How Stress Treatments Influence the Performance of Biodegradable Poly(Butylene Succinate)-Based Copolymers with Thioether Linkages for Food Packaging Applications




Abstract
1. Introduction
2. Results and Discussion
2.1. Molecular Weight Determination
2.2. Thermal Properties
2.3. Mechanical Characterization
2.4. Barrier Properties
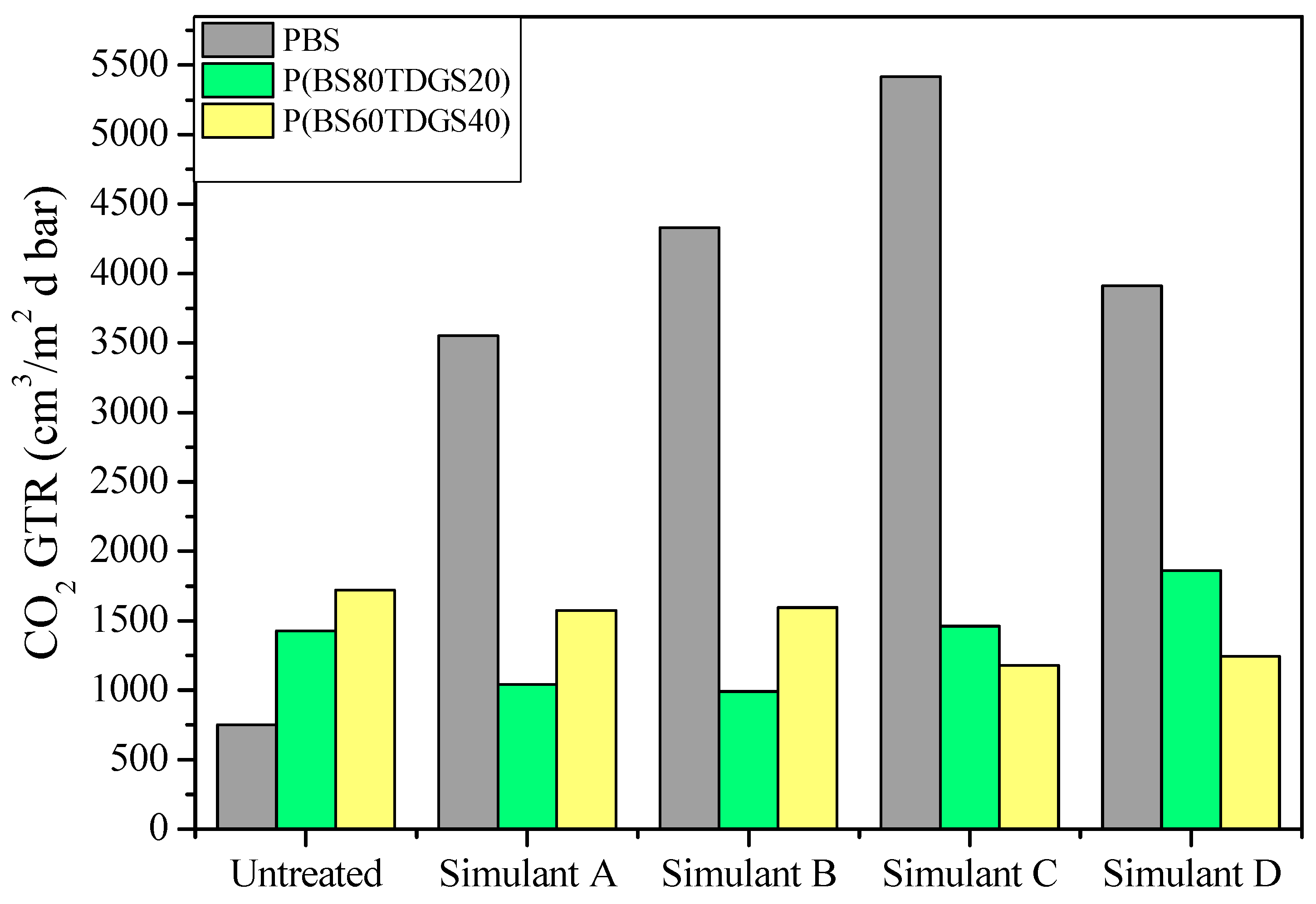
2.4.1. Simulant Liquids
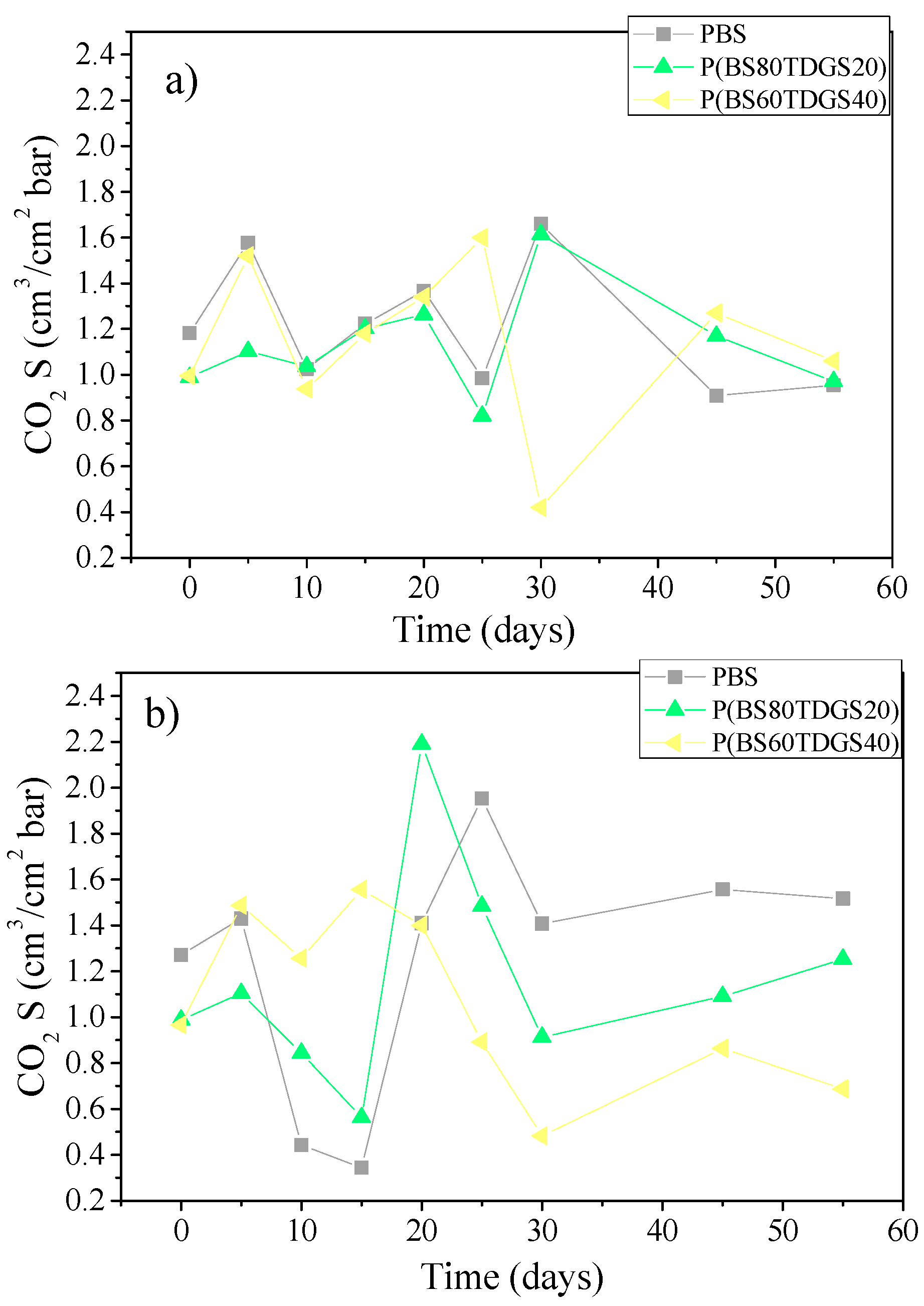
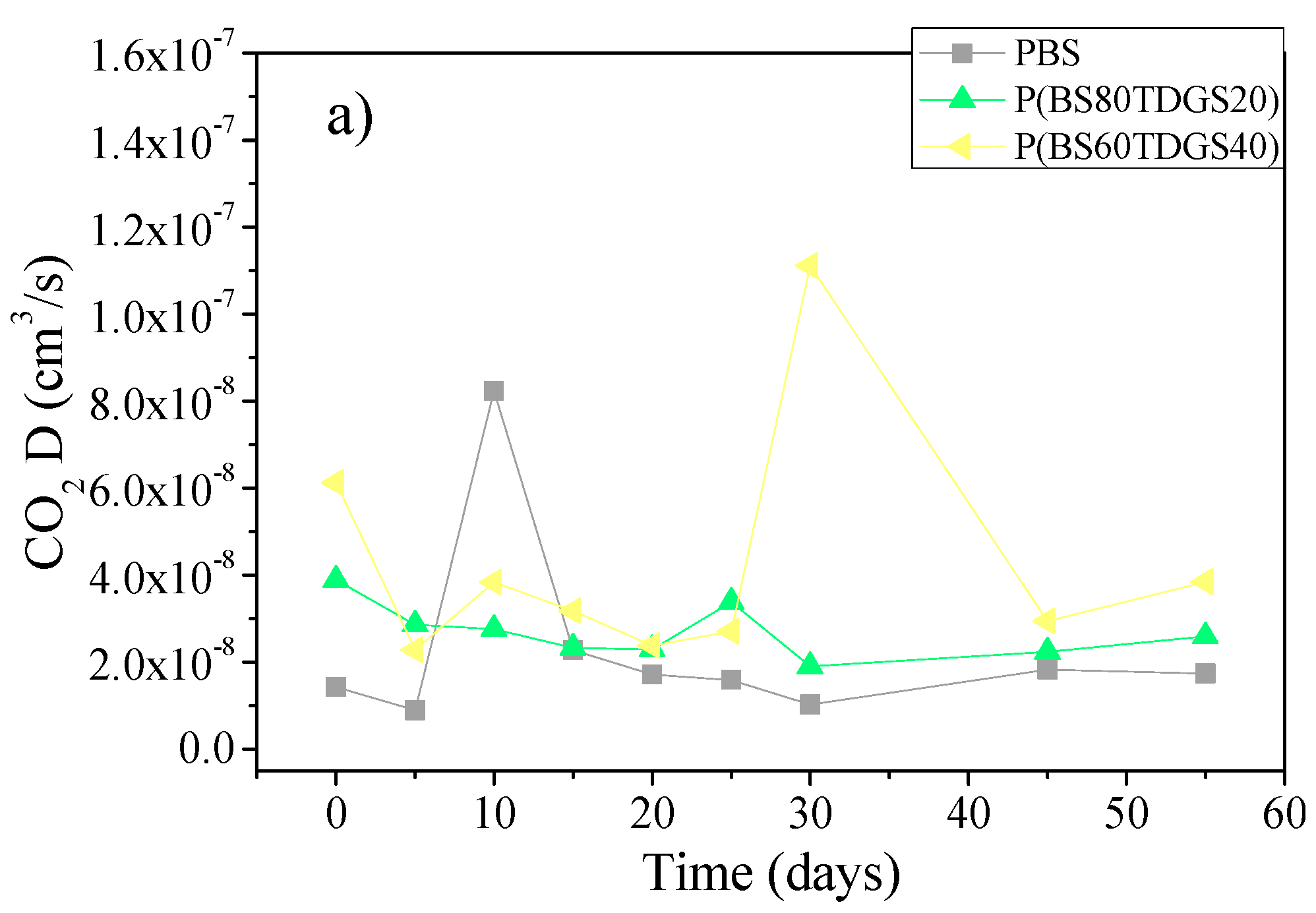
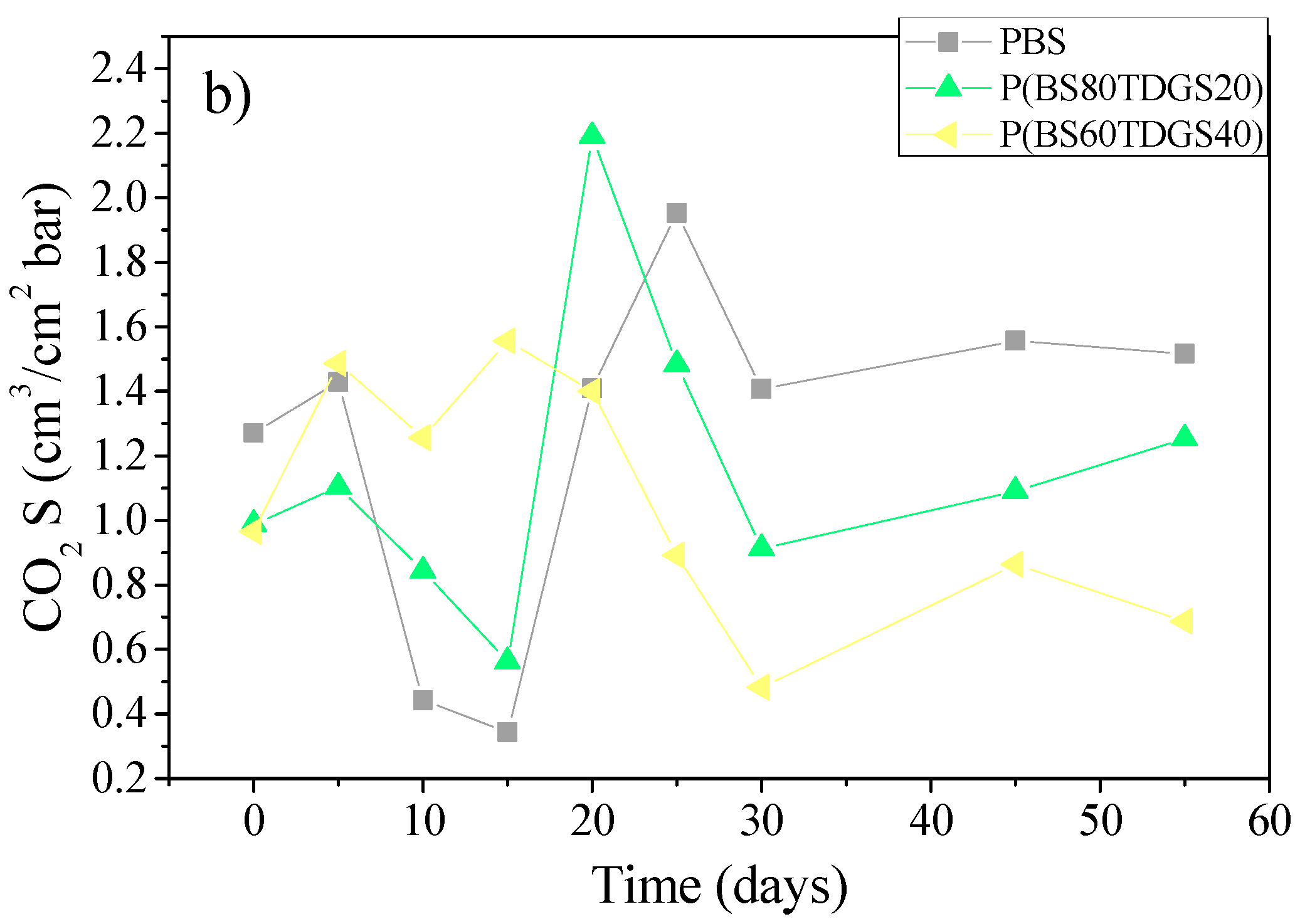
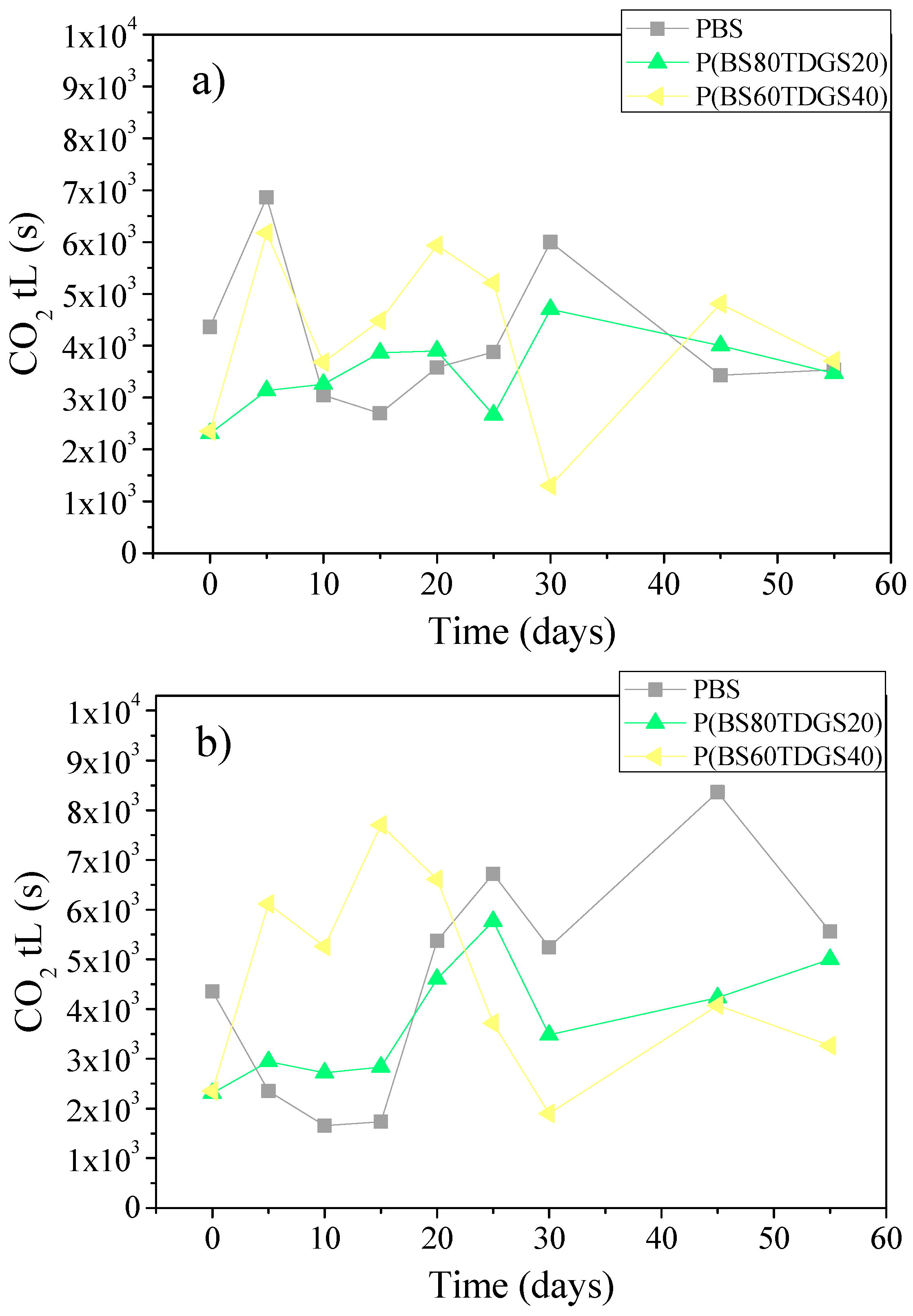
2.4.2. Thermal Aging and Photoaging
2.5. FT-IR Spectroscopic Data
3. Materials and Methods
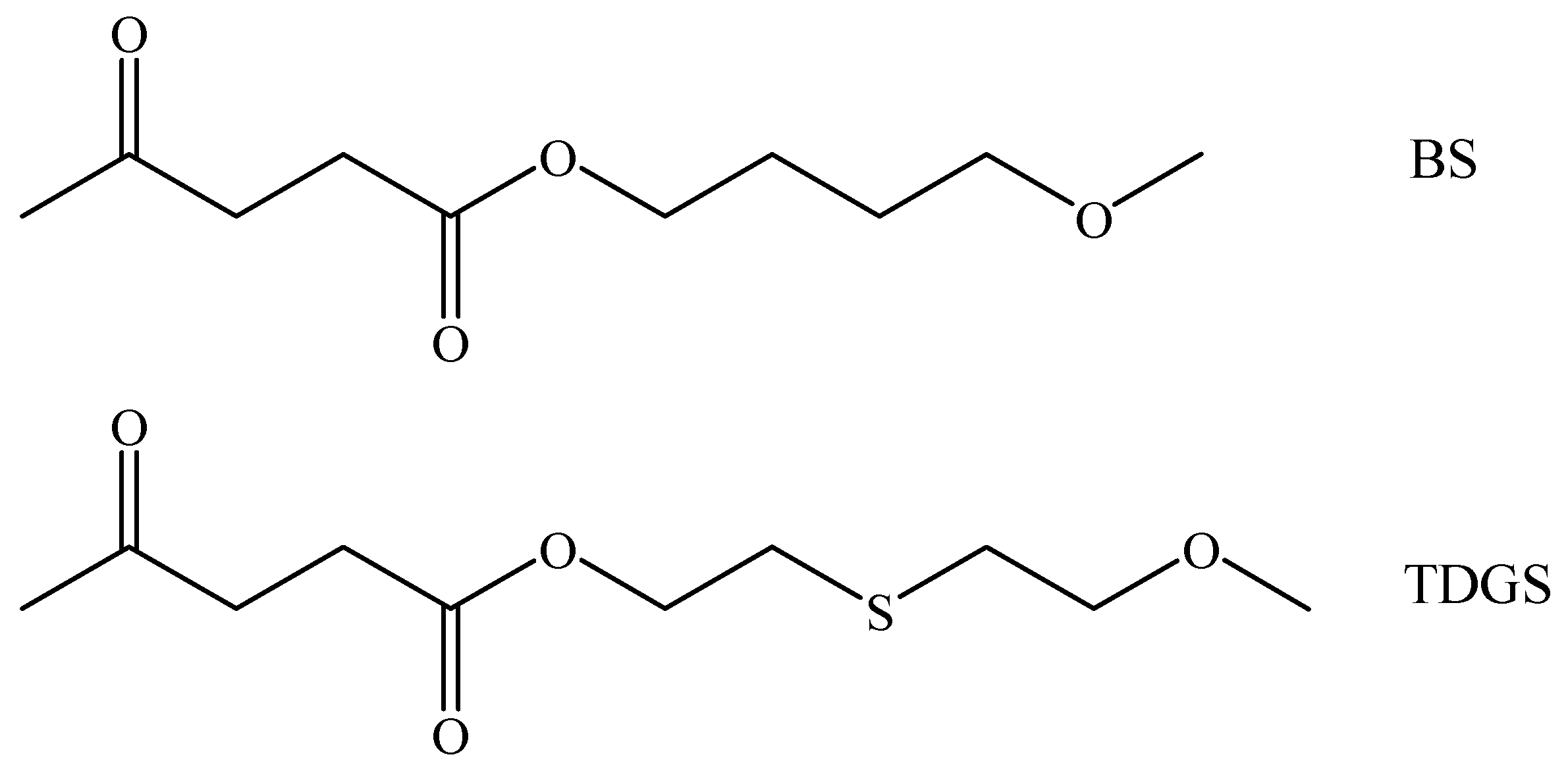
3.1. Synthesis of Poly(Butylene Succinate) Homopolymer and Poly(Butylene/Thiodiethylene Glycol Succinate) Copolymers
3.2. Film Preparation and Thickness Determination
3.3. Thermal and Photoaging Procedures
3.4. Simulant Liquid
- Simulant A, Distilled Water, 10 days, 40 °C
- Simulant B, Acetic acid 3% (v/v), 10 days, 40 °C
- Simulant C, Ethanol 10% (v/v), 10 days, 40 °C
- Simulant D, Isooctane, 2 days, 20 °C
3.5. Permeability Measurement
3.6. Thermal Analysis
3.7. Stress–Strain Measurements
3.8. Molecular Weight Determination
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Siracusa, V.; Rocculi, P.; Romani, S.; Rosa, M.D. Biodegradable polymers for food packaging: A review. Trends Food Sci. Technol. 2008, 19, 634–643. [Google Scholar] [CrossRef]
- Plastic Europe. Plastics—The Facts 2016, An Analysis of European Plastics Production, Demand and Waste Data, 2016. Available online: http://www.plasticseurope.org/Document/plastics---the-facts-2016-15787.aspx?FolID=2 (accessed on 28 August 2017).
- Mülhaupt, R. Green Polymer Chemistry and Bio-based Plastics: Dreams and Reality. Macromol. Chem. Phys. 2013, 214, 159–174. [Google Scholar] [CrossRef]
- Miller, S.A. Sustainable Polymers: Opportunities for the Next Decade. ACS Macromol. Lett. 2013, 2, 550–554. [Google Scholar] [CrossRef]
- Miller, A.S. Sustainable Polymers: Replacing polymers derived from fossil fuels. Polym. Chem. 2014, 5, 3117–3118. [Google Scholar] [CrossRef]
- Iwata, T. Biodegradable and Bio-based Polymers: Future Prospects of Eco-Friendly Plastics. Angew. Chem. Int. Ed. 2015, 54, 3210–3215. [Google Scholar] [CrossRef] [PubMed]
- Peelman, N.; Ragaert, P.; De Meulenaer, B.; Adons, D.; Peeters, R.; Cardon, L.; Van Impe, F.; Devlieghere, F. Application of bioplastics for food packaging. Trends Food Sci. Technol. 2013, 32, 128–141. [Google Scholar] [CrossRef]
- Garrison, T.F.; Murawski, A.; Quirino, R.L. Bio-based Polymers with Potential for Biodegradability. Polymers 2016, 8, 262. [Google Scholar] [CrossRef]
- Reddy, M.M.; Vivekanandhan, S.; Misra, M.; Bathia, S.K.; Mohanty, A.K. Biobased plastics bionanocomposites: Current status and future opportunities. Prog. Polym. Sci. 2013, 38, 1653–1689. [Google Scholar] [CrossRef]
- European Bioplastics in “Bioplastics, Facts and Figures”, 2017. 12th European Bioplastics Conference, 28/29 November 2017, MARITIM proArte Hotel, Berlin, Germany. Available online: http://www.european-bioplastics.org/events/eubp-conference/ (accessed on 30 August 2017).
- Genovese, L.; Lotti, N.; Gazzano, M.; Siracusa, V.; Dalla Rosa, M.; Munari, A. Novel biodegradable aliphatic copolyesters based on poly(butylenesuccinate) containing thioether-linkages for sustainable food packaging applications. Polym. Degrad. Stab. 2016, 132, 191–201. [Google Scholar] [CrossRef]
- Soccio, M.; Lotti, N.; Gazzano, M.; Govoni, M.; Giordano, E.; Munari, A. Molecular architecture and solid-state properties of novel biocompatible PBS-based copolyesters containing sulphur atoms. React. Funct. Polym. 2012, 72, 856–867. [Google Scholar] [CrossRef]
- Gigli, M.; Lotti, N.; Gazzano, M.; Finelli, L.; Munari, A. Novel eco-friendly random copolyesters of poly (buthylene succinate) containing ether-linkage. React. Funct. Polym. 2012, 72, 303–310. [Google Scholar] [CrossRef]
- Gigli, M.; Genovese, L.; Lotti, N.; Munari, A.; Dalla Rosa, M.; Siracusa, V. Gas Barrier and Thermal Behavior of Long Chain Aliphatic Polyesters after Stressed Treatments. Polym. Plast. Technol. Eng. 2017, 56, 71–82. [Google Scholar] [CrossRef]
- Gigli, M.; Lotti, N.; Gazzano, M.; Siracusa, V.; Finelli, L.; Munari, A.; Dalla Rosa, M. Biodegradable aliphatic copolyesters containing PEG-like sequences for sustainable food packaging applications. Polym. Degrad. Stab. 2014, 105, 96–106. [Google Scholar] [CrossRef]
- Badia, J.D.; Gil-Castel, O.; Ribes-Greus, A. Long-term properties and end-of-life of polymers from renewable resources. Polym. Degrad. Stab. 2017, 137, 35–57. [Google Scholar] [CrossRef]
- Zia, K.M.; Noreen, A.; Zuber, M.; Tabasum, S.; Mujahid, M. Recent developments and future prospects on bio-based polyesters derived from renewable resources: A review. Int. J. Biol. Macromol. 2016, 82, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.S. Bioconversion of fumarate to succinicate using glycerol as a carbon source. Appl. Biochem. Biotechnol. 1999, 78, 511–520. [Google Scholar]
- Chen, G.Q.; Patel, M.K. Plastics Derived from Biological Sources: Present and Future: A Technocal and Environmental Review. Chem. Rev. 2012, 112, 2082–2099. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Guo, B.H. Microbial Succinic acid, its polymer poly(butylene succinate), and application. In Plastics from Bacteria-Natural Functions Applications; Chen, G.Q., Ed.; Springer: Heidelberg, Germany, 2010; pp. 347–388. [Google Scholar]
- Ryu, H.W.; Wee, Y.-J. Characterization of bioconversion of fumarate to succinate by alginate immobilized Enterococcus faecalis RKY1. Appl. Biochem. Technol. 2001, 91, 525–535. [Google Scholar] [CrossRef]
- Gigli, M.; Negroni, A.; Zanaroli, G.; Lotti, N.; Fava, F.; Munari, A. Environmentally friendly PBS-based copolyesters containing PEG-like subunit: Effect of block length on solid-state properties and enzymatic degradation. React. Funct. Polym. 2013, 73, 764–771. [Google Scholar] [CrossRef]
- Genovese, L.; Gigli, M.; Lotti, N.; Gazzano, M.; Siracusa, V.; Munari, A.; Dalla Rosa, M. Biodegradable long chain aliphatic polyesters containing ether-linkages: Synthesis, solid-state and barrier properties. Ind. Eng. Chem. Res. 2014, 53, 10965–10973. [Google Scholar] [CrossRef]
- Arvanitoyannis, I.S. Totally and Partially biodegradable polymer blends based on natural synthetic macromolecules: Preparation, physical properties, and potential as food packaging materials. J. Macromol. Sci. Rev. Macromol. Chem. Phys. 1999, 39, 205–271. [Google Scholar] [CrossRef]
- Gigli, M.; Negroni, A.; Soccio, M.; Zanaroli, G.; Lotti, N.; Fava, F.; Munari, A. Influence of chemical and architectural modifications on the enzymatic hydrolysis of poly(butylene succinate). Green Chem. 2012, 14, 2885–2893. [Google Scholar] [CrossRef]
- Gigli, M.; Negroni, A.; Soccio, M.; Zanaroli, G.; Lotti, N.; Fava, F.; Munari, A. Enzymatic hydrolysis studies on novel eco-friendly aliphatic thiocopolyesters. Polym. Degrad. Stab. 2013, 98, 934–942. [Google Scholar] [CrossRef]
- Mendikute, G.; Irusta, L.; Fernández-Berridi, M.J. Infrared study of the photochemical behaviour of aromatic poly(ether urethanes): Effect of various stabilizers. e-Polymer 2009, 125, 1–10. [Google Scholar] [CrossRef]
- Sarwade, B.D.; Singh, R.P. Structural changes in semicrystalline poly(ether ester) on UV irradiation. J. Polym. Eng. 2003, 23, 43–54. [Google Scholar] [CrossRef]
- George, S.C.; Thomas, S. Transport phenomena through polymeric systems. Prog. Polym. Sci. 2001, 26, 985–1017. [Google Scholar] [CrossRef]
- Siracusa, V.; Lotti, N.; Munari, A.; Dalla Rosa, M. Poly(butylene succinate) and poly(butylene succinate-co-adipate) for food packaging applications: Gas barrier properties after stressed treatments. Polym. Degrad. Stab. 2015, 119, 35–45. [Google Scholar] [CrossRef]
- Gauthier, E.; Laycock, B.; Cuoq, F.J.J.M.; Halley, P.J.; George, K.A. Correlation between chain microstructural changes and embrittlement of LLDPE-based films during photo- and thermo-oxidative degradation. Polym. Degrad. Stab. 2013, 98, 2301–2312. [Google Scholar] [CrossRef]
- Halpi, J.C.; Kardos, K.L. Moduli of crystalline polymers employing composite theory. J. Appl. Phys. 1972, 43, 2235–2241. [Google Scholar] [CrossRef]
- Dusunceli, N.; Colak, O.U. Modelling effects of degree of crystallinity on mechanical behavior of semicrystalline polymers. Int. J. Plast. 2008, 24, 1224–1242. [Google Scholar] [CrossRef]
- Siracusa, V. Food Packaging Permeability Behaviour: A report. Int. J. Polym. Sci. 2012, 2012, 1–11. [Google Scholar] [CrossRef]
- Robertson, G.L. Food Packaging: Principles and Practice, 2nd ed.; Marcel Dekker: New York, NY, USA, 2006. [Google Scholar]
- Lee, D.S.; Yam, K.L.; Piergiovanni, L. Food Packaging Science and Technology; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2008. [Google Scholar]
- Siracusa, V.; Ingrao, C. Correlation amongst gas barrier behaviour, temperature and thickness in BOPP films for food packaging usage: A lab-scale testing experience. Polym. Test. 2017, 59, 277–289. [Google Scholar] [CrossRef]
- Schmid, M.; Zillinger, W.; Muller, K.; Sangerlaub, S. Permeation of water vapour, nitrogen, oxygen and carbon dioxide through whey protein isolated based films and coatings—Permselectivity and activation energy. Food Packag. Shelf Life 2015, 6, 21–29. [Google Scholar] [CrossRef]
- Müller, K. Multilayer films for bag-in-container systems used in disposable kegs: Basic principles of possible barrier concepts. Brew. Sci. 2013, 66, 1–2. [Google Scholar]
- Jamshidian, M.; Tehrany, E.A.; Cleymand, F.; Leconte, S.; Falher, T.; Desobry, S. Effect of synthetic phenolic antioxidants on physical, structural, mechanical and barrier properties of poly lactic acid film. Carbohydr. Polym. 2012, 87, 1763–1773. [Google Scholar] [CrossRef]
- Liu, R.Y.F.; Hu, Y.S.; Schiraldi, D.A.; Hiltner, A.; Baer, E. Crystallinity and oxygen transport properties of PET bottle walls. J. Appl. Polym. Sci. 2004, 94, 671–677. [Google Scholar] [CrossRef]
- De Leiris, P. Water activity and permeability. In Food Packaging and Preservation; Mathlouthi, M., Ed.; Elsevier Applied Science: New York, NY, USA, 1986; pp. 213–233. [Google Scholar]
- Jakubowicz, I. Evaluation of degradability of biodegradable polyethylene (PE). Polym. Degrad. Stab. 2003, 80, 39–43. [Google Scholar] [CrossRef]
- Koutny, M.; Lemaire, J.; Delort, A.M. Biodegradation of polyethylene films with prooxidant additives: A review. Chemosphere 2006, 64, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Calligaris, S.; Manzocco, L.; Kravina, G.; Nicoli, M.C. Shelf—Life Modeling of Bakery Products by Using Oxidation Indices. J. Agric. Food Chem. 2007, 55, 2004–2009. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.X.; Xu, F. Effects of light-barrier property of packaging film on the photo-oxidation and shelf life of cookies based on accelerated tests. Packag. Technol. Sci. 2009, 22, 107–113. [Google Scholar] [CrossRef]
- Romeo, F.V.; De Luca, S.; Pscopo, A.; Santisi, V.; Poiana, M. Shelf-life of almond pastry coockis with different types of packaging and levels of temperature. Food Sci. Technol. Int. 2010, 16, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Regulation (EC) No 1935/2004 of the European parliament and of the Council of 27 October 2004 on Materials and Articles Intended to Come into Contact with Food and Repealing, Directives 80/590/EEC and 89/109/EEC. Available online: https://www.fsai.ie/uploadedFiles/Reg1935_2004.pdf (accessed on 28 August 2017).
- Regulation (EU) No 10/2011 of 14 January 2011 on Plastic Materials and Articles Intended to Come into Contact with Food. Available online: https://www.fsai.ie/uploadedFiles/Reg10_2011.pdf (accessed on 28 August 2017).
- Thompson, A.; Taylor, B.N. (Eds.) Guide for the Use of the International System of Units (SI), Special Publication 811; National Institute of Standards and Technology, U.S. Department of Commerce: Gaithersburg, MD, USA, 2008.
- Siracusa, V.; Blanco, I.; Romani, S.; Tylewicz, U.; Dalla Rosa, M. Gas Permeability and Thermal Behavior of Polypropylene Films Used for Packaging Minimally Processed Fresh-Cut Potatoes: A Case Study. J. Food Sci. 2012, 77, E264–E272. [Google Scholar] [CrossRef] [PubMed]
- Mrkić, S.; Galić, K.; Ivanković, M.; Hamin, S.; Ciković, N. Gas transport and thermal characterization of mono- and Di-polyethylene films used for food packaging. J. Appl. Polym. Sci. 2012, 99, 1590–1599. [Google Scholar] [CrossRef]
- Siracusa, V.; Blanco, I.; Romani, S.; Tylewicz, U.; Rocculi, P.; Dalla Rosa, M. Poly(lactic acid)-Modified films for Food Packaging Application: Physical, Mechanical and barrier Behavior. J. Appl. Polym. Sci. 2012, 125, E390–E401. [Google Scholar] [CrossRef]
- Gajdoš, J.; Galić, K.; Kurtanjek, Ž.; Ciković, N. Gas permeability and DSC charactheristics of polymers used in food packaging. Polym. Test. 2001, 20, 49–57. [Google Scholar] [CrossRef]
- Burgess, S.K.; Kriegel, R.M.; Koros, W.J. Carbon Dioxide Sorption in Amorphous Poly(ethylene furanoate). Macromolecules 2015, 48, 2184–2193. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Mn | PDI | TDGS (mol %) by 1H NMR | WCA (°) | Tm (°C) | ΔHm (J/mol) | Χc (%) by XRD | Thickness (μm) |
|---|---|---|---|---|---|---|---|---|
| PBS | 51,000 | 2.7 | 0 | 90 ± 2 | 114 | 81 | 45 | 192 ± 4 |
| P(BS80TDGS20) | 54,000 | 2.8 | 20 | 83 ± 3 | 94 | 52 | 36 | 231 ± 20 |
| P(BS60TDGS40) | 51,000 | 2.8 | 36 | 80 ± 2 | 72 | 12 | 31 | 291 ± 7 |
| Polymer | Simulant A | Simulant B | Simulant C | Simulant D |
|---|---|---|---|---|
| PBS | 91 | 77 | 71 | 100 |
| P(BS80TDGS20) | 95 | 93 | 81 | 97 |
| P(BS60TDGS40) | 78 | 90 | 84 | 100 |
| Polymer | Thermal | Photo | Thermal | Photo | Thermal | Photo |
|---|---|---|---|---|---|---|
| 10 Days | 20 Days | 55 Days | ||||
| PBS | 86 | 79 | 85 | 70 | 82 | 63 |
| P(BS80TDGS20) | 87 | 79 | 84 | 76 | 82 | 60 |
| P(BS60TDGS40) | 80 | 56 | 78 | 36 | 73 | n.d. |
| Polymer | Simulant A | Simulant B | Simulant C | Simulant D |
|---|---|---|---|---|
| PBS | 74 | 68 | 75 | 64 |
| P(BS80TDGS20) | 67 | 67 | 65 | 65 |
| P(BS60TDGS40) | 308 | 375 | 342 | 333 |
| Polymer | Thermal | Photo | Thermal | Photo | Thermal | Photo |
|---|---|---|---|---|---|---|
| 10 Days | 20 Days | 55 Days | ||||
| PBS | 102 | 97 | 98 | 93 | 95 | 92 |
| P(BS80TDGS20) | 89 | 107 | 82 | 93 | 82 | 84 |
| P(BS60TDGS40) | 78 | 78 | 71 | 98 | 71 | n.d. |
| Polymer | Untreated * | Simulant A | Simulant B | Simulant C | Simulant D | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| E (MPa) | εb (%) | E (MPa) | εb (%) | E (MPa) | εb (%) | E (MPa) | εb (%) | E (Ma) | εb (%) | |
| PBS | 440 ± 30 | 17 ± 2 | 431 ± 12 | 15 ± 2 | 428 ± 11 | 13 ± 1 | 413 ± 7 | 15 ± 2 | 378 ± 5 | 12 ± 1 |
| P(BS80TDGS20) | 260 ± 10 | 580 ± 70 | 213 ± 1 | 388 ± 15 | 251 ± 8 | 376 ± 14 | 265 ± 9 | 382 ± 17 | 264 ± 4 | 386 ± 7 |
| P(BS60TDGS40) | 160 ± 3 | 810 ± 20 | 178 ± 12 | 211 ± 17 | 181 ± 7 | 175 ± 15 | 173 ± 15 | 181 ± 13 | 188 ± 16 | 201 ± 4 |
| Polymer | Untreated * | Thermal | Thermal | Thermal | ||||
|---|---|---|---|---|---|---|---|---|
| 0 Days | 10 Days | 20 Days | 55 Days | |||||
| E (MPa) | εb (%) | E (MPa) | εb (%) | E (MPa) | εb (%) | E(MPa) | εb (%) | |
| PBS | 440 ± 30 | 17 ± 2 | 428 ± 22 | 16 ± 3 | 402 ± 12 | 14 ± 1 | 389 ± 21 | 14 ± 2 |
| P(BS80TDGS20) | 260 ± 10 | 580 ± 70 | 233 ± 27 | 567 ± 32 | 203 ± 15 | 556 ± 13 | 213 ± 11 | 560 ± 11 |
| P(BS60TDGS40) | 160 ± 3 | 810 ± 20 | 155 ± 8 | 802 ± 15 | 143 ± 1 | 789 ± 7 | 134 ± 2 | 713 ± 13 |
| Polymer | Untreated * | Photo | Photo | Photo | ||||
|---|---|---|---|---|---|---|---|---|
| 0 Days | 10 Days | 20 Days | 55 Days | |||||
| E (MPa) | εb (%) | E (MPa) | εb (%) | E (MPa) | εb (%) | E (MPa) | εb (%) | |
| PBS | 440 ± 30 | 17 ± 2 | 418 ± 12 | 15 ± 1 | 402 ± 9 | 11 ± 1 | 378 ± 23 | 9 ± 2 |
| P(BS80TDGS20) | 260 ± 10 | 580 ± 70 | 241 ± 17 | 535 ± 16 | 178 ± 16 | 478 ± 9 | 165 ± 10 | 413 ± 19 |
| P(BS60TDGS40) | 160 ± 3 | 810 ± 20 | 133 ± 3 | 767 ± 23 | 111 ± 3 | 213 ± 7 | ||
| Permeability Parameters | Untreated * | Simulant A | Simulant B | Simulant C | Simulant D |
|---|---|---|---|---|---|
| PBS | |||||
| GTR | 750 ± 1 | 914 ± 1 (>) | 772 ± 2 (>) | 909 ± 1 (>) | 1023 ± 9 (>) |
| S | 1.2 × 10−1 ± 0.1 | 1.1 ± 0.1 (>) | 1.5 ± 0.0 (>) | 6.4 ± 0.0 (>) | n.a. |
| D | 1.4 × 10−8 ± 0.0 | 1.8 × 10−8 ± 0.0 (>) | 8.4 × 10−9 ± 0.0 (<) | 3.2 × 10−8 ± 0.0 (>) | n.a. |
| tL | 4358 ± 529 | 3356 ± 279 (<) | 5217 ± 16 (>) | 1942 ± 10 (<) | n.a. |
| P(BS80TDGS20) | |||||
| GTR | 1429 ± 2 | 1040 ± 0.0 (<) | 990 ± 1 (<) | 1460 ± 0 (>) | 1860 ± 8 (>) |
| S | 9.9 × 10−1 ± 0.0 | 1.4 ± 0.0 (>) | 1.0 ± 0.1 (=) | 1.0 ± 0.0 (=) | 6.9 × 10−1 ± 0.0 (<) |
| D | 3.9 × 10−8 ± 0.0 | 2.0 × 10−8 ± 0.0 (<) | 2.6 × 10−8 ± 0.0 (<) | 3.8 × 10−8 ± 0.0 (<) | 7.3 × 10−8 ± 0.0 (<) |
| tL | 2309 ± 122 | 4393 ± 17 (>) | 3418 ± 207 (>) | 2386 ± 5 (>) | 1230 ± 30 (>) |
| P(BS60TDGS40) | |||||
| GTR | 1722 ± 3 | 1573 ± 5 (<) | 1596 ± 1 (<) | 1178 ± 0 (<) | 1243 ± 5 (<) |
| S | 9.7 × 10−1 ± 0.1 | 1.0 ± 0.0 (>) | 7.5 × 10−1 ± 0.1 (<) | 1.4 ± 0.0 (>) | 1.0 ± 0.0 (>) |
| D | 6.1 × 10−8 ± 0.0 | 5.3 × 10−8 ± 0.0 (<) | 7.2 × 10−8 ± 0.0 (>) | 2.8 × 10−8 ± 0.0 (<) | 4.1 × 10−8 ± 0.0 (<) |
| tL | 6762 ± 130 | 2672 ± 4 (<) | 1977 ± 214 (<) | 4979 ± 15 (<) | 3439 ± 11 (<) |
| Polymer | GTR CO2/O2 | S CO2/O2 | D CO2/O2 | tL CO2/O2 |
|---|---|---|---|---|
| PBS | 3.2 | 271.7 | 0.02 | 40.9 |
| P(BS80TDGS20) | 6.5 | 136.9 | 0.05 | 24.9 |
| P(BS60TDGS40) | 8.1 | 13.7 | 0.61 | 4.9 |
| Polymer | Thermal | Photo | Thermal | Photo | Thermal | Photo | Thermal | Photo |
|---|---|---|---|---|---|---|---|---|
| GTR | GTR | S | S | D | D | tL | tL | |
| PBS | 0.036 | 0.060 | 0.164 | 0.167 | 0.043 | 0.471 | 0.058 | 0.456 |
| P(BS80TDGS20) | 0.546 | 0.338 | 0.006 | 0.027 | 0.243 | 0.065 | 0.217 | 0.469 |
| P(BS60TDGS40) | 0.038 | 0.347 | 0.009 | 0.378 | 0.001 | 0.032 | 0.004 | 0.097 |
| Chemical Group | Peak Position, cm−1 |
|---|---|
| –OH stretch (free) | 3571 |
| CH-stretch (of CH2) | 2918 (νas CH2), 2856 (νs CH2) |
| –C=O normal carbonyl stretch | 1712 |
| –CH-deformation symmetric and asymmetric bending | 1472 (δs CH2) |
| C-O-H in-plane bend | 1424 |
| –CH2-scissoring | 1387 |
| –C=O bending | 1245 |
| –C-O stretching | 1178, 1153 |
| –OH bending | 1046 |
| –CH2 wagging and twisting | 1244, 1178 |
| –CH2 rocking | 751 |
| O-H out-of-plane | 994 (as), 955(s) |
| C-C stretch | 920, 809 |
| C-S stretch | 743 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siracusa, V.; Genovese, L.; Munari, A.; Lotti, N. How Stress Treatments Influence the Performance of Biodegradable Poly(Butylene Succinate)-Based Copolymers with Thioether Linkages for Food Packaging Applications. Materials 2017, 10, 1009. https://doi.org/10.3390/ma10091009
Siracusa V, Genovese L, Munari A, Lotti N. How Stress Treatments Influence the Performance of Biodegradable Poly(Butylene Succinate)-Based Copolymers with Thioether Linkages for Food Packaging Applications. Materials. 2017; 10(9):1009. https://doi.org/10.3390/ma10091009
Chicago/Turabian StyleSiracusa, Valentina, Laura Genovese, Andrea Munari, and Nadia Lotti. 2017. "How Stress Treatments Influence the Performance of Biodegradable Poly(Butylene Succinate)-Based Copolymers with Thioether Linkages for Food Packaging Applications" Materials 10, no. 9: 1009. https://doi.org/10.3390/ma10091009
APA StyleSiracusa, V., Genovese, L., Munari, A., & Lotti, N. (2017). How Stress Treatments Influence the Performance of Biodegradable Poly(Butylene Succinate)-Based Copolymers with Thioether Linkages for Food Packaging Applications. Materials, 10(9), 1009. https://doi.org/10.3390/ma10091009