1. Introduction
Because petroleum plays a major role in the world economy, rapid increasing consumption of fossil fuel and depletion of total crude oil reserves have led to a global energy crisis [
1,
2]. Research has found that biomass based energy can be utilized to replace fossil fuel. To utilize biomass feedstock, microwave pyrolysis has been applied on biomass conversion to produce liquid bio-oil as an innovative thermo-chemical technology [
3,
4,
5,
6]. The liquid fuel produced by pyrolysis can be converted to fuels and value-added chemicals which are fully compatible with existing petroleum infrastructure via different upgrading methods [
7]. However, the bio-oil produced from biomass pyrolysis is a complex compound mixture containing alkenes, aromatics, phenolics, guaiacols, furans, esters, aldehydes, ketones, alcohols, sugars, and acids, which has to be upgraded before being used as a liquid fuel or chemical product [
8]. In our prior research, it was observed that liquid-liquid extraction using chloroform solvent on water phase has shown a significant result in eliminating acid, alcohol and sugar compounds from chloroform solvent phase [
9]. These small molecular like acid, alcohol and aldehyde occupy 30 wt % of total organics in bio-oil, and are also seen as the main reason for catalyst coking by polymerization. To make the most of these organics, the water phase can undergo upgrading with different upgrading routes such as esterification, acetalisation and condensation.
Esterification is a process to neutralize the organic acids and convert them into esters with alcohol via catalysts. Taking esterification in water-phase does not only fully utilize the 30 wt % of total organics, but also contributes to reducing the corrosion and deactivation of catalyst. The process extends the carbon chain of the small molecular, which further converted into alkane during upgrading by other pathways. Besides, it is well known that bio-fuel produced by ZSM−5 catalyst cracking method breaks oxygen compounds in form of carbon monoxide and water, accompanied by high energy loss, and results in multi-rings aromatic production [
9,
10]. The esterification process utilizing high energy content alcohol as a feedstock increases the heating value of the bio-oil product [
10].
Based on literature reviews, both homogenous and heterogeneous catalysts can be used in esterification for acid rich feedstock derived from biomass [
11]. Although homogeneous catalysts have been well studied and commercially used, heterogeneous catalysts are still being researched for their better performances on reactant-product separation and catalyst recovery. Various solid catalysts have been applied on heterogeneous catalysis esterification, including silica-supported sulfuric acids [
11,
12,
13], metal oxides [
14,
15,
16], heteropolyacids [
17], and zeolites [
10,
18,
19]. Among all the solid catalysts, pentasil zeolite catalysts with framework type known as ZSM−5 and MFI draws much attention as it has been widely used on both catalytic cracking and esterification processes in commercial refinery already. Bedard
et al. [
10] investigated the zeolite catalyst activities on esterification of acetic acid with ethanol at 60–120 °C, and observed over 90% of ester formation selectivity on all zeolite catalysts. Krumakki
et al. [
18] worked on esterification of acetic acid with C3 and C4 alcohols using BEA, FAU and MFI zeolite at temperatures from 110 to 130 °C, and found that inhabitation was accompanied by increasing alcohol concentrations.
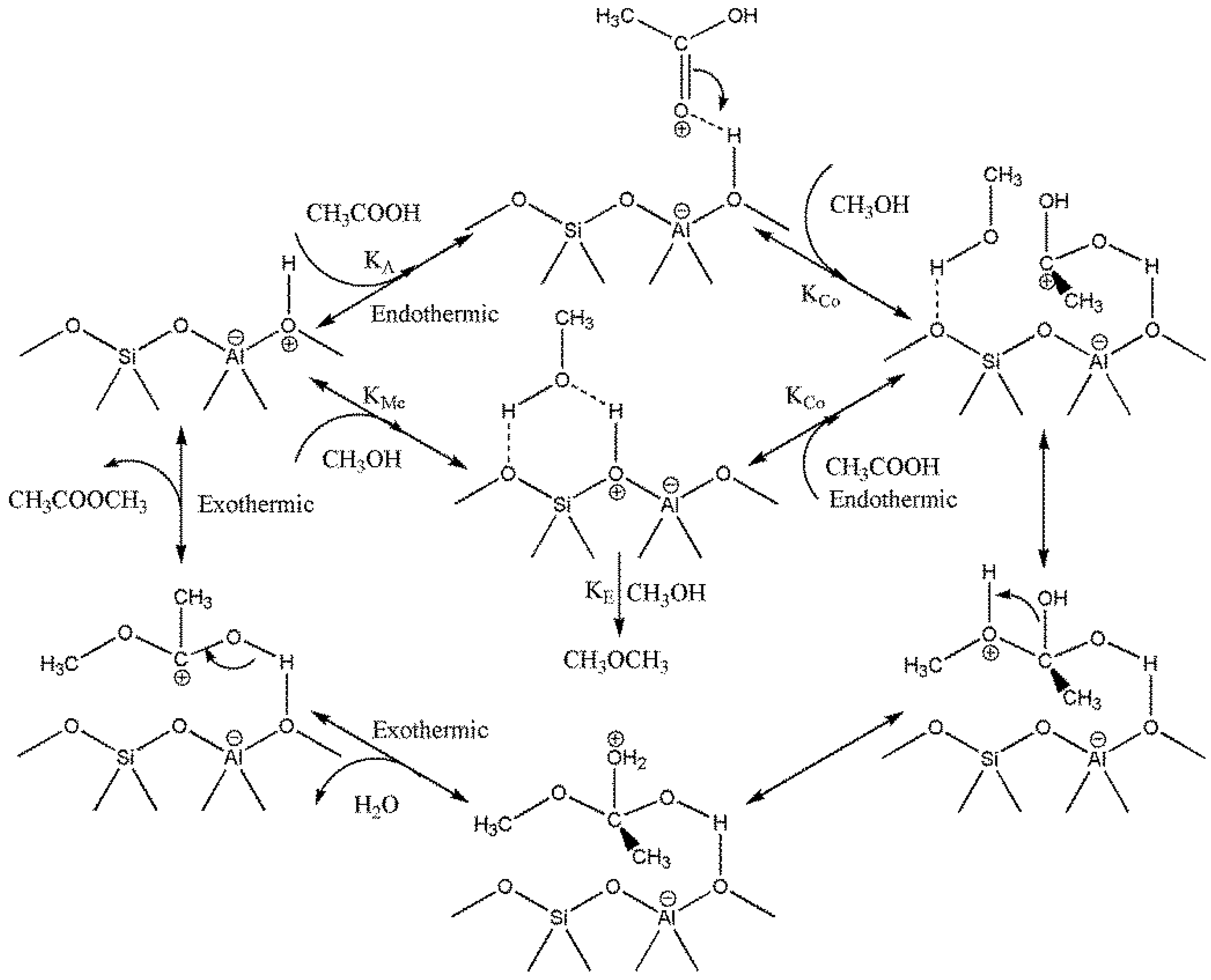
Though the mechanism of heterogeneous catalytic esterification is still debated, literature data had observed two contrary mechanisms for esterification based on acetic acid: One is dual sites Langmuir–Hinshelwood mechanism; the other one is single site Eley–Rideal mechanism [
11,
12,
13,
17]. Miao
et al. [
12] proposed dual site mechanism referring to their results on esterification of acetic acid and methanol using propyl sulfonic acid functionalized SBA-15 catalyst at temperatures ranged from 30 to 60 °C. Chu
et al. [
17], using heteropolyacids SiW12 catalysts at temperatures from 85 to 160 °C, also found that the esterification of acetic acid and ethanol followed the dual site Langmuir−Hinshelwood mechanism. However, Goodwin’s group [
11,
13] found that acetic acid esterifying with methanol and ethanol on silica supported SAC-13 catalyst performed as a single site mechanism at 90–140 °C. This was also supported by Chu and his co-authors [
17] conducting acetic acid esterification with 1 butanol. Besides, Kirumakki
et al. [
18] research described no mass transfer limitation on the zeolite surface during the esterification process, indicating the process should follow single site Eley–Rideal mechanism.
Compared with directly using model compound acids, there are quite a few works conducted on esterification of pyrolysis oil or pyrolysis oil fraction directly due to their complex compounds. Li
et al. [
20] group investigated the esterification of fast pyrolysis oil and alcohols on amberlyst 70 catalyst and obtained the best reaction condition for the process at mild temperature and catalyst loading status (100 °C, 3 wt % catalyst loading). Based on this result, Hu
et al. [
21] in the same group sought reactions on other non-acid organic compounds in bio-oils using amberlyst 70 catalysts. Referring to their conclusions, anhydrosugar in bio-oils converted to methyl levulinate as furanmethanol. They also found that N-containing compounds in bio-oils obtained from mallee eucalypts leaves deactivated the catalyst and led to low conversions [
21]. The use of zeolite catalyst to esterify the acid and furfural rich phase of microwave pyrolysis has not been found in the literature yet.
Herein, the esterification of acid, alcohol, and furfural alcohol compounds in extracted bio-oil water phase was investigated with methanol using activated commercial solid acid catalyst ZSM−5. To seek the optimum reaction conditions for esterification, catalyst loading and reaction temperature were used as variable factors in the research based on the results of a full factorial design. Kinetic and mechanism study have been conducted independently to have a clear understanding of the esterification pathway on ZSM-5 with methanol, by fitting two different models. Also, to achieve better understanding of property changes of bio-oils during the esterification process, the organic compounds in esterified bio-oil fraction were characterized by gas chromatography-mass spectrometry (GC–MS).
2. Materials and Methods
2.1. Material
Methanol (Extra dry, SC, 99.8%) was purchased from Fisher Scientific. Zeolite (CBV 5524G) was purchased from Zeolyst International. The characteristics of zeolite catalyst are shown in
Table 1. ZSM−5, comparing to the parent untreated ZSM−5, had improved BET surface area and acidity (
Table 1).
Table 1.
Characteristics of zeolites in the study.
Table 1.
Characteristics of zeolites in the study.
| Catalyst | Activation | Si/Al | Area BET (m2g−1) | Acidity (mmol g−1) |
|---|
| ZSM−5 | Untreated | 50.00 | 386.87 | |
| | Treated | 47.00 | 400.40 | 0.601 |
Bio-oil fraction (water phase) was obtained by a liquid-liquid extraction [
9]. The bio-oil was produced via microwave pyrolysis at 450 °C, 25 min and a fixed microwave power input of 700 W on a Sineo MAS-II batch microwave oven (Shanghai, China) with a rated power of 1000 W. The bio-oil obtained from microwave pyrolysis was collected via liquid-liquid extraction process (chloroform solvent) and the water phase was stored in a sealed bottle in a freezer until being used as feedstock.
The feedstock contained 13.79% acid, 6.65% ketone, 7.31% alcohol, 11.91% phenols, 24.02% guaiacols, 20.38% furan ring compounds, 1.87% ester, and 3.79% unreacted sugar by GC–MS, with a moisture content of 85%. The rests were the compounds with nitrogen or which cannot be characterized by GC−MS, which were about 10%.
2.2. Experimental Design
A full factorial design (two factors with 4 × 5 factorial treatment structure with duplicates) was used to optimize the esterification. Four different catalyst loadings (0, 1, 2, and 5 wt %) and five different temperatures (60, 80, 100, 120, 135 °C) were employed as independent variables in the design. Reaction time was tested as another variable with 4 levels (0.5, 1, 2, 4 h) beside the design.
2.3. Esterification
The experiments were conducted in an autoclave (SANYO, MLS-3751 loading autoclave, 50 L). Generally, 10 g of liquid water phase and 10 g of methanol was mixed in a 100 mL pressure vessel (Chemglass Life Sciences, 100 mL HW Pressure Vessel). For each experiment, different mass of ZSM−5 catalyst was loaded in the vessel according to the experimental design of catalyst loadings. Prior to heating, the nitrogen was purged into the vessel to displace the residual air. After that, the pressure vessels were sealed and loaded in the autoclave, heated to desired temperatures by a programmed controller of autoclave. Samples were taken immediately after programmed esterification and were tested by GC−MS. The conversion of acid and selectivity of the catalyst (for monoprotic acids) was calculated as below:
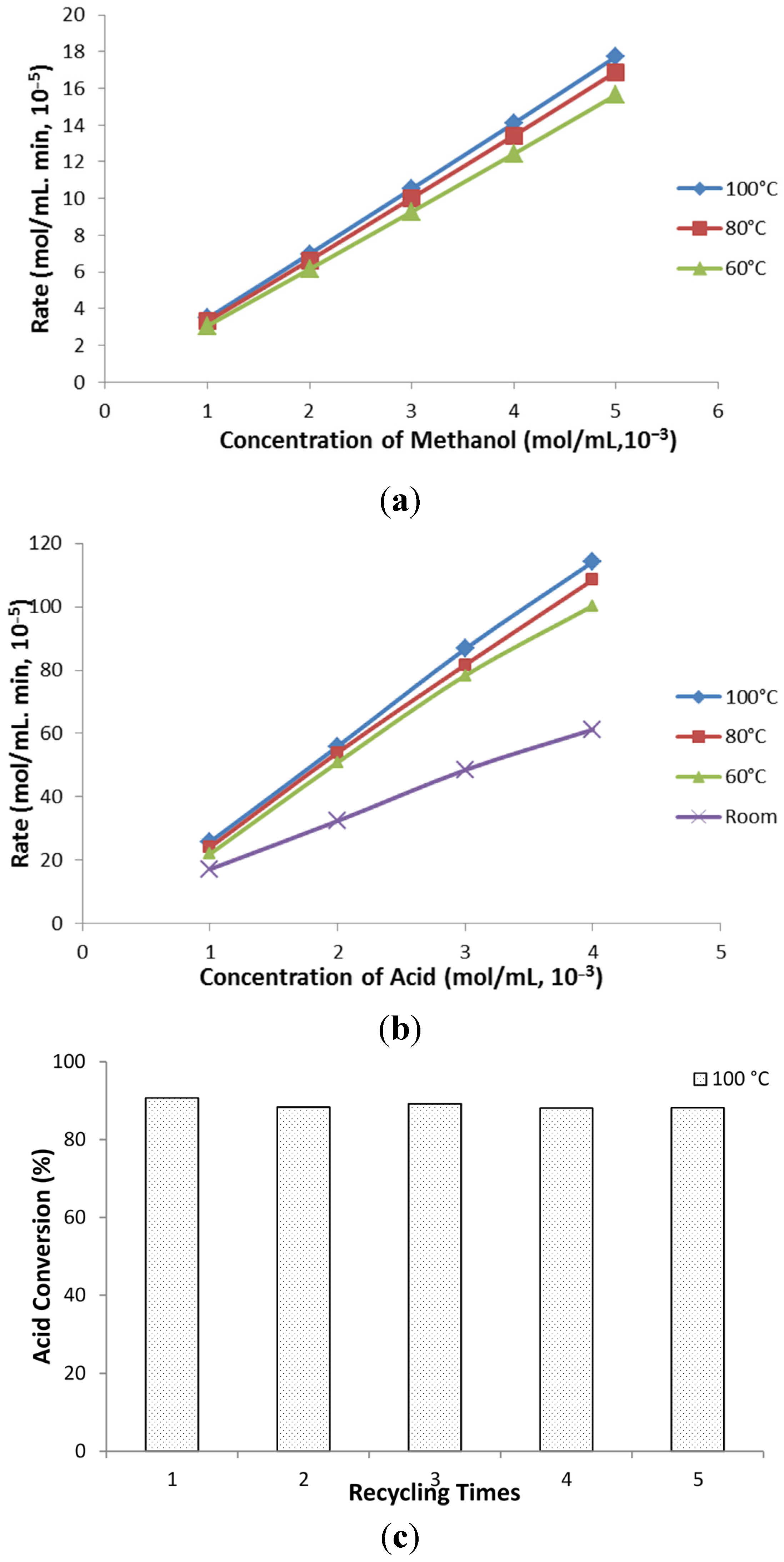
2.4. Kinetic Study
The experiments were conducted in an autoclave (SANYO, MLS-3751 loading autoclave, 50 L). Each time, pure acetic acid and methanol was mixed in a 100 mL pressure vessel (Chemglass Life Sciences, 100 mL HW Pressure Vessel) according to the specific concentrations and ratios. For each experiment, 2 wt % of ZSM−5 catalyst was loaded in the vessel. Prior to heating, the nitrogen was purged into the vessel to displace the residual air. After that, the pressure vessels were sealed and loaded in the autoclave, heated to desired temperatures by a programmed controller of autoclave, running with a reaction time ranging from 30 to 120 min. Samples were taken immediately after programmed esterification and were tested by HPLC. The results obtained from this research was applied to construct kinetic models, which further supported the reaction mechanism of the process.
2.5. HPLC Analysis
The acid conversion was determined by Alliance HPLC (Alliance 2695) and a refractive index detector with Bio-Rad Aminex HPX-87P column. The HPLC was programmed by maintaining column temperature of 60 °C. The injection took place at 20 °C, with sample injection size of 1 μL. The flow rate of the carrier solvent (diluted sulfuric acid) was 0.6 mL/min.
2.6. GC−MS Analysis
The chemical composition of bio-oil was determined by Agilent GCMS (GC–MS; GC, Agilent 7890A; MS, Agilent 5975C) with DB-5 capillary column. The GC was programmed by maintaining at 45 °C for 3 min followed by heating to 300 °C at a heating rate of 10 °C/min. The injection took place at 300 °C, with sample injection size of 1 μL. The flow rate of the carrier gas (helium) was 0.6 mL/min. The ion source temperature was 230 °C for the mass selective detector. Acetic acid and ester was calibrated by an external standard method. The other compounds were identified by comparing the spectral data with the NIST Mass Spectral library.
It was difficult to obtain standards for all the compounds identified in the GC–MS chromatograms. In those cases, the signal intensity (peak area) of the compound was used as a measure of the changes in their concentration as a function of reaction conditions. The standard deviation of the experimental data was within ±5%.
3. Results and Discussion
3.1. Effect of Reaction Time on Product Yield and Conversion
Series of experiments were conducted to investigate the effects of reaction time on ZSM−5 catalyzed esterification from extracted bio-oil and methanol with fixed catalyst loadings and temperatures prior to the full factorial design analysis. Generally, the bio-oil water phase underwent esterification reactions during the process. Acid and alcohol in water phase was converted into ester. It was found that the concentration of ester increased significantly from the beginning of reactions, and approached its peak concentration after 1 h reaction. However, the production of ester was only slightly increased after 2 h. For the control group without catalysts, the concentration of ester was slowly increased in 8 h. The ester concentration of control group at 8 h only reached 40% of the 1 wt % group, which indicated the strong effect of the ZSM−5 catalyst. A similar trend had also been found on acid conversion, as acid nearly reached their peak conversion at 2 h, particularly for the catalyst loading of 5 wt %, while the control group still had less than 9% of conversion on acid. As a result, the highest conversion of acid could be reached within 2 h, which indicated that 2 h were the adequate reaction time for the process at temperature of 60 °C. Based on temperature, higher temperature improved the speed of ester formation. With the reaction temperature above 100 °C, the concentration of esters achieved over 95% of its highest yield within 1 h of reactions, at fixed catalyst loading of 2 wt %. After 2 h running of the experiment, the ester obtained in all five temperature levels reached over 98% of the highest ester concentration from 8 h reactions. Therefore, reaction time of 2 h was considered as the adequate reaction time for the esterification processes.
3.2. Full Factorial Design Analysis
The full factor design using five levels of temperatures and four levels of catalyst loadings was used in this research.
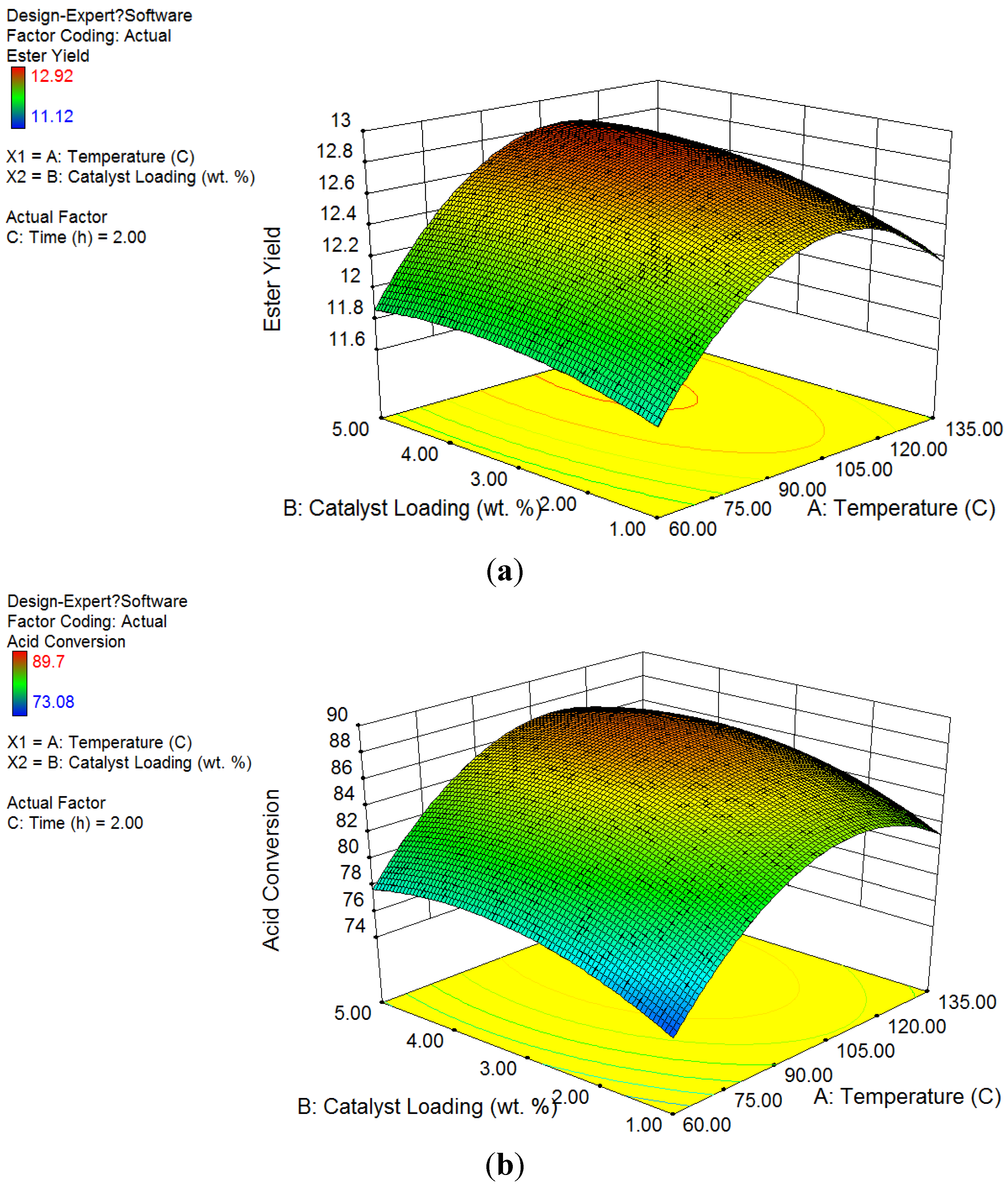
The
Figure 1 showed the ester yield and acid conversion based on factor changes. According to the data, ZSM−5 catalysts had high efficiency and selectivity on conversions of acid compounds in esterification process. The ester concentration increased from 1.87% to over 11.53% on catalyst groups, while control group had less than 6.6% of esters. The acid concentration was significantly reduced. The conversion of acids in the groups using ZSM−5 ranged from 73.08% to 89.39%, while the conversion in the control group kept below 17.48%. Not only higher conversion was achieved in products (around 75%), but also distinct change on catalyst surface was observed. An average ester selectivity of 90% had been obtained during the reaction.
Figure 1.
Factor design results of product concentration and conversion by temperature and catalyst loading with bio-oil water phase to methanol ratio of 1:1 (mass/mass) at 2 h: (a) Ester yield in product; (b) Acid conversion; (c) Optimization analysis.
Figure 1.
Factor design results of product concentration and conversion by temperature and catalyst loading with bio-oil water phase to methanol ratio of 1:1 (mass/mass) at 2 h: (a) Ester yield in product; (b) Acid conversion; (c) Optimization analysis.
3.2.1. Response Surface Analysis on Ester Yield
According to the results of the experiment, the regression Equation (1) showing the yield changes of esters was obtained as a function of the temperature (A) and catalyst loading (B):
According to ANOVA test, the p-value of Equation (1) was 0.0001 < α = 0.05, which is significant and can be used to describe the variation in concentrations of esters obtained by esterification. The interaction of both factors had a p-value = 0.12 > α = 0.05, which means there was no significant interaction between factors of catalyst loadings and temperatures. The p-values of catalyst loadings were less than 0.0001, and the p-value of temperatures was 0.03, which was also less than α = 0.05, indicating that both factors were significant to affect ester yields. In addition, the coefficient of determination for Equation (1) was 0.90, which meant the regression model was suitable to describe the ester yield from its significant factors. According to Fisher test, the p-values of both catalyst loading and temperature factors were less than 0.001, as both of them had significant influence on ester yield.
Figure 1a illustrates the surface response of ester concentrations by temperatures and catalyst loadings. Based on color legend shown on the left, high ester yield was achieved on the temperatures ranging from 80 to 120 °C and over 2 wt % of catalyst loadings. The peak desirability with the highest ester yield was obtained at the temperature of 95 °C and catalyst loading around 4 wt %. As a result, the optimized reaction conditions for the highest ester yield in this study was at the temperature of 100 °C and catalyst loadings of 2 wt % and 5 wt %.
3.2.2. Response Surface Analysis on Acid Conversion
The relationship between conversion of acid and factors of the temperature (A) and catalyst loading (B) was were obtained in the regression Equation (2) below:
The conversion of acid was closely related to the concentration of ester, as acids were esterified with methanol to form esters. According to ANOVA test, the p-value of Equation (2) was less than 0.0001, <α = 0.05, which shows the test is significant and can describe the variation of the conversion of acids. The p-value of interaction was 0.18, which proved no strong interaction between two factors on acid conversion. According to Fisher test, the p-values of both catalyst loading and temperature factors were less than 0.0001, as both of them had significant influence on acid conversions. The coefficient of determination for Equation (2) was 0.91, which meant the regression model was suitable to describe the ester yield from its significant factors.
Figure 1b shows the factor design results of acid conversion on surface response by temperatures and catalyst loadings. Other than ester yield, based on color legend shown on the left, high acid conversion was achieved at temperatures less than 100 °C and catalyst loadings over 2 wt %. The most desirable reaction condition for acid conversion was found around 80 °C with 5 wt % ZSM−5 loading. Consequently, temperature level of 80 °C and catalyst loading level of 5 wt % were chosen as the optimized reaction conditions for acid conversion.
3.3. Effect of Single Factor on Product Yield and Conversion
3.3.1. Effect of Temperature
Table 2 displays the temperature effect on the ZSM−5 esterification.
Table 2.
Effect of single factor on ester yield and acid conversion (2 h).
Table 2.
Effect of single factor on ester yield and acid conversion (2 h).
| Temperature (°C) | Catalyst Loading (wt %) | Ester Yield (%) | Conversion (%) | Selectivity (%) |
|---|
| 60 | 2 | 11.89 | 77.72 | 86.30 |
| 80 | 2 | 12.31 | 80.25 | 91.18 |
| 100 | 0 | 3.65 | 10.91 | ---- |
| 100 | 1 | 12.66 | 85.21 | 88.68 |
| 100 | 2 | 12.79 | 87.16 | 91.86 |
| 100 | 5 | 12.92 | 88.32 | 90.39 |
| 120 | 2 | 12.7 | 88.63 | 90.92 |
| 135 | 2 | 12.38 | 82.65 | 90.00 |
The acid in the water-phase of bio-oil decreased from high abundance (13.79%) to less than 2.7% via esterification, along with ester yield increased from 1.87% to over 9.8%. High productivity of ester was presented from 80 to 120 °C, while it decreased sharply at temperatures above 135 °C. The peak yield of ester was achieved (12.92%) at 100 °C. ZSM−5 catalysts showed a strong affinity to acid conversion and ester production, while control experiments without catalyst had very low concentration of esters and insignificant conversion of acids. Referring to
Table 2, as acids underwent esterification reactions to form esters, the temperature level of the highest conversion also ranged from 80 to 120 °C, which met the temperature at which ester had the highest yield. Nevertheless, even with catalysts, the experimental group running at lower temperature could not compete with experiments at high temperatures on reaction rate. Lower reaction temperatures resulted in lower ester concentration, while high temperature had a negative influence on ester formation, which is discussed in
Section 4. The ester selectivity of the catalyst trended to high ester yield accompanied with temperature increases below 100 °C but dramatically reduced after that. Besides, according to prior research, the unpyrolyzed sugar also underwent other parallel reactions and produced acids such as levulinic acid during the esterification process, which reinforced the ester production higher than conversion rate calculated by acid concentration.
3.3.2. Effect of Catalyst Loading
The concentrations of ester and conversion of acid after 2 h of reaction by catalyst loading factor have also been presented in
Table 2. Comparing with control group, the ZSM−5 catalyst gave an outstanding performance on converting acid compounds. Accordingly, 2.66%–10.7% of ester yield was observed. The conversion of acid achieved over 81.87% in catalyst groups, while these two numbers were reduced to around 11% in control groups, respectively. As mentioned before, both 2 wt % and 5 wt % catalysts loading obtained higher yield of ester and considerable acid conversion. Thus, additional experiments were conducted on seeking the correlation between catalyst loading and initial reaction rate. Generally, reaction rate of catalyst exhibited lower trend due to interaction and depression between catalyst and product. The effect of catalyst loading on estimating initial reaction rates indicated strong increase in initial reaction rate was obtained accompanied by more catalyst added in the reaction. The phenomenon is probably traceable in enhancing surface area and acid sites for esterification process. However, the selectivity indicated slight inhabitation on ester selectivity as more undesirable reactions take place in acid. Taking both conversion rate and selectivity, though contributing to higher initial reaction rate, the 5 wt % of catalyst had less effect on ester yield after 2 h reaction. Thus, catalyst loading of 2 wt % was chosen as the optimized catalyst loading for extracted bio-oil water-phase esterification and acetalisation upgrading.
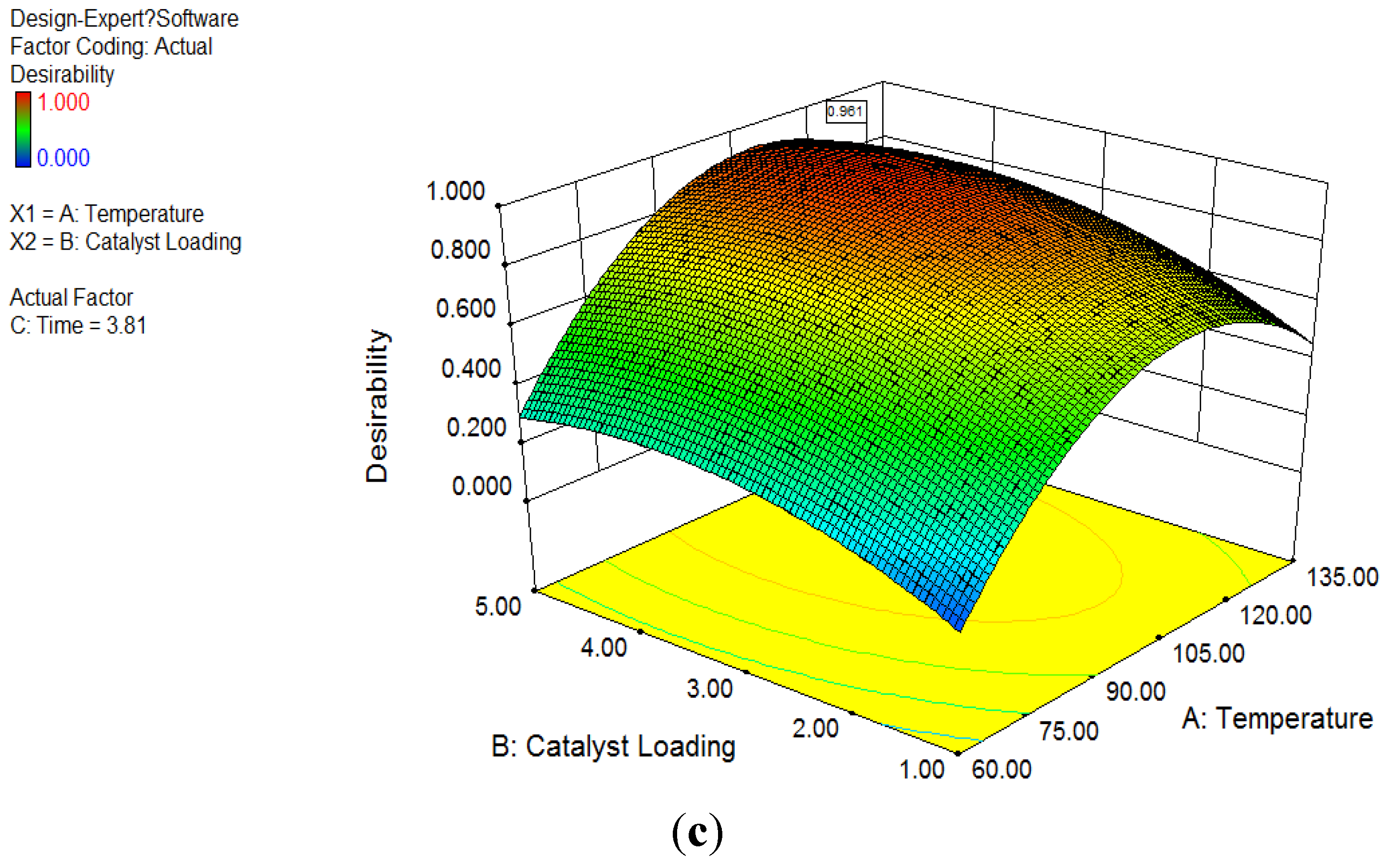
3.4. Optimization Analysis
The result of optimization analysis is shown in
Figure 1c. The optimization of the response method makes use of an objective function, called the desirability function. It reflects the desirable ranges for each response. The desirable ranges are from zero to one (least to most desirable, respectively). It is obvious that 100 °C was selected as the optimum reaction temperature, as the desirability of optimization analysis at 100 °C is always higher than other temperature levels at the same catalyst loading. Generally, higher catalyst loading always achieved higher ester concentration at all temperature levels. 5 wt % catalysts loading had distinguished better performance than 1 wt % and 2 wt % levels at temperature levels of 60 and 140 °C. Nevertheless, the desirability at 2 wt % catalysts loading was close to 5 wt % between 80 and 120 °C, particularly at 100 °C. Referring to
Figure 1c, after considering all four responses, the peak desirability was obtained for a value of 0.96, at 100.1 °C with 3.98 wt % catalyst. In summary, the adequate reaction condition by this test was obtained at temperature of 100.1 °C and catalyst loading of 3.98 wt %.
3.5. Effect on Other Chemical Compounds
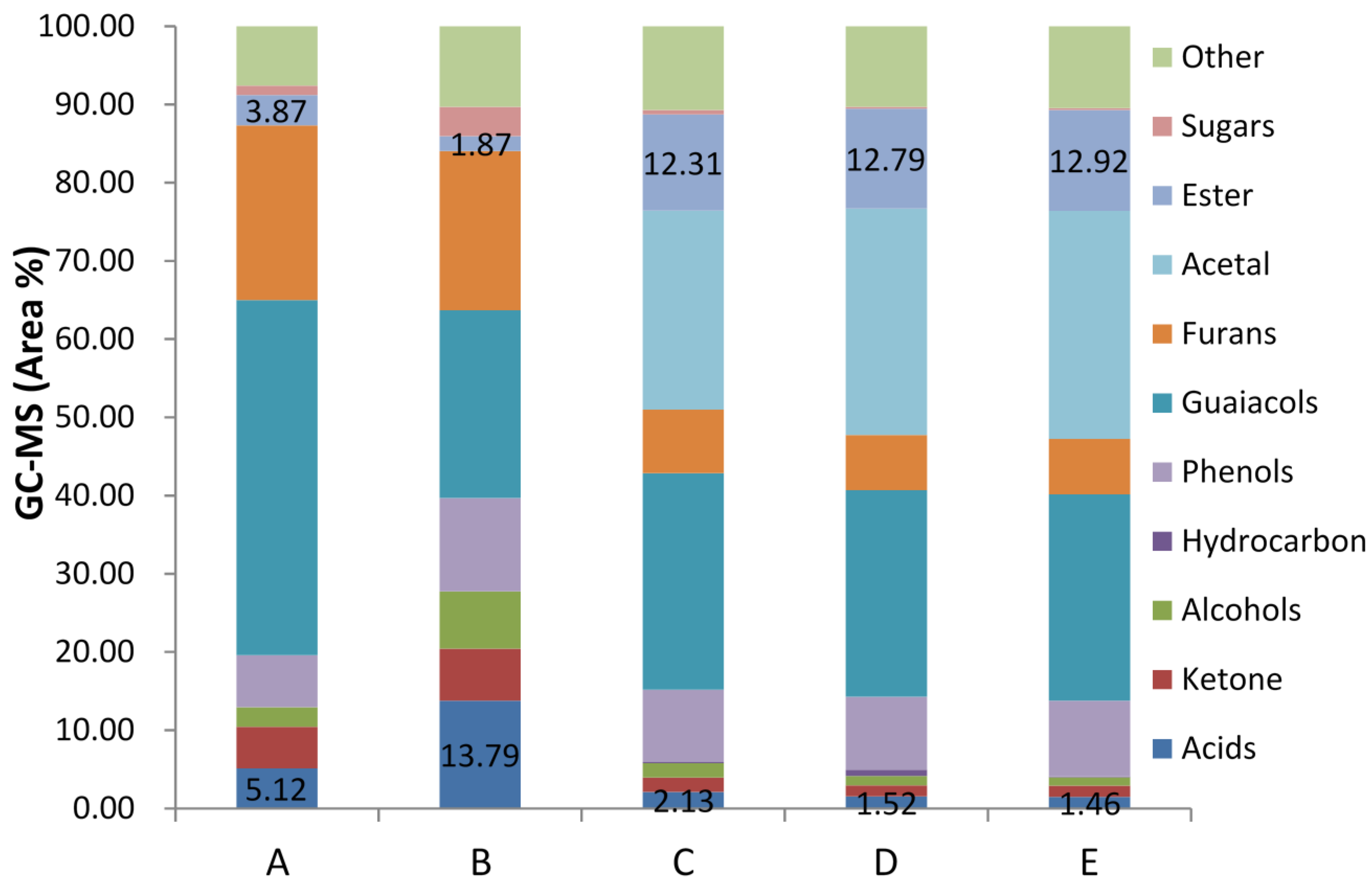
Figure 2 presented the chemical distribution of water-phase before and after esterification on ZSM−5 catalyst.
Figure 2.
Chemical distribution of water-phase esterification process on ZSM−5 catalyst with methanol: (A) Water-phase after water separation; (B) Water phase after Liquid-liquid extraction process (feedstock); (C) Esterification result with 2% ZSM−5 at temperature of 80 °C; (D) Esterification result with 2% ZSM−5 at temperature of 100 °C; (E) Esterification result with 5% ZSM−5 at temperature of 100 °C.
Figure 2.
Chemical distribution of water-phase esterification process on ZSM−5 catalyst with methanol: (A) Water-phase after water separation; (B) Water phase after Liquid-liquid extraction process (feedstock); (C) Esterification result with 2% ZSM−5 at temperature of 80 °C; (D) Esterification result with 2% ZSM−5 at temperature of 100 °C; (E) Esterification result with 5% ZSM−5 at temperature of 100 °C.
Obviously, concentration of acid reduced sharply from over 13% to less than 2%, by huge growth concentration of ester, indicating excellent esterification effect. Concentration of alcohol also dropped from 7% to around 1% because of esterification with acid. As mentioned before, unpyrolyzed sugar underwent other parallel reactions and produced acids such as levulinic acid during the esterification process, which lead to a sugar yield decrease of 85%. The reason for furan compounds and ketone reduction was acetalisation. It had been observed that furfural and acetaldehyde took acetalisation reaction with methanol immediately after ZSM−5 catalyst was added into the mixture at room temperature. Thus, it was possible that furan compounds with aldehyde function groups undertaking the acetalisation process caused the furan concentration to be lower than the initial situation. Similar to furan compounds, small molecular aldehyde in the mixture, which was the principal cause for catalyst deactivation, had been converted into corresponding acetals. Although the concentration of phenol and guaiacol compounds were kept constant during the esterification in the study, prior research in our group had found high efficiency for phenol and guaiacol compounds relied on the catalytic cracking process to produce aromatic hydrocarbon on ZSM−5 catalyst at high temperature. To see if there was any inhabitation for these reactants during the esterification process, an independent experiment conducted on guaiacols and phenols model compounds (2-methoxy-4-methyphenol and phenol) with ZSM−5 catalyst, showed no significant reaction and concentration changes at such low temperatures. Thus, we could conclude that these two compounds did not take any reactions at the temperatures for esterification process on ZSM−5 catalyst.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}