
Carbonization of MOF-5/Polyaniline Composites to N,O-Doped Carbon/ZnO/ZnS and N,O-Doped Carbon/ZnO Composites with High Specific Capacitance, Specific Surface Area and Electrical Conductivity

 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
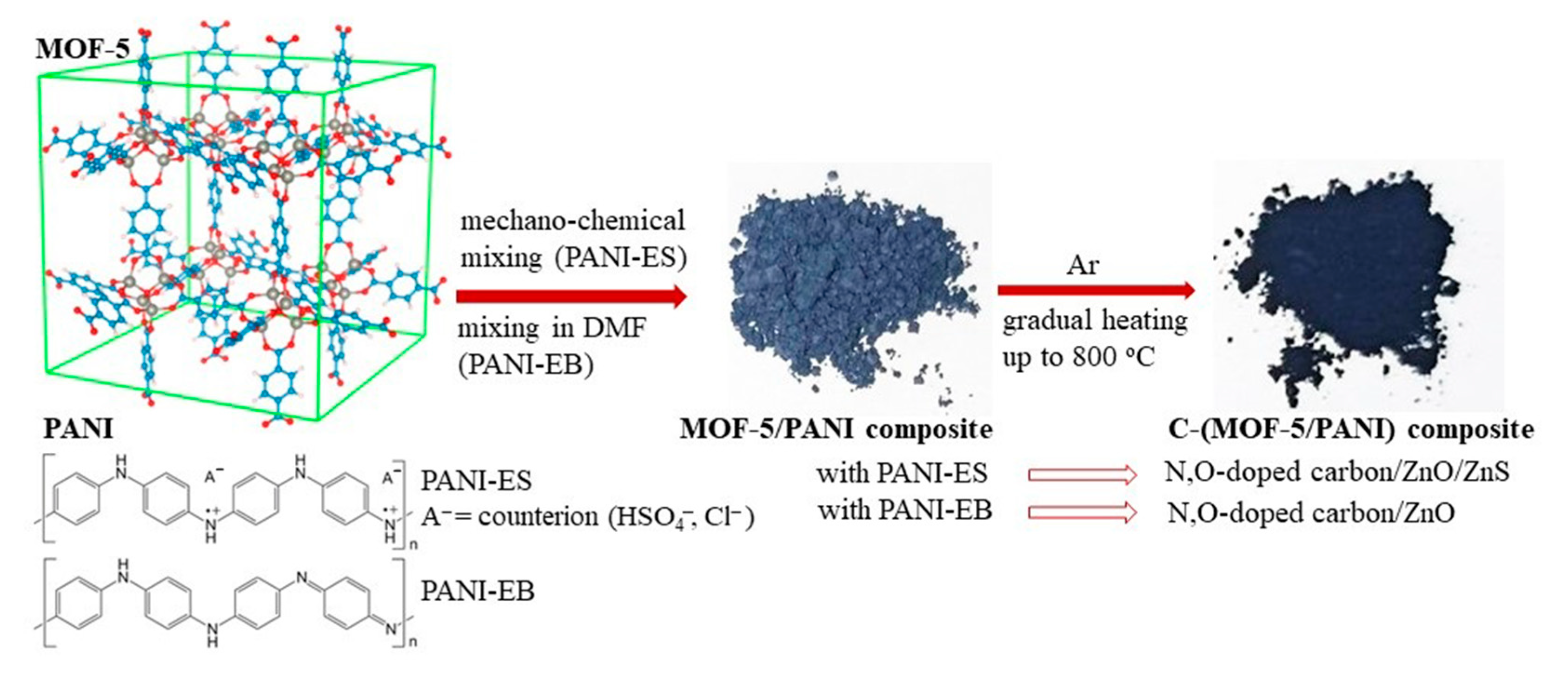
2.2. Syntheses of MOF-5/PANI Composite Precursors
2.3. Syntheses of C-(MOF-5/PANI) Composites, C-MOF, C-PANI-ES, and C-PANI-EB
2.4. Postsynthetic Acid Etching Treatment
2.5. Characterization of Synthesized Materials
3. Results
3.1. Carbonization Yield and the Bulk Elemental Composition of C-(MOF-5/PANI) Samples Determined by the Elemental Microanalysis and FAAS
3.2. The Elemental Composition of C-(MOF-5/PANI) Samples Determined by EDX
3.3. Morphology of C-(MOF-5/PANI) Composites—SEM
3.4. Crystalline Structure of C-(MOF-5/PANI) Samples—XRPD
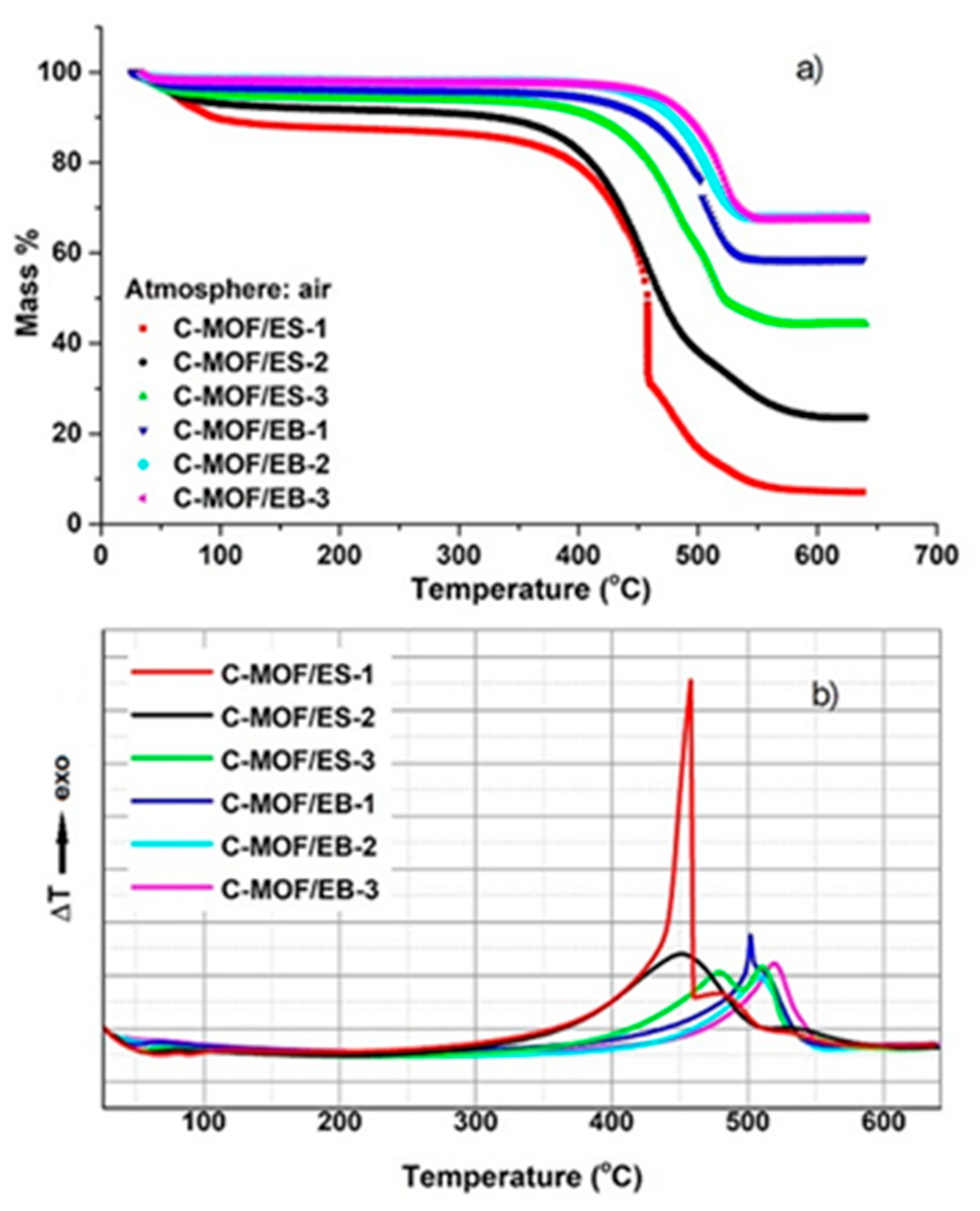
3.5. Thermal Analysis of C-(MOF-5/PANI) Samples—TGA and DTA
3.6. Electrical Conductivity of C-(MOF-5/PANI) Composites and Carbonized Individual Components of Their Precursors
3.7. Specific Surface Area of C-(MOF-5/PANI) Composites and Carbonized Individual Components of Their Precursors—N2 Physisorption Measurements
3.8. Raman Spectra of C-(MOF-5/PANI) Composites
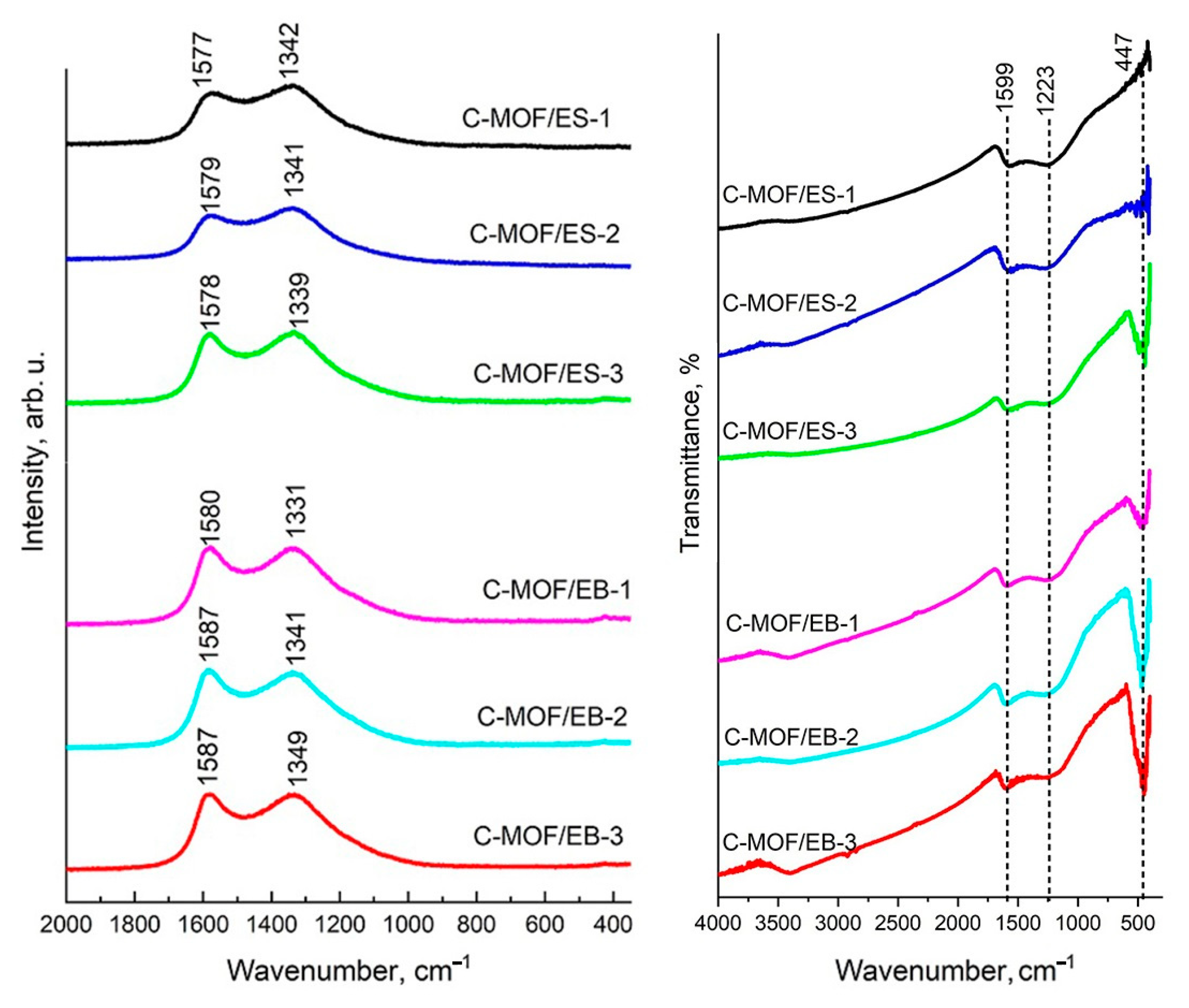
3.9. FTIR Spectra of C-(MOF-5/PANI) Composites
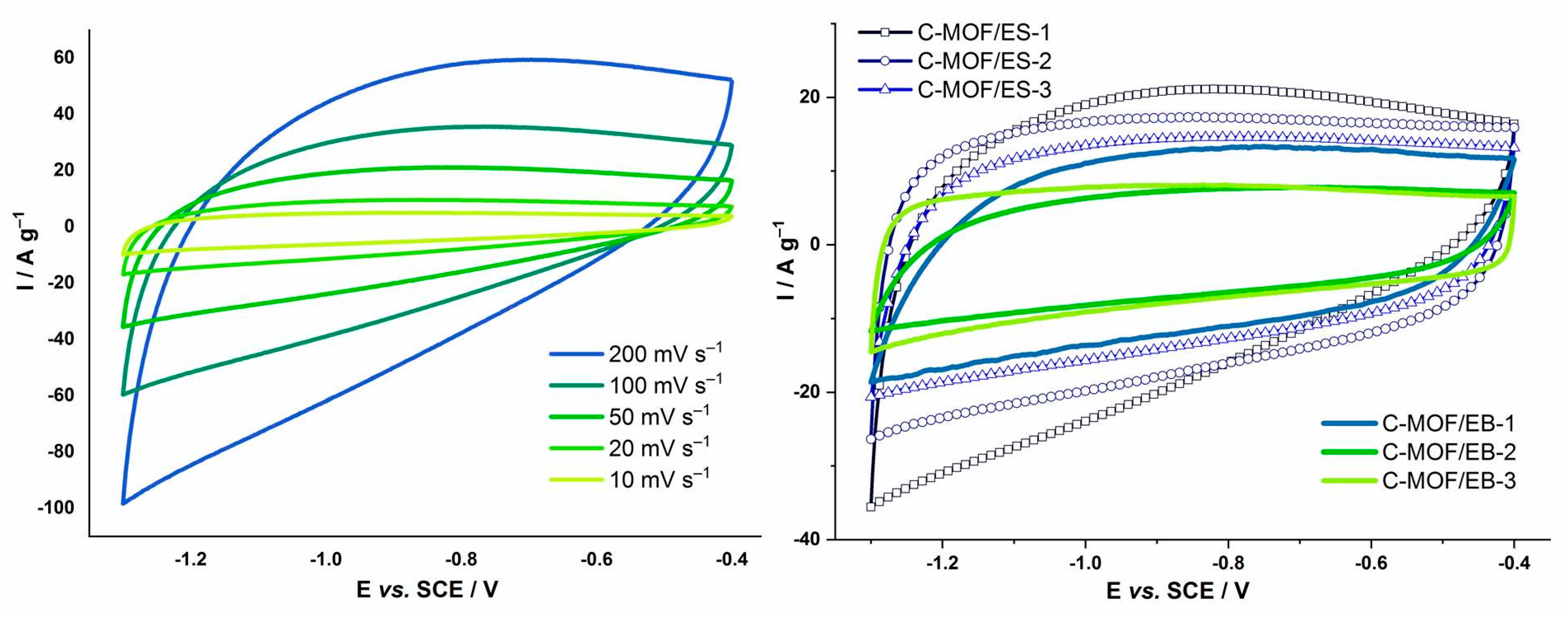
3.10. Cyclic Voltammetry and Specific Capacitance of C-(MOF-5/PANI) Composites
3.11. Characterization of the Acid-Etched Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deng, Y.; Xie, Y.; Zou, K.; Ji, X. Review on recent advances in nitrogen-doped carbons: Preparations and applications in supercapacitors. J. Mater. Chem. A 2015, 4, 1144–1173. [Google Scholar] [CrossRef]
- Zhou, W.; Xie, L.; Gao, J.; Nazari, R.; Zhao, H.; Meng, X.; Sun, F.; Zhao, G.; Ma, J. Selective H2O2 electrosynthesis by O-doped and transition-metal-O-doped carbon cathodes via O2 electroreduction: A critical review. Chem. Eng. J. 2021, 410, 128368. [Google Scholar] [CrossRef]
- Ćirić-Marjanović, G.; Pašti, I.; Mentus, S. One-dimensional nitrogen-containing carbon nanostructures. Prog. Mater. Sci. 2015, 69, 61–182. [Google Scholar] [CrossRef]
- Park, S.; Kim, J.; Kwon, K. A review on biomass-derived N-doped carbons as electrocatalysts in electrochemical energy applications. Chem. Eng. J. 2022, 446, 137116. [Google Scholar] [CrossRef]
- Zhao, S.; Wang, D.; Amal, R.; Dai, L. Carbon-Based Metal-Free Catalysts for Key Reactions Involved in Energy Conversion and Storage. Adv. Mater. 2018, 31, e1801526. [Google Scholar] [CrossRef]
- Kim, J.-I.; Park, S.-J. Effect of nitrogen-containing groups on enhanced capacitive behaviors of multi-walled carbon nanotubes. J. Solid State Chem. 2011, 184, 2184–2189. [Google Scholar] [CrossRef]
- Li, L.; Liu, Y.; Geng, X.; An, B. Synthesis and electrochemical performance of nitrogen-doped carbon nanotubes. Acta Phys.–Chim. Sin. 2011, 27, 443–448. [Google Scholar]
- Yang, J.; Ju, Z.; Jiang, Y.; Xing, Z.; Xi, B.; Feng, J.; Xiong, S. Enhanced Capacity and Rate Capability of Nitrogen/Oxygen Dual-Doped Hard Carbon in Capacitive Potassium-Ion Storage. Adv. Mater. 2017, 30, 1700104. [Google Scholar] [CrossRef]
- Huang, F.; Liu, W.; Wang, Q.; Wang, F.; Yao, Q.; Yan, D.; Xu, H.; Xia, B.Y.; Deng, J. Natural N/O-doped hard carbon for high performance K-ion hybrid capacitors. Electrochim. Acta 2020, 354, 136701. [Google Scholar] [CrossRef]
- Liu, Q.; Sun, S.; Zhang, L.; Luo, Y.; Yang, Q.; Dong, K.; Fang, X.; Zheng, D.; Alshehri, A.A.; Sun, X. N, O-doped carbon foam as metal-free electrocatalyst for efficient hydrogen production from seawater. Nano Res. 2022, 15, 8922–8927. [Google Scholar] [CrossRef]
- Jia, N.; Weng, Q.; Shi, Y.; Shi, X.; Chen, X.; Chen, P.; An, Z.; Chen, Y. N-doped carbon nanocages: Bifunctional electrocatalysts for the oxygen reduction and evolution reactions. Nano Res. 2017, 11, 1905–1916. [Google Scholar] [CrossRef]
- Ni, W.; Gao, Y.; Zhang, Y.; Younus, H.A.; Guo, X.; Ma, C.; Zhang, Y.; Duan, J.; Zhang, J.; Zhang, S. O-Doping Boosts the Electrochemical Oxygen Reduction Activity of a Single Fe Site in Hydrophilic Carbon with Deep Mesopores. ACS Appl. Mater. Interfaces 2019, 11, 45825–45831. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Chen, M.; Xu, R.; Zeng, S.; Yang, H.; Ye, S.; Liu, F.; Wu, X.; Yu, Y. CNT Interwoven Nitrogen and Oxygen Dual-Doped Porous Carbon Nanosheets as Free-Standing Electrodes for High-Performance Na-Se and K-Se Flexible Batteries. Adv. Mater. 2018, 30, e1805234. [Google Scholar] [CrossRef] [PubMed]
- Hilal, M.E.; Younus, H.A.; Chaemchuen, S.; Dekyvere, S.; Zen, X.; He, D.; Park, J.; Han, T.; Verpoort, F. Sacrificial ZnO nanorods drive N and O dual-doped carbon towards trifunctional electrocatalysts for Zn–air batteries and self-powered water splitting devices. Catal. Sci. Technol. 2021, 11, 4149–4161. [Google Scholar] [CrossRef]
- Pašti, I.A.; Ležaić, A.J.; Gavrilov, N.M.; Ćirić-Marjanović, G.; Mentus, S.V. Nanocarbons derived from polymers for electrochemical energy conversion and storage–A review. Synth. Met. 2018, 246, 267–281. [Google Scholar] [CrossRef]
- Cui, H.; Xu, J.; Shi, J.; Yan, N.; Liu, Y.; Zhang, S. Zinc Nitrate as an Activation Agent for the Synthesis of Nitrogen-Doped Porous Carbon and Its Application in CO2 Adsorption. Energy Fuels 2020, 34, 6069–6076. [Google Scholar] [CrossRef]
- Ćirić-Marjanović, G. Recent advances in polyaniline research: Polymerization mechanisms, structural aspects, properties and applications. Synth. Met. 2013, 177, 1–47. [Google Scholar] [CrossRef]
- Ćirić-Marjanović, G.; Pašti, I.; Gavrilov, N.; Janošević, A.; Mentus, S. Carbonised polyaniline and polypyrrole: Towards advanced nitrogen-containing carbon materials. Chem. Pap. 2013, 67, 781–813. [Google Scholar] [CrossRef]
- Meng, Y.; Voiry, D.; Goswami, A.; Zou, X.; Huang, X.; Chhowalla, M.; Liu, Z.; Asefa, T. N-, O-, and S-Tridoped Nanoporous Carbons as Selective Catalysts for Oxygen Reduction and Alcohol Oxidation Reactions. J. Am. Chem. Soc. 2014, 136, 13554–13557. [Google Scholar] [CrossRef]
- Gavrilov, N.; Pašti, I.A.; Mitrić, M.; Travas-Sejdić, J.; Ćirić-Marjanović, G.; Mentus, S.V. Electrocatalysis of oxygen reduction reaction on polyaniline-derived nitrogen-doped carbon nanoparticle surfaces in alkaline media. J. Power Sources 2012, 220, 306–316. [Google Scholar] [CrossRef]
- Kyotani, M.; Goto, H.; Suda, K.; Nagai, T.; Matsui, Y.; Akagi, K. Tubular-Shaped Nanocarbons Prepared from Polyaniline Synthesized by a Self-Assembly Process and Their Electrical Conductivity. J. Nanosci. Nanotechnol. 2008, 8, 1999–2004. [Google Scholar] [CrossRef]
- Mentus, S.; Ćirić-Marjanović, G.; Trchová, M.; Stejskal, J. Conducting carbonized polyaniline nanotubes. Nanotechnology 2009, 20, 245601. [Google Scholar] [CrossRef] [PubMed]
- Trchová, M.; Konyushenko, E.N.; Stejskal, J.; Kovářová, J.; Ćirić-Marjanović, G. The conversion of polyaniline nanotubes to nitrogen-containing carbon nanotubes and their comparison with multi-walled carbon nanotubes. Polym. Degrad. Stab. 2009, 94, 929–938. [Google Scholar] [CrossRef]
- Janošević, A.; Pašti, I.; Gavrilov, N.; Mentus, S.; Ćirić-Marjanović, G.; Krstić, J.; Stejskal, J. Micro/mesoporous conducting carbonized polyaniline 5-sulfosalicylate nanorods/nanotubes: Synthesis, characterization and electrocatalysis. Synth. Met. 2011, 161, 2179–2184. [Google Scholar] [CrossRef]
- Janošević, A.; Pašti, I.; Gavrilov, N.; Mentus, S.; Krstić, J.; Mitrić, M.; Travas-Sejdic, J.; Ćirić-Marjanović, G. Microporous conducting carbonized polyaniline nanorods: Synthesis, characterization and electrocatalytic properties. Microporous Mesoporous Mater. 2012, 152, 50–57. [Google Scholar] [CrossRef]
- Ćirić-Marjanović, G.; Mentus, S.; Pašti, I.; Gavrilov, N.; Krstić, J.; Travas-Sejdic, J.; Strover, L.T.; Kopecká, J.; Moravková, Z.; Trchová, M.; et al. Synthesis, Characterization, and Electrochemistry of Nanotubular Polypyrrole and Polypyrrole-Derived Carbon Nanotubes. J. Phys. Chem. C 2014, 118, 14770–14784. [Google Scholar] [CrossRef]
- Gavrilov, N.; Pašti, I.A.; Vujković, M.; Travas-Sejdic, J.; Ćirić-Marjanović, G.; Mentus, S.V. High-performance charge storage by N-containing nanostructured carbon derived from polyaniline. Carbon 2012, 50, 3915–3927. [Google Scholar] [CrossRef]
- Czaja, A.U.; Trukhan, N.; Müller, U. Industrial applications of metal–organic frameworks. Chem. Soc. Rev. 2009, 38, 1284–1293. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-C.; Kitagawa, S. Metal–Organic Frameworks (MOFs). Chem. Soc. Rev. 2014, 43, 5415–5418. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Liang, F.; Li, Y.; Wu, J.; Guan, S.; Wu, M.; Xie, S.; Luo, M.; Ma, D. A 2D Porous Zinc-Organic Framework Platform for Loading of 5-Fluorouracil. Inorganics 2022, 10, 202. [Google Scholar] [CrossRef]
- Qin, L.; Li, Y.; Liang, F.; Li, L.; Lan, Y.; Li, Z.; Lu, X.; Yang, M.; Ma, D. A microporous 2D cobalt-based MOF with pyridyl sites and open metal sites for selective adsorption of CO2. Microporous Mesoporous Mater. 2022, 341, 112098. [Google Scholar] [CrossRef]
- Yap, M.H.; Fow, K.L.; Chen, G.Z. Synthesis and applications of MOF-derived porous nanostructures. Green Energy Environ. 2017, 2, 218–245. [Google Scholar] [CrossRef]
- Kim, H.-C.; Huh, S. Porous Carbon-Based Supercapacitors Directly Derived from Metal–Organic Frameworks. Materials 2020, 13, 4215. [Google Scholar] [CrossRef]
- Zhi, M.; Xiang, C.; Li, J.; Li, M.; Wu, N. Nanostructured carbon–metal oxide composite electrodes for supercapacitors: A review. Nanoscale 2013, 5, 72–88. [Google Scholar] [CrossRef]
- Chaikittisilp, W.; Torad, N.; Li, C.; Imura, M.; Suzuki, N.; Ishihara, S.; Ariga, K.; Yamauchi, Y. Synthesis of Nanoporous Carbon-Cobalt-Oxide Hybrid Electrocatalysts by Thermal Conversion of Metal-Organic Frameworks. Chem.–A Eur. J. 2014, 20, 4217–4221. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Eddaoudi, M.; O’Keeffe, M.; Yaghi, O.M. Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nature 1999, 402, 276–279. [Google Scholar] [CrossRef] [Green Version]
- Ming, Y.; Purewal, J.; Liu, D.; Sudik, A.; Xu, C.; Yang, J.; Veenstra, M.; Rhodes, K.; Soltis, R.; Warner, J.; et al. Thermophysical properties of MOF-5 powders. Microporous Mesoporous Mater. 2014, 185, 235–244. [Google Scholar] [CrossRef]
- Bisercic, M.S.; Marjanović, B.; Vasiljević, B.N.; Mentus, S.; Zasońska, B.A.; Ćirić-Marjanović, G. The quest for optimal water quantity in the synthesis of metal-organic framework MOF-5. Microporous Mesoporous Mater. 2018, 278, 23–29. [Google Scholar] [CrossRef]
- Tranchemontagne, D.J.; Hunt, J.R.; Yaghi, O.M. Room temperature synthesis of metal-organic frameworks: MOF-5, MOF-74, MOF-177, MOF-199, and IRMOF-0. Tetrahedron 2008, 64, 8553–8557. [Google Scholar] [CrossRef]
- Yang, S.J.; Kim, T.; Im, J.H.; Kim, Y.S.; Lee, K.; Jung, H.; Park, C.R. MOF-Derived Hierarchically Porous Carbon with Exceptional Porosity and Hydrogen Storage Capacity. Chem. Mater. 2012, 24, 464–470. [Google Scholar] [CrossRef]
- Hu, J.; Wang, H.; Gao, Q.; Guo, H. Porous carbons prepared by using metal–organic framework as the precursor for supercapacitors. Carbon 2010, 48, 3599–3606. [Google Scholar] [CrossRef]
- Khan, I.A.; Badshah, A.; Haider, N.; Ullah, S.; Anjum, D.H.; Nadeem, M.A. Porous carbon as electrode material in direct ethanol fuel cells (DEFCs) synthesized by the direct carbonization of MOF-5. J. Solid State Electrochem. 2014, 18, 1545–1555. [Google Scholar] [CrossRef]
- Biserčić, M.S.; Marjanović, B.; Zasońska, B.A.; Stojadinović, S.; Ćirić-Marjanović, G. Novel microporous composites of MOF-5 and polyaniline with high specific surface area. Synth. Met. 2020, 262, 116348. [Google Scholar] [CrossRef]
- Nascimento, G.M.D.; Silva, C.H.; Temperini, M.L. Spectroscopic characterization of the structural changes of polyaniline nanofibers after heating. Polym. Degrad. Stab. 2008, 93, 291–297. [Google Scholar] [CrossRef]
- Nasseh, N.; Arghavan, F.S.; Rodriguez-Couto, S.; Panahi, A.H.; Esmati, M.; A-Musawi, T.J. Preparation of activated carbon@ZnO composite and its application as a novel catalyst in catalytic ozonation process for metronidazole degradation. Adv. Powder Technol. 2019, 31, 875–885. [Google Scholar] [CrossRef]
- Trchová, M.; Šeděnková, I.; Konyushenko, E.N.; Stejskal, J.; Holler, P.; Ćirić-Marjanović, G. Evolution of Polyaniline Nanotubes: The Oxidation of Aniline in Water. J. Phys. Chem. B 2006, 110, 9461–9468. [Google Scholar] [CrossRef] [PubMed]
- Marjanović, B.; Juranić, I.; Ćirić-Marjanović, G. Revised Mechanism of Boyland−Sims Oxidation. J. Phys. Chem. A 2011, 115, 3536–3550. [Google Scholar] [CrossRef]
- Kataria, N.; Garg, V.; Jain, M.; Kadirvelu, K. Preparation, characterization and potential use of flower shaped Zinc oxide nanoparticles (ZON) for the adsorption of Victoria Blue B dye from aqueous solution. Adv. Powder Technol. 2016, 27, 1180–1188. [Google Scholar] [CrossRef]
- Devi, P.G.; Velu, A.S. Synthesis, structural and optical properties of pure ZnO and Co doped ZnO nanoparticles prepared by the co-precipitation method. J. Theor. Appl. Phys. 2016, 10, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.-N.; Ching, W.Y. Electronic, optical, and structural properties of some wurtzite crystals. Phys. Rev. B 1993, 48, 4335–4351. [Google Scholar] [CrossRef]
- Kisi, E.H.; Elcombe, M.M. u parameters for the wurtzite structure of ZnS and ZnO using powder neutron diffraction. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1989, 45, 1867–1870. [Google Scholar] [CrossRef]
- Sayeed, A.; Bhattacharyya, S.; Subramanyam, S.D.C. conductivity measurements on amorphous conducting carbon. Mater. Sci. Eng. C 1995, 3, 231–233. [Google Scholar] [CrossRef]
- Yin, J.; Xia, X.; Xiang, L.; Zhao, X. Conductivity and polarization of carbonaceous nanotubes derived from polyaniline nanotubes and their electrorheology when dispersed in silicone oil. Carbon 2010, 48, 2958–2967. [Google Scholar] [CrossRef]
- Wahab, R.; Ansari, S.G.; Kim, Y.-S.; Dhage, M.S.; Seo, H.K.; Song, M.; Shin, H.-S. Effect of annealing on the conversion of ZnS to ZnO nanoparticles synthesized by the sol-gel method using zinc acetate and thiourea. Met. Mater. Int. 2009, 15, 453–458. [Google Scholar] [CrossRef]
- Caglar, M.; Ilican, S.; Caglar, Y.; Yakuphanoglu, F. Electrical conductivity and optical properties of ZnO nanostructured thin film. Appl. Surf. Sci. 2009, 255, 4491–4496. [Google Scholar] [CrossRef]
- Goktas, A.; Aslan, F.; Tumbul, A. Nanostructured Cu-doped ZnS polycrystalline thin films produced by a wet chemical route: The influences of Cu doping and film thickness on the structural, optical and electrical properties. J. Sol-Gel Sci. Technol. 2015, 75, 45–53. [Google Scholar] [CrossRef]
- Ferrari, A.C. Raman spectroscopy of graphene and graphite: Disorder, electron–phonon coupling, doping and nonadiabatic effects. Solid State Commun. 2007, 143, 47–57. [Google Scholar] [CrossRef]
- Sadezky, A.; Muckenhuber, H.; Grothe, H.; Niessner, R.; Pöschl, U. Raman microspectroscopy of soot and related carbonaceous materials: Spectral analysis and structural information. Carbon 2005, 43, 1731–1742. [Google Scholar] [CrossRef]
- Ćirić-Marjanović, G.; Trchová, M.; Stejskal, J. The chemical oxidative polymerization of aniline in water: Raman spectroscopy. J. Raman Spectrosc. 2008, 39, 1375–1387. [Google Scholar] [CrossRef]
- Rodríguez, N.A.; Parra, R.; Grela, M.A. Structural characterization, optical properties and photocatalytic activity of MOF-5 and its hydrolysis products: Implications on their excitation mechanism. RSC Adv. 2015, 5, 73112–73118. [Google Scholar] [CrossRef]
- Najib, S.; Bakan, F.; Abdullayeva, N.; Bahariqushchi, R.; Kasap, S.; Franzò, G.; Sankir, M.; Sankir, N.D.; Mirabella, S.; Erdem, E. Tailoring morphology to control defect structures in ZnO electrodes for high-performance supercapacitor devices. Nanoscale 2020, 12, 16162–16172. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Rho, H.; Choi, Y.-J.; Park, J.-G. Raman spectroscopy of ZnS nanostructures. J. Raman Spectrosc. 2012, 43, 906–910. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd ed.; Wiley: Chichester, UK, 2001; pp. 101–326. [Google Scholar]
- Ţucureanu, V.; Matei, A.; Avram, A.M. FTIR Spectroscopy for Carbon Family Study. Crit. Rev. Anal. Chem. 2016, 46, 502–520. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Cheng, B.; Song, H.; Chen, X. Preparation and electrochemical performance of polyaniline-based carbon nanotubes as electrode material for supercapacitor. Electrochim. Acta 2010, 55, 7021–7027. [Google Scholar] [CrossRef]
- Tang, S.; Sun, G.; Qi, J.; Sun, S.; Guo, J.; Xin, Q.; Haarberg, G.M. Review of New Carbon Materials as Catalyst Supports in Direct Alcohol Fuel Cells. Chin. J. Catal. 2010, 31, 12–17. [Google Scholar] [CrossRef]
- Sahu, V.; Shekhar, S.; Ahuja, P.; Gupta, G.; Singh, S.K.; Sharma, R.K.; Singh, G. Synthesis of hydrophilic carbon black; role of hydrophilicity in maintaining the hydration level and protonic conduction. RSC Adv. 2013, 3, 3917–3924. [Google Scholar] [CrossRef]
- Shrivastav, V.; Sundriyal, S.; Goel, P.; Saha, A.; Tiwari, U.K.; Deep, A. A novel zinc sulfide impregnated carbon composite derived from zeolitic imidazolate framework-8 for sodium-ion hybrid solid-state flexible capacitors. Nanoscale Adv. 2021, 3, 6164–6175. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Z.; Yuan, R.; Lin, Y.; Yan, J.; Zhang, J.; Lu, Z.; Luo, D.; Pietrasik, J.; Bockstaller, M.R.; et al. ZnO/carbon hybrids derived from polymer nanocomposite precursor materials for pseudocapacitor electrodes with high cycling stability. Polymer 2018, 137, 370–377. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, X.; Pan, L.; Li, H.; Sun, Z.; Sun, C.; Tay, B.K. Carbon nanotube–ZnO nanocomposite electrodes for supercapacitors. Solid State Ion. 2009, 180, 1525–1528. [Google Scholar] [CrossRef]
- Yu, J.; Fu, N.; Zhao, J.; Liu, R.; Li, F.; Du, Y.; Yang, Z. High Specific Capacitance Electrode Material for Supercapacitors Based on Resin-Derived Nitrogen-Doped Porous Carbons. ACS Omega 2019, 4, 15904–15911. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Li, B.; Yang, H.; Zhou, Y.; Xu, X.; Li, J. Metal-organic framework–derived carbon/N-doped three-dimensional reduced graphene oxide composite with high capacitive deionization performance. Mater. Today Sustain. 2022, 20, 100228. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C-(MOF-5/PANI) Sample | m (g) | Yield (%) | Content (wt.%) | ||||
|---|---|---|---|---|---|---|---|
| C | N | H | Zn | O | |||
| C-MOF/ES-1 | 0.243 | 48.6 | 65.61 | 9.95 | 1.53 | 7.76 | 15.15 |
| C-MOF/ES-2 | 0.222 | 44.4 | 53.97 | 8.30 | 1.42 | 15.96 | 20.35 |
| C-MOF/ES-3 | 0.201 | 40.2 | 43.41 | 4.64 | 1.22 | 31.34 | 19.39 |
| C-MOF/EB-1 | 0.216 | 43.2 | 35.82 | 2.43 | 1.05 | 41.53 | 19.17 |
| C-MOF/EB-2 | 0.235 | 47.0 | 28.89 | 1.41 | 0.90 | 45.05 | 23.75 |
| C-MOF/EB-3 | 0.241 | 48.2 | 28.84 | 0.95 | 0.77 | 44.90 | 24.54 |
| C-(MOF-5/PANI) Sample | Content (wt.%) | |||||
|---|---|---|---|---|---|---|
| C | Zn | N | O | Cl | S | |
| C-MOF/ES-1 | 64.20 | 8.85 | 18.00 | 7.74 | 0.43 | 0.78 |
| C-MOF/ES-2 | 59.32 | 13.48 | 16.40 | 9.55 | 0.57 | 0.68 |
| C-MOF/ES-3 | 57.87 | 27.55 | 7.09 | 6.60 | 0.34 | 0.55 |
| C-MOF/EB-1 | 74.77 | 5.72 | 7.40 | 11.68 | / | 0.43 |
| C-MOF/EB-2 | 67.12 | 17.73 | 2.90 | 11.95 | / | 0.30 |
| C-MOF/EB-3 | 57.05 | 33.03 | 2.03 | 7.77 | / | 0.12 |
| C-(MOF-5/PANI) Sample | Content (wt.%) | wC/ZnO,ZnS | ||
|---|---|---|---|---|
| Carbonaceous Part | Zn-Containing Part (ZnO, ZnO+ZnS) | H2O | ||
| C-MOF/ES-1 | 77.83 | 6.97 | 15.20 | 11.17 |
| C-MOF/ES-2 | 65.67 | 23.52 | 10.81 | 2.79 |
| C-MOF/ES-3 | 50.10 | 42.28 | 7.62 | 1.19 |
| C-MOF/EB-1 | 35.66 | 58.50 | 5.84 | 0.61 |
| C-MOF/EB-2 | 28.98 | 67.83 | 3.19 | 0.43 |
| C-MOF/EB-3 | 28.19 | 67.61 | 4.20 | 0.42 |
| Precursors | Products of Carbonization | ||||
| MOF-5/PANI | σ a (S cm−1) | C-(MOF-5/PANI) | σ (S cm−1) | SBET (m2 g−1) | ID/IG |
| MOF/ES-1 | 1.0 × 10−3 | C-MOF/ES-1 | 0.16 | 47 | 3.56 |
| MOF/ES-2 | 1.4 × 10−4 | C-MOF/ES-2 | 0.10 | 109 | 3.67 |
| MOF/ES-3 | 7.9 × 10−7 | C-MOF/ES-3 | 0.21 | 393 | 3.43 |
| MOF/EB-1 | 4.3 × 10−7 | C-MOF/EB-1 | 0.18 | 609 | 3.42 |
| MOF/EB-2 | 3.9 × 10−7 | C-MOF/EB-2 | 0.08 | 470 | 3.65 |
| MOF/EB-3 | 4.0 × 10−7 | C-MOF/EB-3 | 0.24 | 412 | 3.61 |
| Carbonized Individual Components | |||||
| C-MOF | 0.92 | 553 | |||
| C-PANI-ES | 0.66 | 351 | |||
| C-PANI-EB | 0.87 | 273 | |||
| Acid-Etched Samples | |||||
| C-MOF/ES-1-a | 0.42 | 45.2 | |||
| C-MOF/ES-2-a | 0.41 | 106 | |||
| C-MOF/EB-1-a | 0.42 | 1148 | |||
| C-MOF-a | 0.13 | 1994 | |||
| C-(MOF-5/PANI) | Cspec (F g−1) | ||||
|---|---|---|---|---|---|
| 10 mV s−1 | 20 mV s−1 | 50 mV s−1 | 100 mV s−1 | 200 mV s−1 | |
| C-MOF/ES-1 | 238.2 | 219.2 | 187.7 | 152.4 | 130.2 |
| C-MOF/ES-2 | 176.5 | 169.4 | 158.8 | 143.2 | 129.0 |
| C-MOF/ES-3 | 146.3 | 139.4 | 127.5 | 111.2 | 92.6 |
| C-MOF/EB-1 | 136.2 | 127.6 | 110.5 | 90.3 | 68.3 |
| C-MOF/EB-2 | 80.4 | 75.3 | 65.2 | 54.1 | 41.5 |
| C-MOF/EB-3 | 91.2 | 83.3 | 74.7 | 63.4 | 54.1 |
| Carbonized Individual Components | 10 mV s−1 | 20 mV s−1 | 50 mV s−1 | 100 mV s−1 | 200 mV s−1 |
| C-MOF | 72.1 | 69.2 | 67.4 | 61.1 | 60.5 |
| C-PANI-ES | 96.1 | 81.5 | 67.7 | 55.8 | 46.1 |
| C-PANI-EB | 144.7 | 139.0 | 126.4 | 107.7 | 89.7 |
| Acid-Etched Samples | 10 mV s−1 | 20 mV s−1 | 50 mV s−1 | 100 mV s−1 | 200 mV s−1 |
| C-MOF/ES-1-a | 171.5 | 167.0 | 156.5 | 133.5 | 114.0 |
| C-MOF/ES-2-a | 163.9 | 160.0 | 157.1 | 133.2 | 114.1 |
| C-MOF/EB-1-a | 341.0 | 336.4 | 319.8 | 306.4 | 268.2 |
| C-MOF-a | 224.8 | 221.6 | 208.2 | 185.5 | 160.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savić, M.; Janošević Ležaić, A.; Gavrilov, N.; Pašti, I.; Nedić Vasiljević, B.; Krstić, J.; Ćirić-Marjanović, G. Carbonization of MOF-5/Polyaniline Composites to N,O-Doped Carbon/ZnO/ZnS and N,O-Doped Carbon/ZnO Composites with High Specific Capacitance, Specific Surface Area and Electrical Conductivity. Materials 2023, 16, 1018. https://doi.org/10.3390/ma16031018
Savić M, Janošević Ležaić A, Gavrilov N, Pašti I, Nedić Vasiljević B, Krstić J, Ćirić-Marjanović G. Carbonization of MOF-5/Polyaniline Composites to N,O-Doped Carbon/ZnO/ZnS and N,O-Doped Carbon/ZnO Composites with High Specific Capacitance, Specific Surface Area and Electrical Conductivity. Materials. 2023; 16(3):1018. https://doi.org/10.3390/ma16031018
Chicago/Turabian StyleSavić, Marjetka, Aleksandra Janošević Ležaić, Nemanja Gavrilov, Igor Pašti, Bojana Nedić Vasiljević, Jugoslav Krstić, and Gordana Ćirić-Marjanović. 2023. "Carbonization of MOF-5/Polyaniline Composites to N,O-Doped Carbon/ZnO/ZnS and N,O-Doped Carbon/ZnO Composites with High Specific Capacitance, Specific Surface Area and Electrical Conductivity" Materials 16, no. 3: 1018. https://doi.org/10.3390/ma16031018