H.; Choi, K.S. Design and Synthesis of a New Side-Chain Liquid Crystalline Polymer by Metathesis Polymerization. Macromolecules 1994, 27, 309–311. [Google Scholar] [CrossRef]
- Osada, K.; Koike, M.; Tagawa, H.; Tokita, M.; Watanabe, J. Thermotropic Liquid Crystals of Main-Chain Polyesters having a Mesogenic 4,4'-Biphenyldicarboxylate Unit, 14a―Anisotropic Micro-Brownian Motions of Polymer in Smectic CA Phase. Macromol. Chem. Phys. 2004, 205, 1051–1057. [Google Scholar] [CrossRef]
- Nagata, Y.; Chujo, Y. Main-Chain-Type Organoboron Quinolate Polymers: Synthesis and Photoluminescence Properties. Macromolecules 2007, 40, 6–8. [Google Scholar] [CrossRef]
- Walba, M.D.; Keller, P.; Shao, R.; Clark, A.N.; Hillmyer, M.; Grubbs, H.R. Main-Chain Ferroelectric Liquid Crystal Oligomers by Acyclic Diene Metathesis Polymerization. J. Am. Chem. Soc. 1996, 118, 2740–2741. [Google Scholar] [CrossRef]
- Huang, C.; Zhang, Q.; Jákli, A. Nematic Anisotropic Liquid-Crystal Gels―Self-Assembled Nanocomposites with High Electromechanical Response. Adv. Funct. Mater. 2003, 13, 525–529. [Google Scholar] [CrossRef]
- Chen, H.S.; Conger, M.B.; Mastrangelo, C.J.; Kende, S.A.; Kim, U.D. Synthsis and Optical Properties of Thermotropic Polythiophene and Poly(p-phenylene) Derivatives. Macromolecules 1998, 31, 8051–8057. [Google Scholar] [CrossRef]
- Kwak, G.; Jin, H.S.; Park, W.J.; Gal, S.Y. Ionic Polyacetylene with Aromatic Functional Groups: Synthesis and Properties. Macromol. Chem. Phys. 2008, 209, 1769–1777. [Google Scholar] [CrossRef]
- Pemg, Z.; Zhang, J.; Xu, B. New Poly(p-phenylene vinylene) Derivatives Exhibiting High Photoluminescence Quantum Efficiencies. Macromolecules 1999, 32, 5162–5164. [Google Scholar] [CrossRef]
- Samitsu, S.; Iida, T.; Fujimori, M.; Heike, S.; Hasizume, T.; Shimomura, T.; Ito, K. Conductivity Measurements of PEDOT Nanowires on Nanoelectrodes. Synth. Met. 2005, 152, 497–500. [Google Scholar] [CrossRef]
- Kurosawa, S.; Teja, S.A.; Kowalik, J.; Tolbert, L. Supercritical carbon dioxide processing of conducting composites of polypyrrole and porous crosslinked polystyrene. Polymer 2006, 47, 2997–3004. [Google Scholar] [CrossRef]
- Goldberg, B.I.; Crowe, R.H.; Newman, R.P.; Heeger, J.A.; MacDiarmid, G.A. Electron spin resonance of polyacetylene and AsF5-doped polyacetylene. J. Chem. Phys. 1979, 70, 1132–1136. [Google Scholar] [CrossRef]
- Saf, R.; Swoboda, P.; Lechner, A.; Hummel, K. Synthesis and characterization of conjugated polymers with imino nitroxide groups. Macromol. Chem. Phys. 1996, 197, 1439–1447. [Google Scholar] [CrossRef]
- Goto, H.; Nimori, S.; Akagi, K. Synthsis and properties of mono-subatituted liquid crystalline polyacetylene derivatives―doping, magnetic orientation, and photo-isomerization. Synth. Met. 2005, 155, 576–587. [Google Scholar] [CrossRef]
- Goto, H.; Dai, X.; Ueoka, T.; Akagi, K. Synthesis and Properties of Polymers from Monosubstituted Acetylene Derivatives Bearing Ferroelectric Liquid Crystalline Groups. Macromolecules 2004, 37, 4783–4793. [Google Scholar] [CrossRef]
- Ibison, P.; Foot, S.J.P.; Brown, W.J. Preparation and characterization of polypyrrole, N-substituted with liquid crystalline moieties. Synth. Met. 1996, 76, 297–300. [Google Scholar] [CrossRef]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kawabata, K.; Goto, H. Liquid Crystalline π-Conjugated Copolymers Bearing a Pyrimidine Type Mesogenic Group. Materials 2009, 2, 22-37. https://doi.org/10.3390/ma2010022
Kawabata K, Goto H. Liquid Crystalline π-Conjugated Copolymers Bearing a Pyrimidine Type Mesogenic Group. Materials. 2009; 2(1):22-37. https://doi.org/10.3390/ma2010022
Chicago/Turabian StyleKawabata, Kohsuke, and Hiromasa Goto. 2009. "Liquid Crystalline π-Conjugated Copolymers Bearing a Pyrimidine Type Mesogenic Group" Materials 2, no. 1: 22-37. https://doi.org/10.3390/ma2010022
APA StyleKawabata, K., & Goto, H. (2009). Liquid Crystalline π-Conjugated Copolymers Bearing a Pyrimidine Type Mesogenic Group. Materials, 2(1), 22-37. https://doi.org/10.3390/ma2010022