Gas Hydrate Occurrence Inferred from Dissolved Cl− Concentrations and δ18O Values of Pore Water and Dissolved Sulfate in the Shallow Sediments of the Pockmark Field in Southwestern Xisha Uplift, Northern South China Sea

Abstract
:1. Introduction
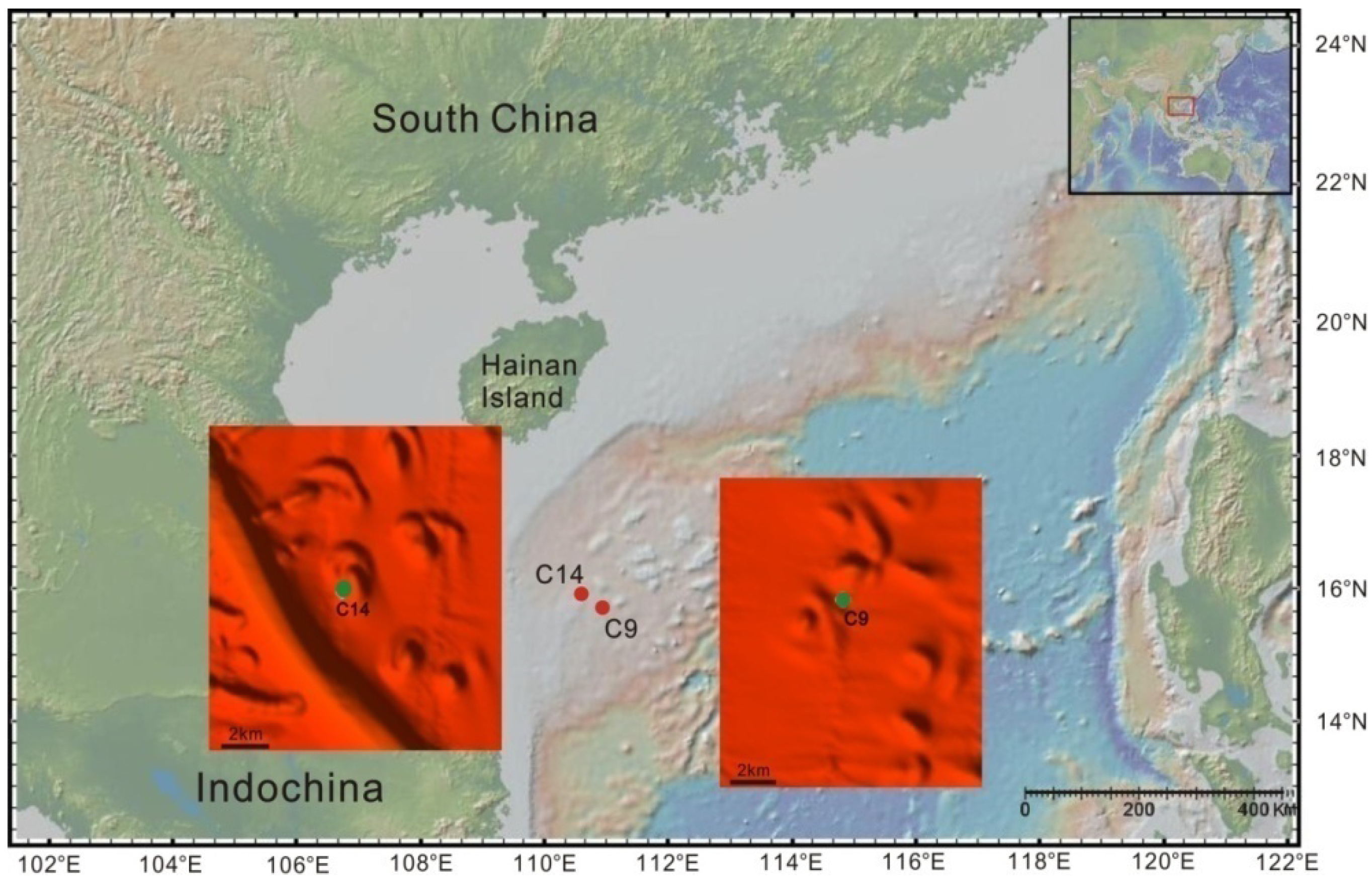
2. Sampling and Methods
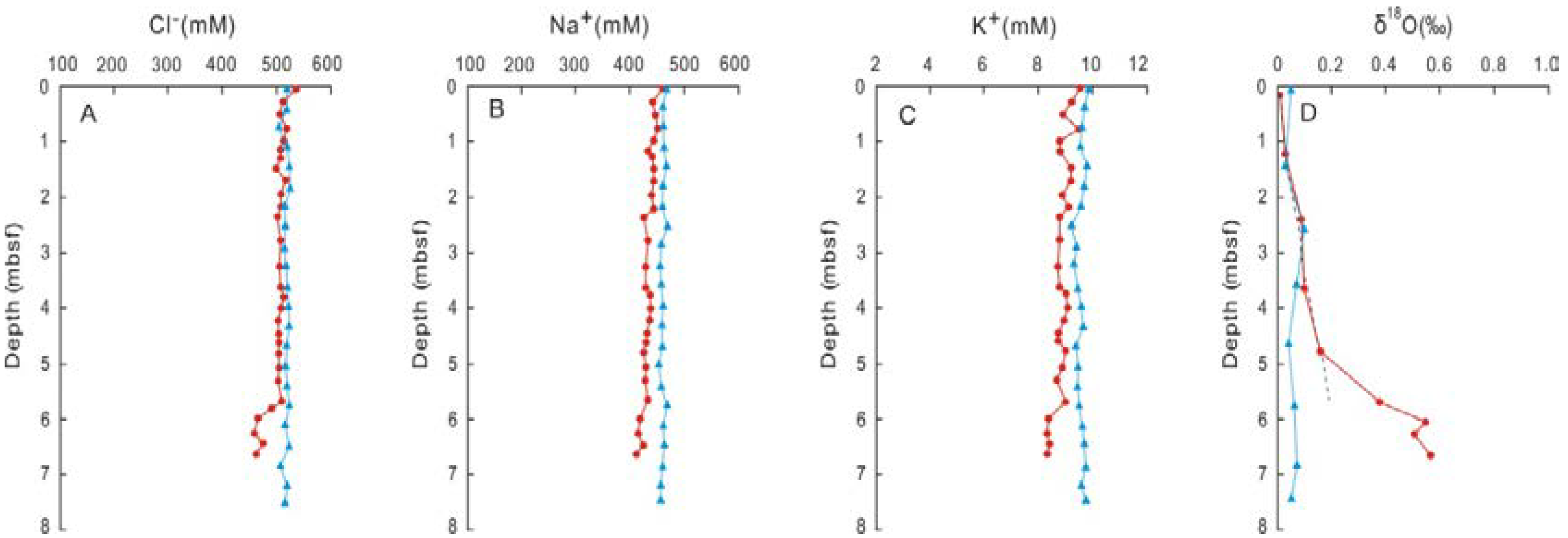
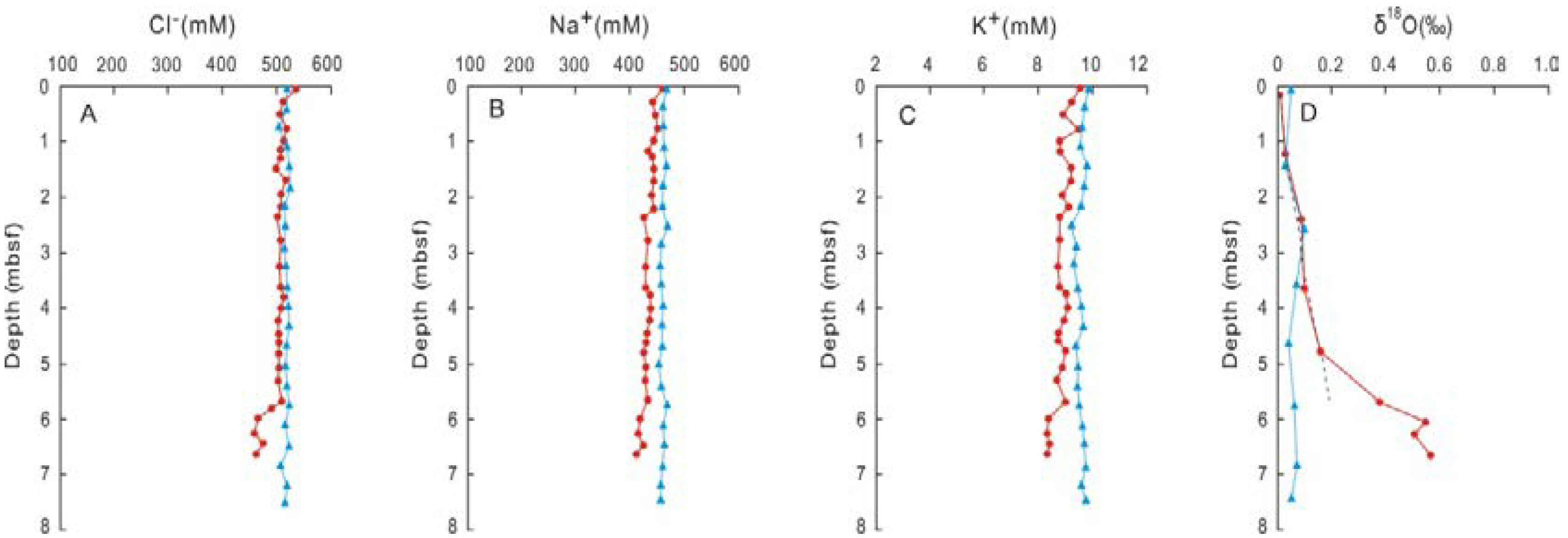
3. Results

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Depth(mbsf) | Cl−(mM) | Na+(mM) | K+(mM) | δ18OH2O (‰ V-SMOW) | δ18OSO4 (‰ V-SMOW) | |||
|---|---|---|---|---|---|---|---|---|
| C9 | ||||||||
| 0.06 | 519.9 | 465.9 | 9.9 | 0.05 | 10.7 | |||
| 0.42 | 517.2 | 463.5 | 9.7 | |||||
| 0.78 | 507.9 | 461.0 | 9.6 | 13.3 | ||||
| 1.14 | 516.7 | 464.5 | 9.6 | |||||
| 1.50 | 523.7 | 468.6 | 9.8 | 0.03 | ||||
| 1.86 | 524.7 | 461.4 | 9.7 | |||||
| 2.22 | 514.7 | 459.7 | 9.6 | 18.4 | ||||
| 2.58 | 516.4 | 468.4 | 9.3 | 0.1 | ||||
| 2.94 | 513.8 | 459.1 | 9.5 | |||||
| 3.30 | 517.6 | 457.1 | 9.3 | |||||
| 3.66 | 519.1 | 466.6 | 9.5 | 0.07 | ||||
| 4.02 | 520.1 | 460.7 | 9.6 | 20.3 | ||||
| 4.38 | 522.8 | 458.8 | 9.8 | |||||
| 4.74 | 518.6 | 459.9 | 9.4 | 0.04 | ||||
| 5.10 | 516.9 | 451.9 | 9.5 | |||||
| 5.46 | 519.1 | 457.1 | 9.5 | |||||
| 5.82 | 523.3 | 468.6 | 9.5 | 0.06 | 20.6 | |||
| 6.18 | 516.6 | 461.3 | 9.7 | |||||
| 6.54 | 523.3 | 463.6 | 9.7 | |||||
| 6.90 | 508.5 | 459.6 | 9.8 | 0.07 | 21.6 | |||
| 7.26 | 518.5 | 455.7 | 9.6 | |||||
| 7.56 | 516.1 | 456.3 | 9.8 | 0.05 | 21.4 | |||
| C14 | ||||||||
| 0.06 | 536.6 | 460.7 | 9.5 | |||||
| 0.18 | 529.3 | 445.4 | 9.3 | 0.03 | 10.9 | |||
| 0.3 | 512.5 | 441.9 | 9.2 | |||||
| 0.54 | 505.5 | 446.8 | 8.9 | 16.6 | ||||
| 0.78 | 519 | 450.6 | 9.5 | |||||
| 1.02 | 513.1 | 443 | 8.8 | 17.3 | ||||
| 1.2 | 504.5 | 433.3 | 8.9 | 0.03 | ||||
| 1.26 | 509.7 | 440.6 | 8.7 | |||||
| 1.5 | 499.4 | 443.8 | 9.2 | |||||
| 1.74 | 515.9 | 443.6 | 9.2 | 17.6 | ||||
| 1.98 | 508.6 | 439.7 | 8.9 | |||||
| 2.22 | 508 | 443.4 | 9.1 | |||||
| 2.4 | 500 | 425.1 | 8.8 | 0.09 | ||||
| 2.82 | 508.4 | 432.8 | 8.8 | 18.5 | ||||
| 3.3 | 504.3 | 429.6 | 8.7 | |||||
| 3.66 | 507.8 | 429.1 | 8.8 | 0.1 | ||||
| 3.78 | 513.9 | 435.6 | 9 | |||||
| 4.02 | 507.5 | 437.6 | 9.1 | |||||
| 4.26 | 502 | 435.8 | 9 | 19.4 | ||||
| 4.5 | 504.5 | 431.5 | 8.8 | |||||
| 4.62 | 504.8 | 430.6 | 8.8 | |||||
| 4.8 | 501.8 | 425.4 | 9 | 0.16 | ||||
| 4.86 | 504.8 | 426.5 | 8.7 | |||||
| 5.1 | 503.6 | 429.4 | 8.9 | |||||
| 5.34 | 502.9 | 428 | 8.7 | 19.6 | ||||
| 5.7 | 509.5 | 433.5 | 9 | 0.37 | ||||
| 5.88 | 478.7 | 431.5 | 8.9 | |||||
| 6.06 | 465.2 | 418.4 | 8.4 | 0.55 | ||||
| 6.3 | 458.6 | 415.9 | 8.3 | 0.51 | ||||
| 6.54 | 469.3 | 417.4 | 8.4 | 19.9 | ||||
| 6.66 | 462.5 | 412.4 | 8.3 | 0.57 | ||||
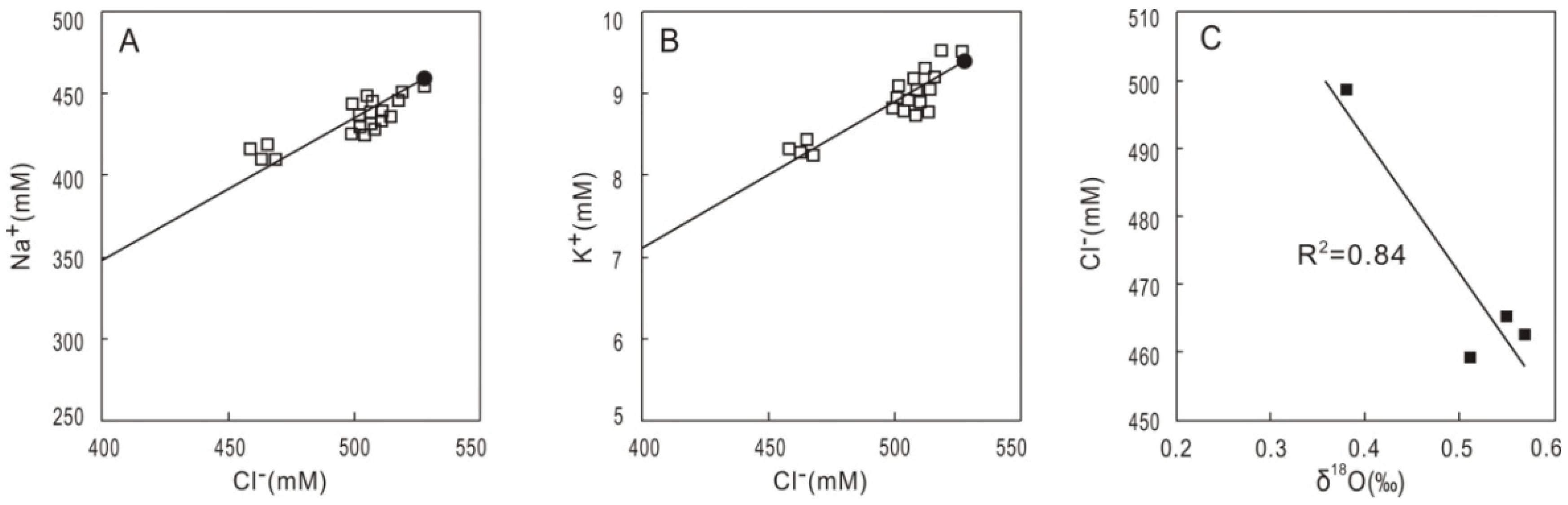
4. Discussion
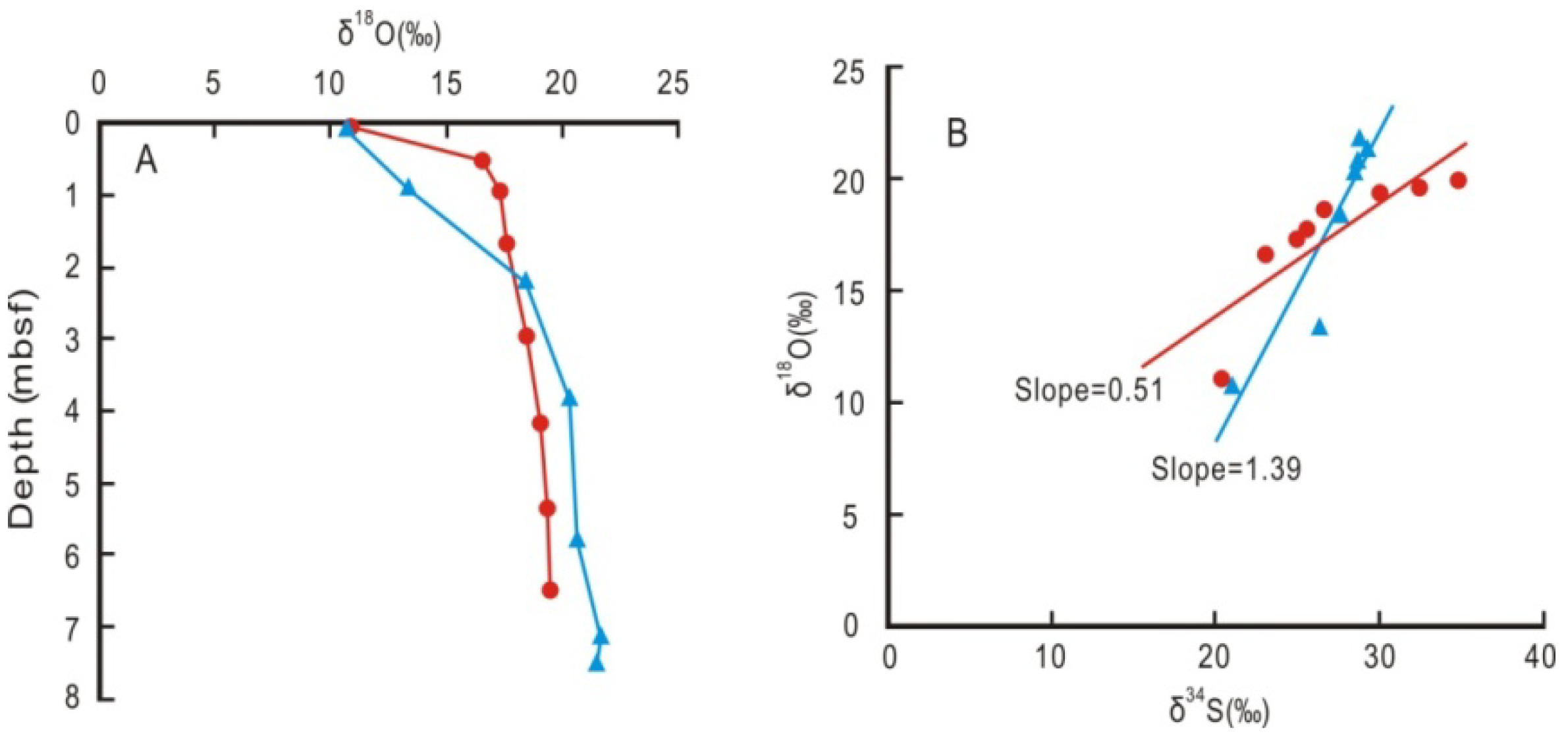
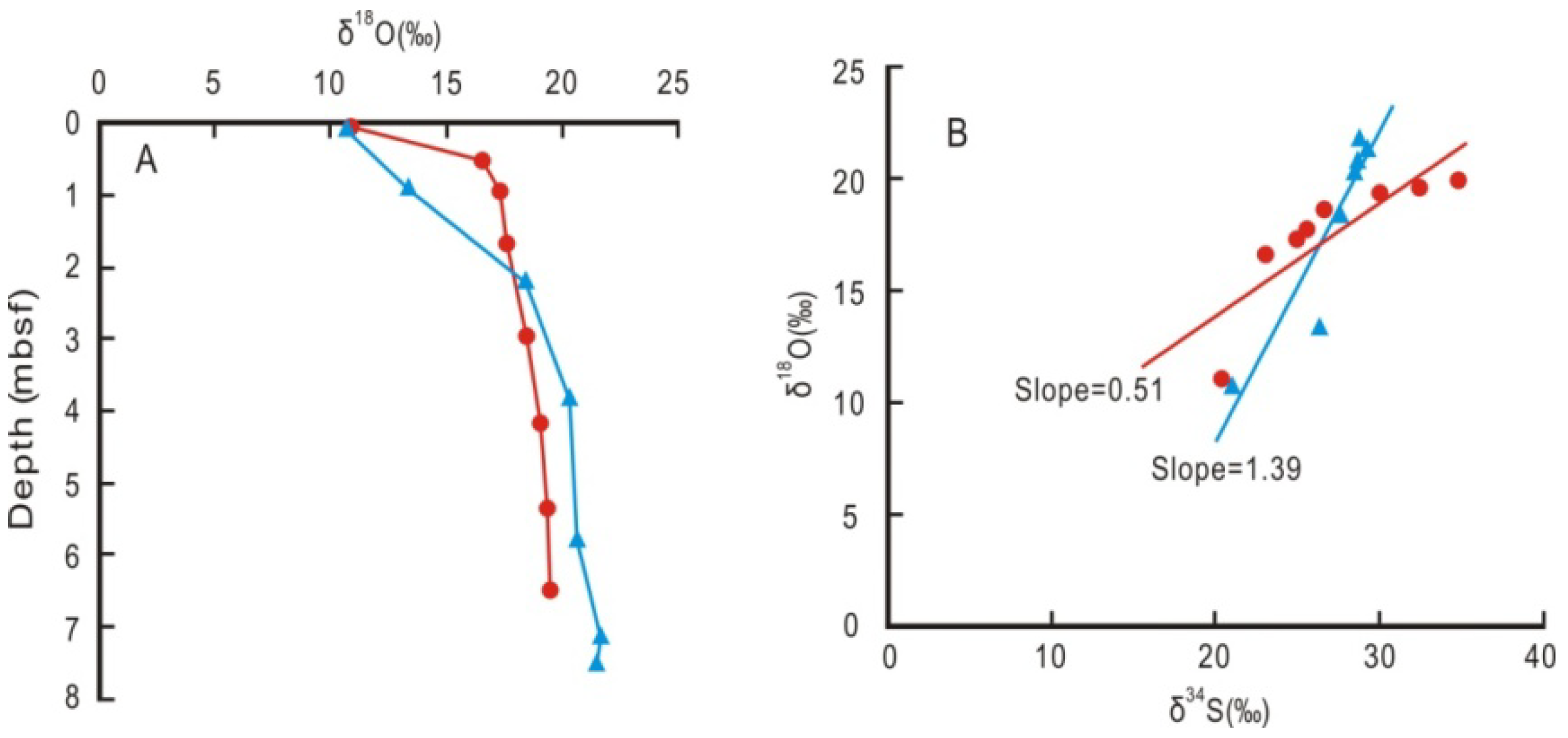
4.1. Potential Occurrence of Gas Hydrates Inferred from Coupled Chlorinity Decrease and δ18O Increase
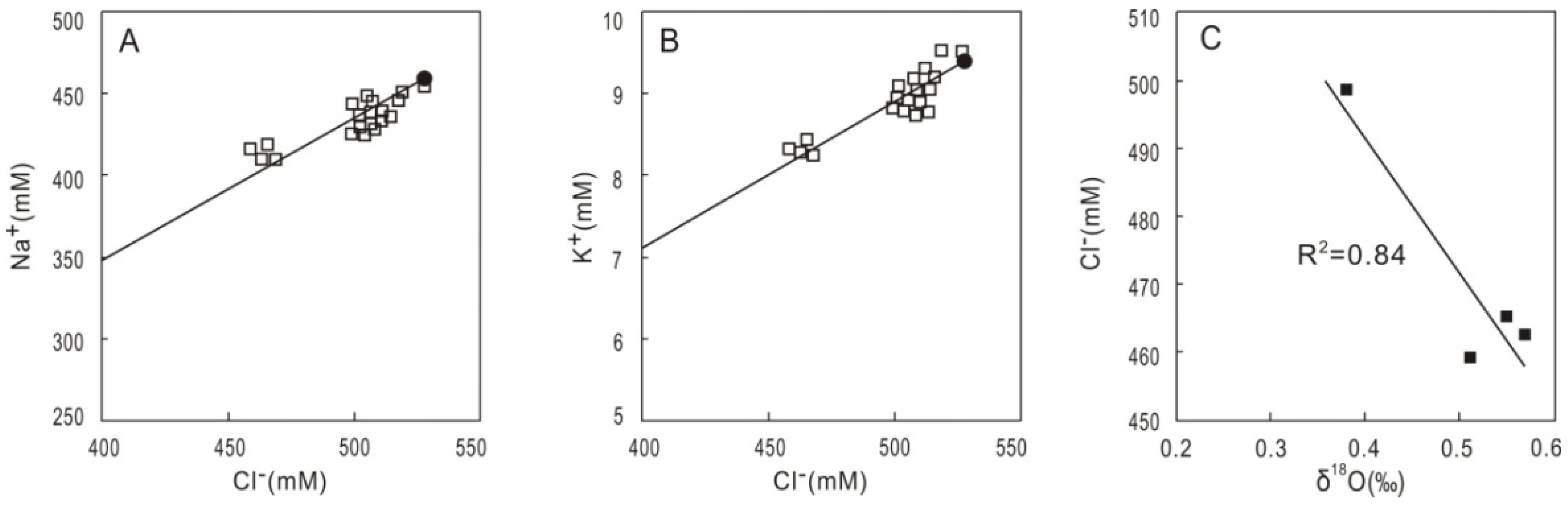
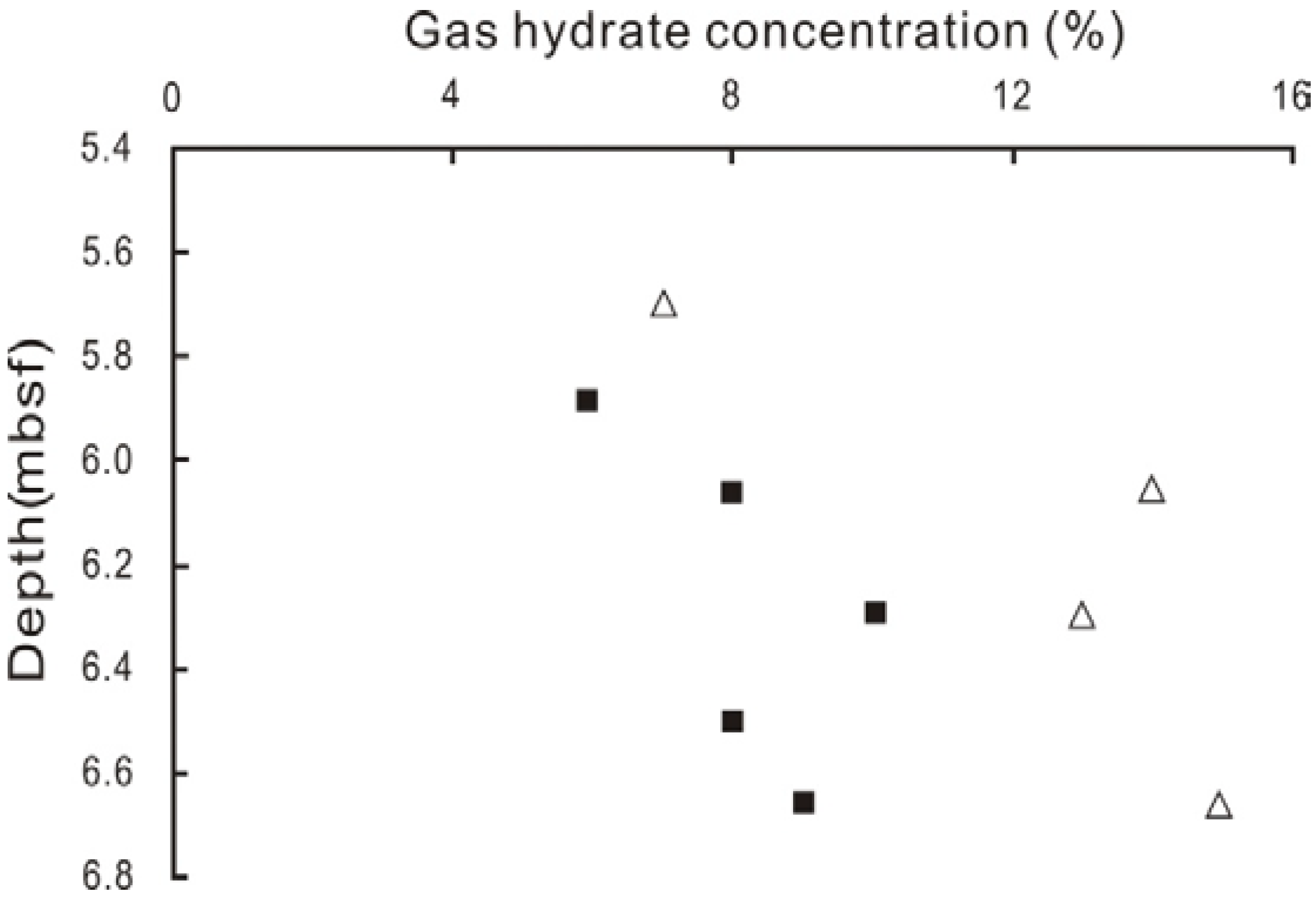
4.3. Gas Hydrate Contents Estimated from the Anomalies of Pore Water Chlorinity and δ18O Values
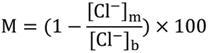

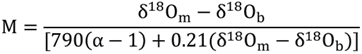
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Judd, A.G.; Hovland, M. Seabed Fluid Flow: The Impact of Geology, Biology and the Marine Environment; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Hovland, M.; Svensen, H. Submarine pingoes: Indicators of shallow gas hydrates in a pockmark at Nyegga, Norwegian Sea. Mar. Geol. 2006, 228, 15–23. [Google Scholar] [CrossRef]
- Sahling, H.; Bohrmann, G.; Spiess, V.; Bialas, J.; Breitzke, M.; Ivanov, M.; Kasten, S.; Krastel, S.; Schneider, R. Pockmarks in the Northern Congo Fan area, SW Africa: Complex seafloor features shaped by fluid flow. Mar. Geol. 2008, 249, 206–225. [Google Scholar] [CrossRef]
- Kastner, M.; Claypool, G.; Robertson, G. Geochemical constraints on the origin of the pore fluids and gas hydrate distribution at Atwater Valley and Keathley Canyon, northern Gulf of Mexico. Mar. Pet. Geol. 2008, 25, 860–872. [Google Scholar] [CrossRef]
- Borowski, W.S.; Paull, C.K.; Ussler, W. Carbon cycling within the upper methanogenic zone of continental rise sediments; an example from the methane-rich sediments overlying the Blake Ridge gas hydrate deposits. Mar. Chem. 1997, 57, 299–311. [Google Scholar] [CrossRef]
- Hesse, R.; Harrison, W.E. Gas hydrates (clathrates) causing pore-water freshening and oxygen-isotope fractionation in deepwater sedimentary sections of terrigenous continental margins. Earth Planet. Sci. Lett. 1981, 55, 453–462. [Google Scholar] [CrossRef]
- Hesse, R. Pore water anomalies of submarine gas-hydrate zones as tool to assess hydrate abundance and distribution in the subsurface—What have we learned in the past decade? Earth-Sci. Rev. 2003, 61, 149–179. [Google Scholar] [CrossRef]
- Ussler, W.; Paull, C.K. Effects of ion-exclusion and isotopic fractionation on pore-water geochemistry during gas hydrate formation and decomposition. Geo-Mar. Lett. 1995, 15, 37–44. [Google Scholar] [CrossRef]
- Matsumoto, R.; Borowski, W. Gas hydrate estimates from newly determined oxygen isotopic fractionation (αGH-IW) and δ18O anomalies of the interstitial waters: Leg 164, Blake Ridge. Proc. Ocean Drill. Program. Sci. Result. 2000, 164, pp. 59–66. Available online: http://www-odp.tamu.edu/publications/164_SR/VOLUME/CHAPTERS/SR164_06.PDF (accessed on 10 May 2013).
- Kastner, M.; Kvenvolden, K.A.; Whiticar, M.J.; Camerlenghi, A.; Lorenson, T.D. Relation between pore fluids chemistry and gas hydrates associated with bottom simulating reflectors at the Cascadia margin, site 889 and 892. Proc. Ocean Drill. Program. Sci. Result. 1995, 146, pp. 175–187. Available online: http://www-odp.tamu.edu/publications/146_1_SR/VOLUME/CHAPTERS/sr146pt1_10.pdf (accessed on 10 May 2013).
- Froelich, P.N.; Klinkhammer, G.P.; Bender, M.L.; Luedtke, N.A.; Heath, G.R.; Cullen, D.; Dauphin, P. Early oxidation of organic matter in pelagic sediments of the eastern equatorial Atlantic: Suboxic diagenesis. Geochim. Cosmochim. Acta 1979, 43, 1075–1090. [Google Scholar]
- Berner, R.A. Early Diagenesis: A Theoretical Approach; Princeton University Press: Princeton, NJ, USA, 1980. [Google Scholar]
- Boetius, A.; Ravenschlag, K.; Schubert, C.J.; Rickert, D.; Widdel, F.; Gieseke, A.; Amann, R.; Jørgensen, B.B.; Witte, U.; Pfannkuche, O. A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 2000, 407, 623–626. [Google Scholar]
- Hoehler, T.M.; Alperin, M.J.; Albert, D.B.; Martens, C.S. Field and laboratory studies of methane oxidation in an anoxic marine sediment: Evidence for a methanogen-sulfate reducer consortium. Glob. Biogeochem. Cycles 1994, 8, 451–463. [Google Scholar] [CrossRef]
- Canfield, D. Biogeochemistry of sulfur isotopes. Rev. Mineral. Geochem. 2001, 43, 607–636. [Google Scholar] [CrossRef]
- Harrison, A.; Thode, H. Mechanism of the bacterial reduction of sulphate from isotope fractionation studies. Trans. Faraday Soc. 1958, 54, 84–92. [Google Scholar] [CrossRef]
- Luo, M.; Wang, H.; Yang, S.; Chen, D. Research advancement of natural gas hydrate in South China Sea. Bull. Mineral. Petrol. Geochem. 2013, 32, 56–69, (in Chinese with English abstract). [Google Scholar]
- Wu, N.; Zhang, G.; Liang, J.; Su, Z.; Wu, D.; Lu, H.; Lu, J.; Sha, Z.; Fu, S.; Gong, Y.; et al. Progress of gas hydrate research in Northern South China Sea. Adv. New Renew. Energy 2013, 1, 80–94, (in Chinese with English abstract). [Google Scholar]
- Xu, H.; Li, L.; Shu, H.; Wen, P.; Zhang, B. The seismic reflecting characteristics of gas hydrate bearing strata and its possible distribution in the South China Sea. Appl. Geophys. 2006, 3, 42–47. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, G.; Huang, Y.; Liang, J.; Wong, H.K. Gas hydrate occurrence on the continental slope of the northern South China Sea. Mar. Pet. Geol. 2005, 22, 403–412. [Google Scholar] [CrossRef]
- Li, L.; Lei, X.; Zhang, X.; Zhang, G. Heat flow derived from BSR and its implications for gas hydrate stability zone in Shenhu Area of northern South China Sea. Mar. Geophys. Res. 2012, 33, 77–87. [Google Scholar] [CrossRef]
- Sun, C.; Wang, H.; Niu, B.; Huang, X. Geochemical prospecting of gas hydrate at Xisha Ocean Trough. Earth Sci.-J. China Univ. Geosci. 2004, 29, 135–140, (in Chinese with English abstract). [Google Scholar]
- Fu, S. Gas origin constraint on the formation of gas hydrate. Earth Sci. Front. 2005, 12, 263–267, (in Chinese with English abstract). [Google Scholar]
- Wu, N.; Zhang, H.; Yang, S.; Zhang, G.; Liang, J. Gas hydrate system of shenhu area, Northern South China Sea: Geochemical results. J. Geophys. Res. 2011, 1, 1–8. [Google Scholar]
- Wu, L.; Yang, S.; Liang, J.; Su, X.; Fu, S.; Sha, Z.; Yang, T. Variations of pore water sulfate gradients in sediments as indicator for underlying gas hydrate in Shenhu Area, the South China Sea. Sci. China 2013, 56, 530–540. [Google Scholar] [CrossRef]
- Yang, T.; Jiang, S.; Ge, L.; Yang, J.; Ling, H.; Wu, N.; Zhang, G.; Liu, J.; Chen, D. Geochemical characteristics of sediment pore water from Site XS-01 in the Xisha Trough of South China Sea and the significance for gas hydrate occurrence. Quat. Sci. 2006, 26, 442–448, (in Chinese with English abstract). [Google Scholar]
- Yang, T.; Jiang, S.; Ge, L.; Yang, J.; Wu, N.; Zhang, G.; Liu, J. Pore water geochemistry in the shallow sediments of Shenhu Area, northern South China Sea and its significance for the occurrence of gas Hydrates. Chin. Sci. Bull. 2009, 54, 3231–3240, (in Chinese with English abstract). [Google Scholar] [CrossRef]
- Han, X.; Suess, E.; Huang, Y.; Wu, N.; Bohrmann, G.; Su, X.; Eisenhauer, A.; Rehder, G.; Fang, Y. Jiulong methane reef: Microbial mediation of seep carbonates in the South China Sea. Mar. Geol. 2008, 249, 243–256. [Google Scholar] [CrossRef]
- Tong, H.; Feng, D.; Cheng, H.; Yang, S.; Wang, H.; Min, A.G.; Lawrence Edwards, R.; Chen, Z.; Chen, D. Authigenic carbonates from seeps on the northern continental slope of the South China Sea: New insights into fluid sources and geochronology. Mar. Pet. Geol. 2013, 43, 260–271. [Google Scholar]
- Sun, Q.L.; Wu, S.G.; Hovland, M.; Luo, P.; Lu, Y.T.; Qu, T.L. The morphologies and genesis of mega-pockmarks near the Xisha Uplift, South China Sea. Mar. Pet. Geol. 2011, 28, 1146–1156. [Google Scholar] [CrossRef]
- Zhu, W.; Huang, B.; Mi, L.; Wilkins, R.W.T.; Fu, N.; Xiao, X. Geochemistry, origin, and deep-water exploration potential of natural gases in the Pearl River Mouth and Qiongdongnan basins, South China Sea. AAPG Bull. 2009, 93, 741–761. [Google Scholar] [CrossRef]
- Huang, B.J.; Xiao, X.M.; Li, X.X. Geochemistry and origins of natural gases in the Yinggehai and Qiongdongnan basins, offshore South China Sea. Org. Geochem. 2003, 34, 1009–1025. [Google Scholar] [CrossRef]
- Luo, M.; Chen, L.; Wang, S.; Yan, W.; Wang, H.; Chen, D. Pockmark activity inferred from pore water geochemistry in shallow sediments of the pockmark field in southwestern Xisha Uplift, northwestern South China Sea. Mar. Pet. Geol. 2013, 48, 247–259. [Google Scholar] [CrossRef]
- Kastner, M.; Elderfield, H.; Martin, J.B.; Suess, E.; Kvenvolden, K.A.; Garrison, R.E. Diagenesis and interstitial-water chemistry at the Peruvian continental margin—major constituents and strontium isotopes. Proc. Ocean Drill. Program. Sci. Result. 1990, 112, pp. 413–440. Available online: http://www-odp.tamu.edu/publications/112_SR/VOLUME/CHAPTERS/sr112_25.pdf (accessed on 17 March 2013).
- Tomaru, H.; Matsumoto, R.; Lu, H.; Uchida, T. Geochemical process of gas hydrate formation in the Nankai Trough based on chloride and isotopic anomalies in interstitial water. Resour. Geol. 2004, 54, 45–51. [Google Scholar] [CrossRef]
- Tomaru, A.; Matsumoto, R.; Torres, M.E.; Borowski, W.S. Geological and geochemical contraints on the isotopic composition of interstitial waters from the Hydrate Ridge region, Cascadia Continental margin. Proc. Ocean Drill. Program. Sci. Result. 2006, 204, pp. 1–20. Available online: http://www-odp.tamu.edu/publications/204_SR/VOLUME/CHAPTERS/109.PDF (accessed on 10 May 2013).
- Chen, Y.; Ussler, W., III; Haflidason, H.; Lepland, A.; Rise, L.; Hovland, M.; Hjelstuen, B.O. Sources of methane inferred from pore-water delta C-13 of dissolved inorganic carbon in Pockmark G11, offshore Mid-Norway. Chem. Geol. 2010, 275, 127–138. [Google Scholar] [CrossRef]
- Sheppard, S.; Gilg, H. Stable isotope geochemistry of clay minerals. Clay Miner. 1996, 31, 1–24. [Google Scholar]
- Dählmann, A.; de Lange, G. Fluid–sediment interactions at Eastern Mediterranean mud volcanoes: A stable isotope study from ODP Leg 160. Earth Planet. Sci. Lett. 2003, 212, 377–391. [Google Scholar] [CrossRef]
- Aloisi, G.; Drews, M.; Wallmann, K.; Bohrmann, G. Fluid expulsion from the Dvurechenskii mud volcano (Black Sea): Part I. Fluid sources and relevance to Li, B, Sr, I and dissolved inorganic nitrogen cycles. Earth Planet. Sci. Lett. 2004, 225, 347–363. [Google Scholar]
- Godon, A.; Jendrzejewski, N.; Castrec-Rouelle, M.; Dia, A.; Pineau, F.; Boulègue, J.; Javoy, M. Origin and evolution of fluids from mud volcanoes in the Barbados accretionary complex. Geochim. Cosmochim. Acta 2004, 68, 2153–2165. [Google Scholar] [CrossRef]
- Martin, J.B.; Kastner, M.; Henry, P.; Le Pichon, X.; Lallement, S. Chemical and isotopic evidence for sources of fluids in a mud volcano field seaward of the Barbados accretionary wedge. J. Geophys. Res. 1996, 101, 20325–20345. [Google Scholar] [CrossRef]
- Hensen, C.; Nuzzo, M.; Hornibrook, E.; Pinheiro, L.M.; Bock, B.; Magalhães, V.H.; Brückmann, W. Sources of mud volcano fluids in the Gulf of Cadiz—indications for hydrothermal imprint. Geochim. Cosmochim. Acta 2007, 71, 1232–1248. [Google Scholar] [CrossRef]
- Wang, X.; Wu, S.; Wang, D.; Ma, Y.; Yao, G.; Gong, Y. The role of polygonal faults in fluid migration and gas hydrate reservoir forming in Southeast Hainan Basin. Oil Geophys. Prospect. 2010, 45, 122–128, (in Chinese with English abstract). [Google Scholar]
- Wang, X.; Wu, S.; Dong, D.; Gong, Y.; Chai, C. Characteristics of gas chimney and its relationship to gas hydrate in Qiongdongnan Basin. Mar. Geol. Quat. Geol. 2008, 28, 103–108, (in Chinese with English abstract). [Google Scholar]
- Wang, X.; Wu, S.; Yuan, S.; Wang, D.; Ma, Y.; Yao, G.; Gong, Y.; Zhang, G. Geophysical signatures associated with fluid flow and gas hydrate occurrence in a tectonically quiescent sequence, Qiongdongnan Basin, South China Sea. Geofluids 2010, 10, 351–368. [Google Scholar] [CrossRef]
- Aharon, P.; Fu, B. Microbial sulfate reduction rates and sulfur and oxygen isotope fractionations at oil and gas seeps in deepwater Gulf of Mexico. Geochim. Cosmochim. Acta 2000, 64, 233–246. [Google Scholar] [CrossRef]
- Fritz, P.; Basharmal, G.; Drimmie, R.; Ibsen, J.; Qureshi, R. Oxygen isotope exchange between sulphate and water during bacterial reduction of sulphate. Chem. Geol. 1989, 79, 99–105. [Google Scholar]
- Brunner, B.; Bernasconi, S.M.; Kleikemper, J.; Schroth, M.H. A model for oxygen and sulfur isotope fractionation in sulfate during bacterial sulfate reduction processes. Geochim. Cosmochim. Acta 2005, 69, 4773–4785. [Google Scholar] [CrossRef]
- Mangalo, M.; Meckenstock, R.U.; Stichler, W.; Einsiedl, F. Stable isotope fractionation during bacterial sulfate reduction is controlled by reoxidation of intermediates. Geochim. Cosmochim. Acta 2007, 71, 4161–4171. [Google Scholar] [CrossRef]
- Antler, G.; Turchyn, A.V.; Rennie, V.; Herut, B.; Sivan, O. Coupled sulfur and oxygen isotope insight into bacterial sulfate reduction in the natural environment. Geochim. Cosmochim. Acta 2013, 118, 98–117. [Google Scholar] [CrossRef]
- Feng, D.; Roberts, H.H. Geochemical characteristics of the barite deposits at cold seeps from the northern Gulf of Mexico continental slope. Earth Planet. Sci. Lett. 2011, 309, 89–99. [Google Scholar]
- Mangalo, M.; Einsiedl, F.; Meckenstock, R.U.; Stichler, W. Influence of the enzyme dissimilatory sulfite reductase on stable isotope fractionation during sulfate reduction. Geochim. Cosmochim. Acta 2008, 72, 1513–1520. [Google Scholar] [CrossRef]
- Wortmann, U.G.; Chernyavsky, B.; Bernasconi, S.M.; Brunner, B.; Böttcher, M.E.; Swart, P.K. Oxygen isotope biogeochemistry of pore water sulfate in the deep biosphere: Dominance of isotope exchange reactions with ambient water during microbial sulfate reduction (ODP Site 1130). Geochim. Cosmochim. Acta 2007, 71, 4221–4232. [Google Scholar] [CrossRef]
- Chiba, H.; Sakai, H. Oxygen isotope exchange rate between dissolved sulfate and water at hydrothermal temperatures. Geochim. Cosmochim. Acta 1985, 49, 993–1000. [Google Scholar] [CrossRef]
- Gamsjager, H.; Murmann, R.K. Advances in Inorganic and Bioinorganic Mechanisms; Sykes, A.G., Ed.; Academic London: London, UK, 1983; pp. 317–381. [Google Scholar]
- Hesse, R.; Frape, S.K.; Egeberg, P.K.; Matsumoto, R. Stable isotopic studies (Cl, O, and H) of interstitial waters from site 997, Blake Ridge gas hydrate field, west Atlantic. Proc. Ocean Drill. Program. Sci. Result. 2000, 164, pp. 129–137. Available online: http://www-odp.tamu.edu/publications/164_SR/VOLUME/CHAPTERS/SR164_12.PDF (accessed on 13 May 2013).
- MacDonald, G.J. The future of methane as an energy resource. Annu. Rev. Energy 1990, 15, 53–83. [Google Scholar]
- Davidson, D.; Leaist, D.; Hesse, R. Oxygen-18 enrichment in the water of a clathrate hydrate. Geochim. Cosmochim. Acta 1983, 47, 2293–2295. [Google Scholar] [CrossRef]
- Pavlova, G.; Pashkina, V. Distribution of halogens in interstitial waters of the Sea of Okhotsk as related to hydrate generation. Oceanology 1989, 29, 329–333. [Google Scholar]
- Hesse, R. Pore-water anomalies in gas hydrate-bearing sediments of the deeper continental margins: Facts and problems. J. Incl. Phenom. Macrocycl. Chem. 1990, 8, 117–138. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Luo, M.; Chen, L.; Tong, H.; Yan, W.; Chen, D. Gas Hydrate Occurrence Inferred from Dissolved Cl− Concentrations and δ18O Values of Pore Water and Dissolved Sulfate in the Shallow Sediments of the Pockmark Field in Southwestern Xisha Uplift, Northern South China Sea. Energies 2014, 7, 3886-3899. https://doi.org/10.3390/en7063886
Luo M, Chen L, Tong H, Yan W, Chen D. Gas Hydrate Occurrence Inferred from Dissolved Cl− Concentrations and δ18O Values of Pore Water and Dissolved Sulfate in the Shallow Sediments of the Pockmark Field in Southwestern Xisha Uplift, Northern South China Sea. Energies. 2014; 7(6):3886-3899. https://doi.org/10.3390/en7063886
Chicago/Turabian StyleLuo, Min, Linying Chen, Hongpeng Tong, Wen Yan, and Duofu Chen. 2014. "Gas Hydrate Occurrence Inferred from Dissolved Cl− Concentrations and δ18O Values of Pore Water and Dissolved Sulfate in the Shallow Sediments of the Pockmark Field in Southwestern Xisha Uplift, Northern South China Sea" Energies 7, no. 6: 3886-3899. https://doi.org/10.3390/en7063886
APA StyleLuo, M., Chen, L., Tong, H., Yan, W., & Chen, D. (2014). Gas Hydrate Occurrence Inferred from Dissolved Cl− Concentrations and δ18O Values of Pore Water and Dissolved Sulfate in the Shallow Sediments of the Pockmark Field in Southwestern Xisha Uplift, Northern South China Sea. Energies, 7(6), 3886-3899. https://doi.org/10.3390/en7063886