1. Introduction
Development of advanced, reliable, and clean electrical storage devices would help to mitigate the diminishing fossil fuel supplies and environmental concerns regarding vehicles using conventional fuel. To address issues with global climate change, energy security, and energy sustainability, the electrification of transportation and large-scale deployment of renewable energy have been considered an indispensable strategy [
1]. However, such a transformation is severely limited by the unsatisfactory performance of current electrical energy storage systems. Although rechargeable lithium-ion batteries, one of the most promising electrical energy storage technologies available so far, are attractive owing to their high energy density and efficiency, they fall far below the requirement for electric vehicle (EV) and grid energy storage applications. For instance, the low energy storage capacity of state-of-the-art Li-ion batteries in EVs limits their driving distance to less than 100 miles per charge.
To make the transition from current hybrid electric vehicles (HEVs) to plug-in hybrid electric vehicles (PHEVs) or EVs, it is critical to develop a battery technology that enables acceptable driving ranges (300 miles per charge). This range requires much higher specific energy and energy density than those available from conventional Li-ion batteries. Therefore, researchers have been devoting an increasing amount of research on electrical energy storage systems that can go beyond the Li-ion battery limits [
2,
3,
4,
5,
6,
7,
8].
Lithium-air cells can be considered the “holy grail” of lithium batteries because they offer, in principle, a significantly superior theoretical energy density compared to conventional lithium-ion systems, as shown in
Table 1. For example, after electrochemical activation, layered lithium-manganese-nickel-cobalt-oxide cathodes, such as Li
1.200Mn
0.525Ni
0.175Co
0.100O
2, when coupled to a lithiated-graphite anode (LiC
6), can provide high capacity (~250 mA h/g) at an average open-circuit voltage of 3.6 V. This lithium-ion cell chemistry, the best to date, has a theoretical specific energy of ~900 W h/kg based on a calculation that uses the masses of the anode and cathode materials alone; in practice, 150–200 W h/kg has been accomplished at the cell level. In contrast, a Li-air cell, when discharged to the peroxide composition lithium peroxide (Li
2O
2) at an average voltage of ~3.0 V, would provide a theoretical specific energy of 3623 W h/kg, or when discharged to lithium oxide (Li
2O) at the same voltage, 5204 W h/kg. Note that gasoline (octane) offers a theoretical energy of ~13,000 W h/kg if the mass of the injected oxygen is not considered in the calculation because gasoline is supplied externally and combusted within, and exhausted from, the engine. By the same token, a Li-air cell would offer a specific energy of ~11,000 W h/kg if the “free” oxygen supplied during discharge and released during charge is ignored in the calculation.
Table 1.
Relative specific energies of Li/O2, lithium-ion and gasoline systems.
Table 1.
Relative specific energies of Li/O2, lithium-ion and gasoline systems.
| System | Reaction | Open circuit voltage (V) | Theoretical specific energy (W h/kg) |
|---|
| Li/O2 [8] | 2Li + O2 → 2Li2O2 | 2.96 | 3,623 (incl. O) |
| 4Li + O2 → 2Li2O | 2.9 | 5,204 (incl. O) |
| 4Li + O2 → 2Li2O | 2.9 | 11,202 (excl. O) |
| Lithium-ion, e.g., C6/LiMO2 (M = Mn, Ni, Co) | Li
xC6 + Li1-xMO2 → C6 + LiMO2 | 3.6 | ~900 |
| Gasoline (octane) | C8H18 + 12.5O2 → 8CO2 + 9H2O | - | ~13,000 (excl. O) |
While the inherent energy potential of lithium metal approaches that of gasoline, today’s battery manufacturers have not yet been able to unlock this potential. While current lithium-ion batteries may provide acceptable power for HEVs and EVs, they do not provide sufficient energy for an acceptable driving distance. This range limitation and the absence of a battery charging infrastructure have limited public interest in electric vehicles, particularly for long distance travel. A breakthrough in Li-air battery technology would significantly increase the possibility of extending the electric range of these vehicles, with the added advantages of reducing battery cost and weight. Recently, the enormous potential that the Li-air battery possesses triggered sharply increased research activity, as evidenced by over 300 research articles, including about 20 review articles, having been published on this technology in the last four years alone. In 2009, Argonne National Laboratory launched a project with the main goal of developing innovative and radically new concepts that will dramatically advance Li-air batteries with an energy density far surpassing that of conventional lithium-ion batteries, achieving the requirement for electrically powered vehicles. Under this project, a multidisciplinary team was established from a broad spectrum of basic and applied scientific disciplines with additional advantages of using Argonne’s unique research facilities. The team worked closely together to develop a Li-air battery system with exceptionally high energy density and charge-discharge reversibility by pursuing new approaches that combine innovation in advanced electrode-, electrolyte-, and catalytic materials research, assisted by extensive computer modeling and advanced characterization techniques.
In this short review article, except for a brief overview of Li-air batteries (
Section 2), we will mainly focus on the research activities with regard to non-aqueous Li-air batteries from Argonne under the above-mentioned project [
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26,
27,
28,
29,
30,
31,
32,
33,
34]. The following sections cover the stability of the electrolyte and its influence on the electrochemical performance of Li-air cells, the structure and magnetic properties of Li
2O
2 and its relevance to the Li-air battery design, the design and optimization of the air cathode structure with different types of catalyst, the oxygen cross-over effect on the Li anode based on both experimental and theoretical modeling results, and the development of a single-crystal silicon membrane with high lithium conductivity. The last section of the article will discuss future challenges with the aim to gain a better understanding of this promising battery technology. Covering the immense body of work published in this field is beyond the scope of the article. For those who are interested, more detail of recent research progress and fundamental understanding from other research groups can be found in many review articles [
3,
5,
8,
29,
35,
36,
37,
38,
39,
40,
41,
42,
43,
44,
45,
46,
47,
48,
49,
50,
51] (and the references cited thereby) published in the last few years.
2. Concept and Challenges for Li-Air Batteries
The Li-air battery chemistry uses the oxidation of lithium at the anode and reduction of oxygen at the cathode to induce current flow. Originally proposed in the 1970s as a possible power source for electric vehicles, Li-air batteries recaptured scientific interest in the late 2000s due to advances in materials technology and an increasing demand for environmentally safe and oil-independent energy sources [
2,
7,
52,
53,
54,
55,
56,
57,
58,
59,
60,
61,
62]. Because oxygen is supplied as a gaseous reactant to the cell during discharge, Li-air cells differ from conventional battery systems such as lead-acid, nickel-metal hydride, and lithium-ion; they can be constructed as part of hybrid battery-fuel cell systems. Compared to other metal-air batteries such as Zn-air and Al-air cells, which have aqueous electrolytes and operate at a relatively low voltage of ~1.4 V and ~1.2 V, respectively, non-aqueous Li-air cells provide ~3 V and, therefore, a significantly higher specific energy [
63]. During electrochemical discharge, the lithium anode is oxidized by releasing electrons to the external circuit to produce lithium ions in the electrolyte, whereas the oxygen is reduced at the catalytic cathode surface to form, in the case of non-aqueous electrolytes, Li
2O
2 or Li
2O [
51]. In addition, because the Zn-air and Al-air electrochemical reactions are not readily reversible, these metal-air cells have to be mechanically recharged with fresh anodes and electrolytes. In contrast, non-aqueous Li-air cells can be recharged internally, particularly when the discharge is restricted to the formation of Li
2O
2.
There are two principal electrochemical reactions of interest in non-aqueous Li-air batteries:
Theoretically, the full reduction of O
2 to Li
2O [Equation (2)] is desired because of its higher specific energy and energy density. However, this reaction involves a four-electron transfer, which is not favored for substrates with weak binding for oxygen, such as carbon, the most widely used cathode material in the Li-air battery. If the cut-off voltage is lowered to 2.0 V or below (
vs. Li/Li
+), Li
2O does appear to be the main discharge product on the cathode, but the subsequent charge of Li
2O, the reverse reaction in Equation (2), cannot proceed under the test condition due to the thermal stability of Li
2O [
64]. Therefore, Li
2O
2 is the reaction product observed in most recent cell tests (cut-off voltage >2.0 V). From a kinetic point of view, the formation of Li
2O
2 during discharge may be beneficial to the rate performance of the cell, since full cleavage of the O–O bond may not be necessary if a suitable catalyst can be identified. During charge, Li
2O
2 can decompose via either a two-electron process, Li
2O
2 → 2Li
+ + 2e
− + O
2, or a one-electron process that involves the formation of lithium superoxide (LiO
2), Li
2O
2 → Li
+ + e
− + LiO
2; these processes permit rechargeability of the non-aqueous Li-air cell. However, the reactivity of the other components of the Li-O
2 cells, including the electrolyte, electrocatalysts, carbon support, and lithium anode has a significant impact on the oxygen reduction reaction on discharge and oxygen evolution reaction on charge and thus on the overall cell performance [
12,
55,
56,
65], as will be detailed in the following sections.
The successful development of non-aqueous Li-air cells has been hampered because of severe materials and technological problems that have limited electrochemical performance: (1) the non-aqueous electrolytes are unstable during the discharge due to presence of reduced oxygen species; at high potential during charge, they are easily oxidized by the oxygen released; thereby seriously limiting cycle life; (2) during discharge, the solid and insoluble Li2O2 products are deposited on the surface or within the pores of the carbon cathode, thereby clogging the pores and restricting oxygen flow; (3) poisoning of the lithium electrode due to oxygen crossover occurs and destroys the integrity and functioning of the cell; and (4) commonly used cathode catalysts, such as MnO2 or Mn metal, do not access the full capacity of the oxygen electrode or enable sufficiently high rates.
3. Effect of Organic Electrolytes
Recently, it has been recognized that the choice of the electrolyte, in particular, the organic solvent, might be the most critical factor in the development of rechargeable non-aqueous Li-air cells [
36,
40]. A good electrolyte should be able to survive from the nucleophilic attack of the superoxide radical (O
2−), which is believed to be the intermediate phase formed from the oxygen reduction reaction upon discharge. Reaction with such highly reactive species (LiO
2, O
2−) to form decomposition products is especially problematic for carbonated-based electrolytes, as will be detailed below. In addition to its stability against superoxide radical, a good electrolyte for Li-air cells should also meet the following criteria: a wide potential window to withstand high oxidation potentials; stable toward reaction with the lithium anode; low viscosity and volatility; and high oxygen solubility and diffusivity [
66]. Ideally, the electrolyte should be able to dissolve the discharge products,
i.e., Li
2O
2, at least partially. Unfortunately, none of the electrolytes investigated so far for Li-air cells meets all of the above requirements, despite significant efforts to that end in the past few years. Understanding the reaction mechanisms between the electrolytes and discharge products will, no doubt, be the key to developing a stable electrolyte for Li-air cells.
A wealth of experimental and theoretical evidence indicates that the organic carbonates [e.g., propylene carbonate (PC), ethylene carbonate, and dimethyl carbonate] commonly used in Li-ion batteries are not stable toward the oxygen reduction products formed during battery discharge [
64]. During discharge, in addition to formation of the desired Li
2O
2, PC decomposes, resulting in the formation of lithium carbonate (Li
2CO
3) and other organic species [
67]. Another challenge besides the decomposition of the electrolyte is the large overpotential observed in the Li-air cell, which affects both power and cycle life [
5].
To understand the source of the large polarization observed in the Li-air cell, Zhang
et al. [
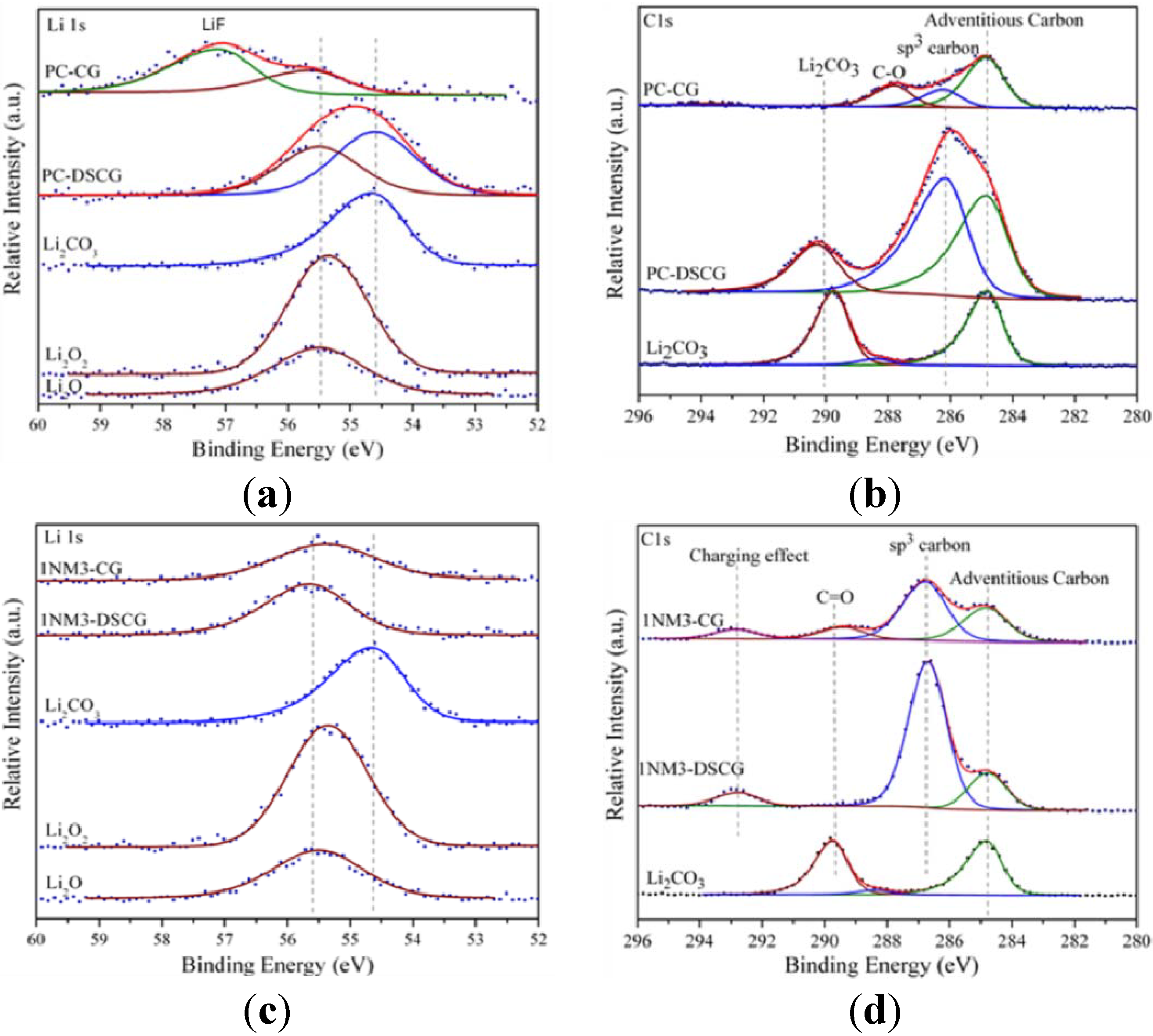
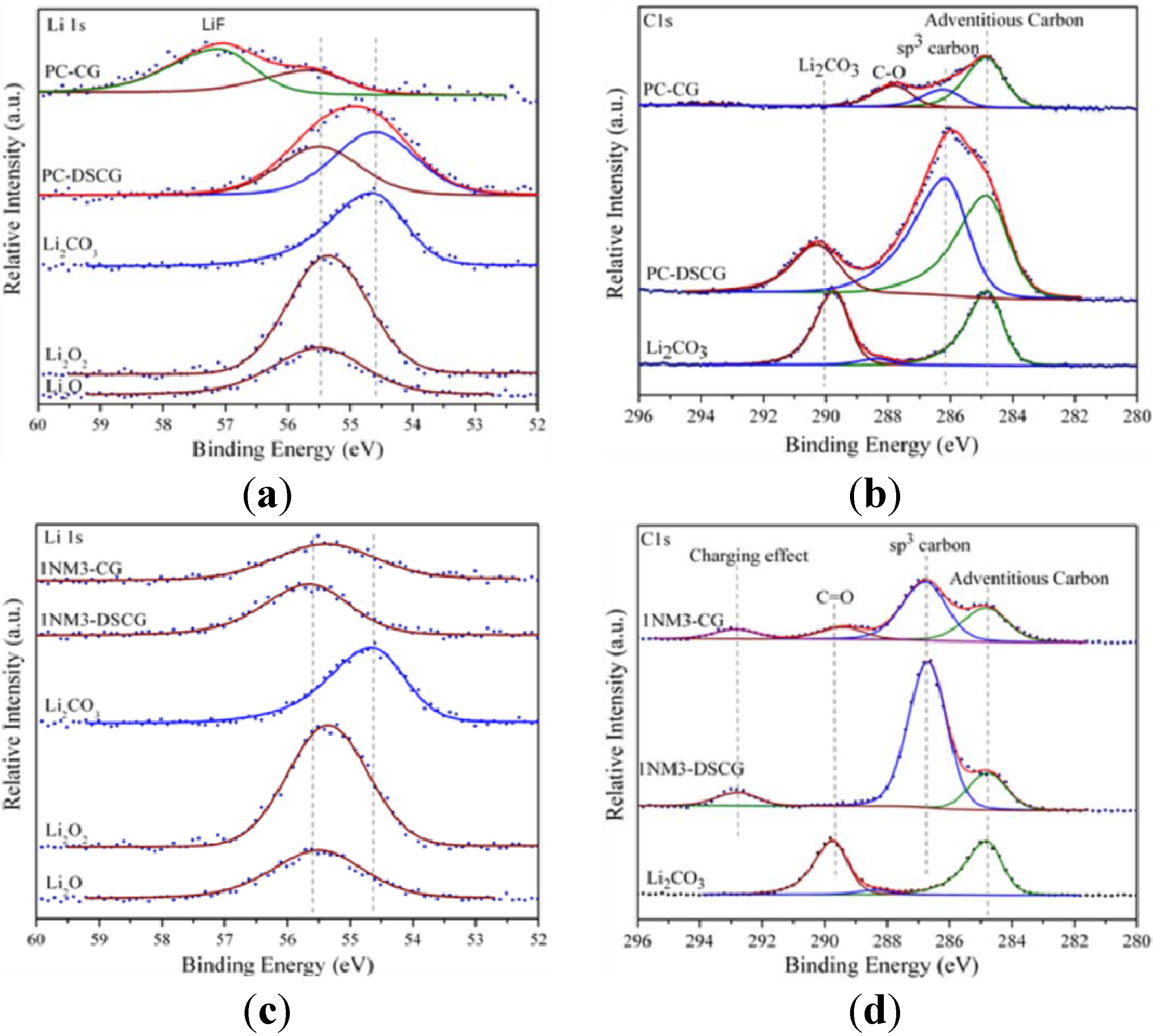
27] carried out a detailed investigation of the charge and discharge products formed in cell systems employing the PC-based electrolyte. They employed X-ray photoelectron spectroscopy (XPS) as a primary tool to characterize the charge and discharge products, as shown by the spectra in
Figure 1.
Figure 1.
X-ray photoelectron spectroscopy (XPS) spectra of Li 1s and C 1s core peaks of the cathode carbon electrodes after discharge and charge: (
a) Li 1s and (
b) C 1s in propylene carbonate (PC); and (
c) Li 1s and (
d) C 1s in tri(ethylene glycol)-substituted methyltrimethyl silane (1NM3). Standard compounds Li
2O
2, Li
2O and Li
2CO
3 are listed at the bottom of each spectrum. Reprinted with permission [
27]; Copyright 2011, American Chemical Society.
Figure 1.
X-ray photoelectron spectroscopy (XPS) spectra of Li 1s and C 1s core peaks of the cathode carbon electrodes after discharge and charge: (
a) Li 1s and (
b) C 1s in propylene carbonate (PC); and (
c) Li 1s and (
d) C 1s in tri(ethylene glycol)-substituted methyltrimethyl silane (1NM3). Standard compounds Li
2O
2, Li
2O and Li
2CO
3 are listed at the bottom of each spectrum. Reprinted with permission [
27]; Copyright 2011, American Chemical Society.
The results indicated that Li2CO3 was the major product formed during discharge, with only minor amounts of lithium oxides being formed. The XPS data also indicate that, during the first charge cycle the Li2CO3 was decomposed. This reaction is believed to be the primary cause of the large overpotential required for charging, owing to the need to oxidize the carbonate or other materials rather than the desired Li2O2.
Zhang
et al. [
27] also employed a density functional theory (DFT) and high level quantum chemistry calculation to address the decomposition of PC in the Li-air cell during discharge. They considered four possible intermediates from the oxygen reduction reaction of O
2,
i.e., O
2−, O
22− (Li
2O
2), LiO
2 and LiO
2−. The modeling results showed that “ring-opening”,
i.e., C–O bond breaking of PC, is the first step in the decomposition of PC to Li
2CO
3 or other lithium alkyl carbonates; this observation is consistent with the findings of Bryantsev
et al. [
68] The calculated energy barriers for all four species are quite small, with LiO
2− being the most reactive, as shown in
Figure 2a. The energy barrier for the subsequent C–O bond breaking is relatively smaller than the first step; therefore, it is thermodynamically favorable. In other words, upon “ring-opening” of PC, the following reactions are thermodynamically downhill, which could lead to the formation of Li
2CO
3 and other products such as formaldehyde and acetaldehyde with the assumption that a second electron transfer can occur. The DFT calculations confirmed that PC is unstable to oxygen reduction species. At present, Li-air cells based upon PC cannot be considered viable as the overall reaction appears to be oxidation of PC to CO
2. The PC decomposition during the discharge has also been reported by other groups [
65,
67].
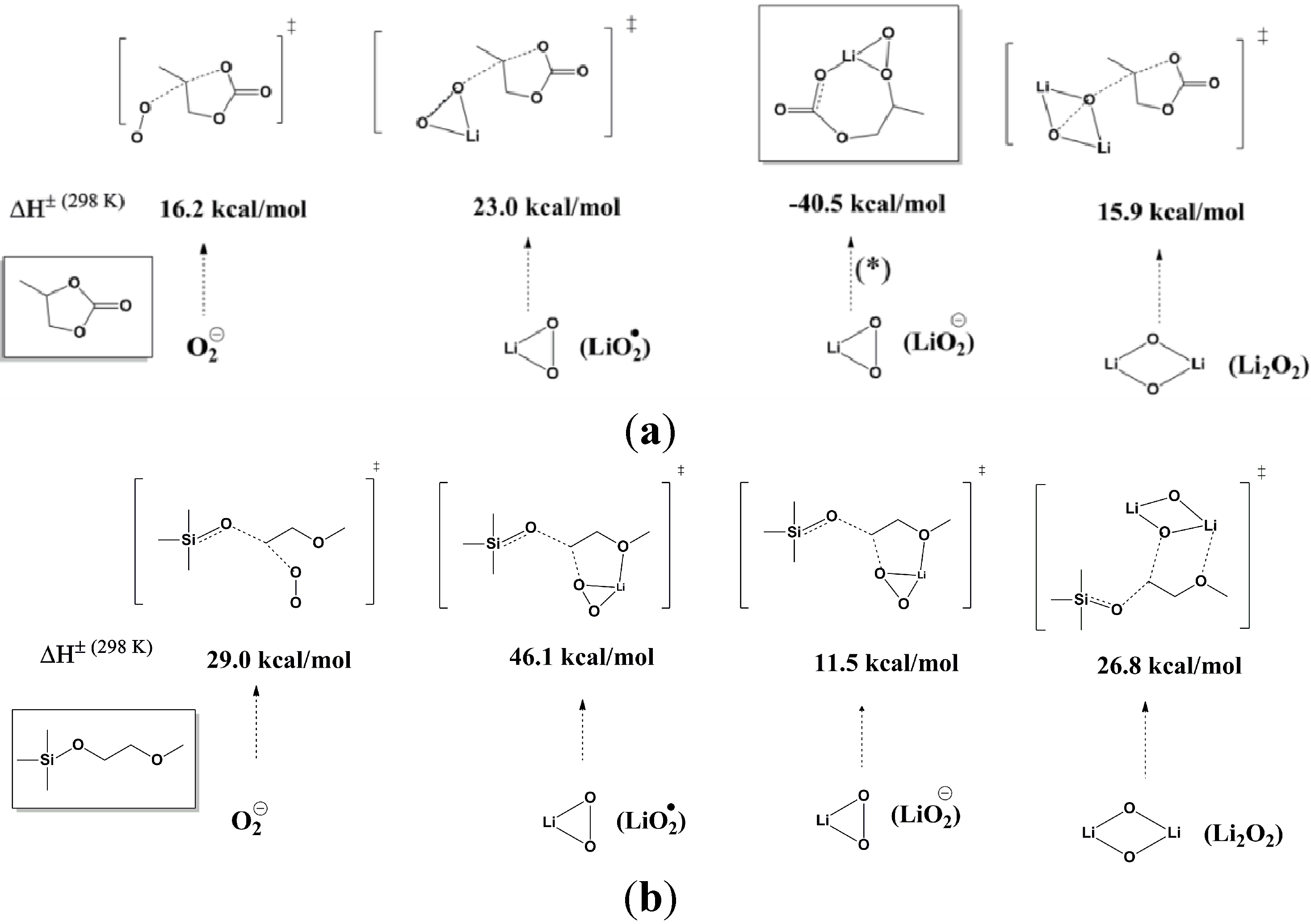
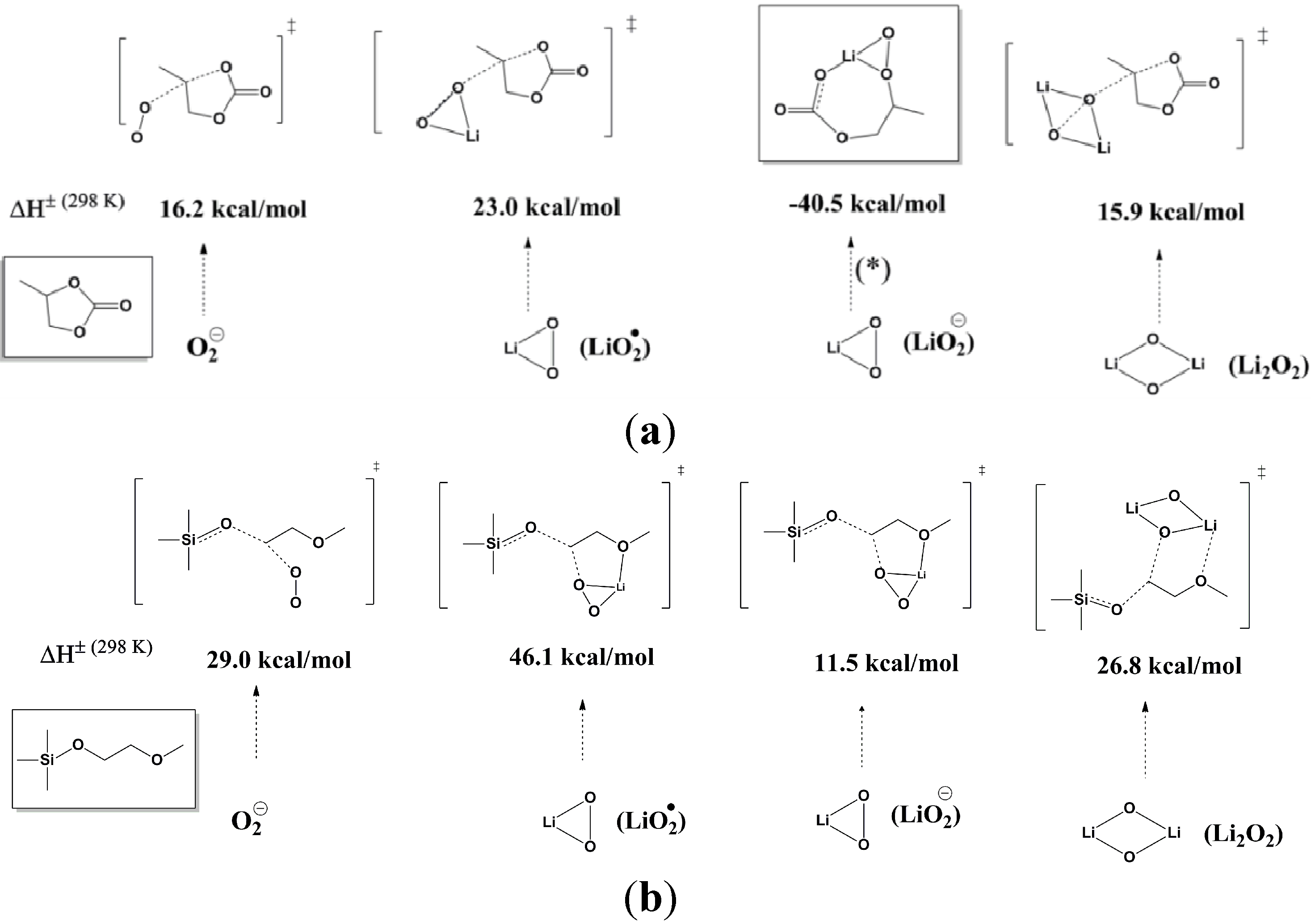
Figure 2.
Comparison of the computed barriers (enthalpies) for activation of (
a) PC decomposition by O
2 anion radical (O
2−), Li
2O radical, Li
2O anion (LiO
2−), and Li
2O
2; and (
b) (ethylene glycol)-substituted methyltrimethyl silane (1NM1) decomposition by O
2 anion radical, Li
2O radical, Li
2O anion, and Li
2O
2. The reaction of Li
2O anion with PC is exothermic by 40.5 kcal/mol and is a barrierless process. The O
2 anion radical barriers at G4MP2 are 12.1 kcal/mol (for PC) and 21.4 kcal/mol (for 1NM3). Reprinted with permission [
27]; Copyright 2011, American Chemical Society.
Figure 2.
Comparison of the computed barriers (enthalpies) for activation of (
a) PC decomposition by O
2 anion radical (O
2−), Li
2O radical, Li
2O anion (LiO
2−), and Li
2O
2; and (
b) (ethylene glycol)-substituted methyltrimethyl silane (1NM1) decomposition by O
2 anion radical, Li
2O radical, Li
2O anion, and Li
2O
2. The reaction of Li
2O anion with PC is exothermic by 40.5 kcal/mol and is a barrierless process. The O
2 anion radical barriers at G4MP2 are 12.1 kcal/mol (for PC) and 21.4 kcal/mol (for 1NM3). Reprinted with permission [
27]; Copyright 2011, American Chemical Society.
To identify more stable electrolytes that do not generate side reactions during the discharge, Zhang
et al. [
27] investigated the reactivity of a new silicon-containing oligo(ethylene oxide) solvent, tri(ethylene glycol)-substituted methyltrimethyl silane (1NM3). The DFT calculations indicated that this new solvent seems to be more stable to the highly active oxygen reduction species when compared to PC, as shown in
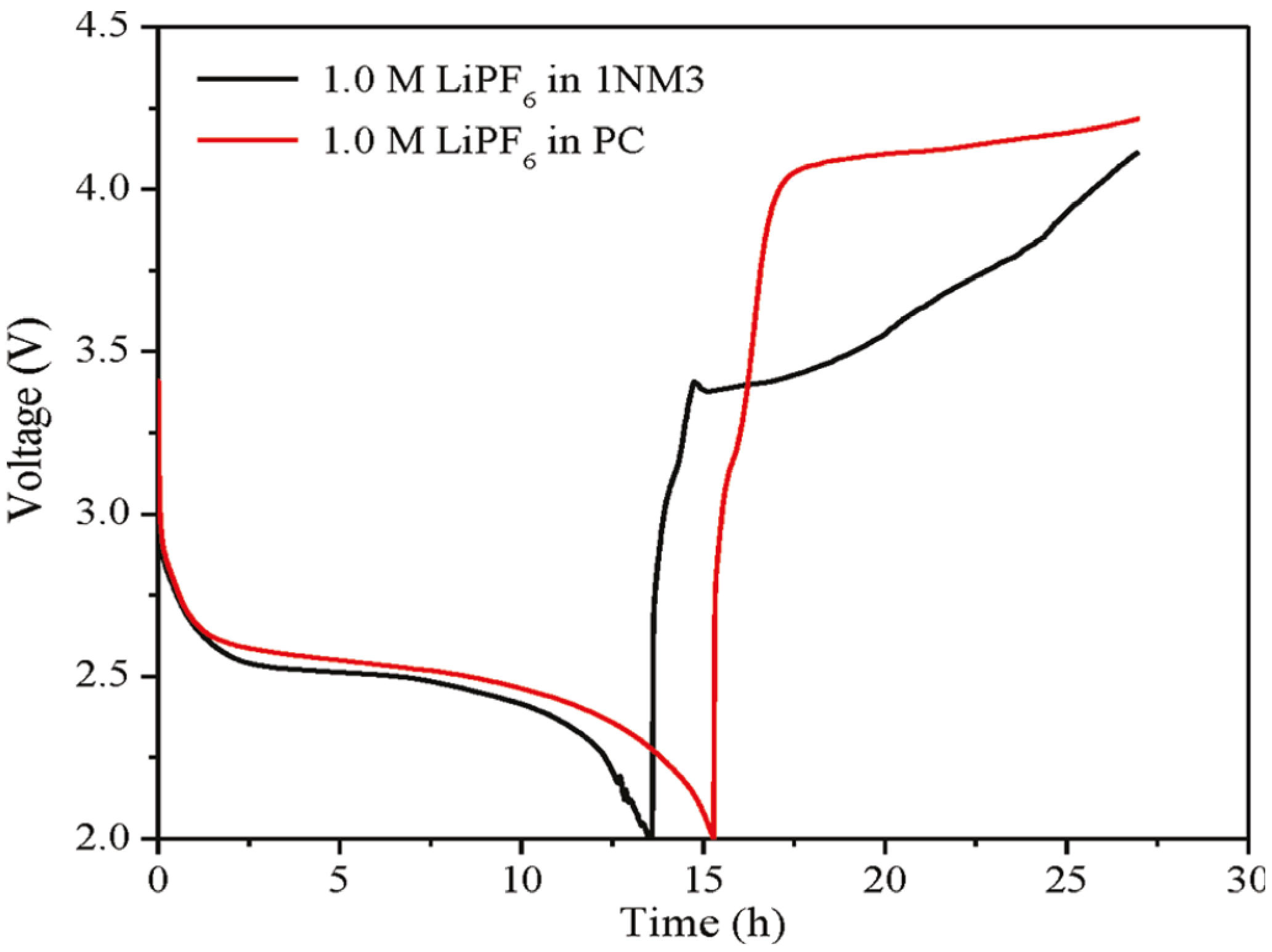
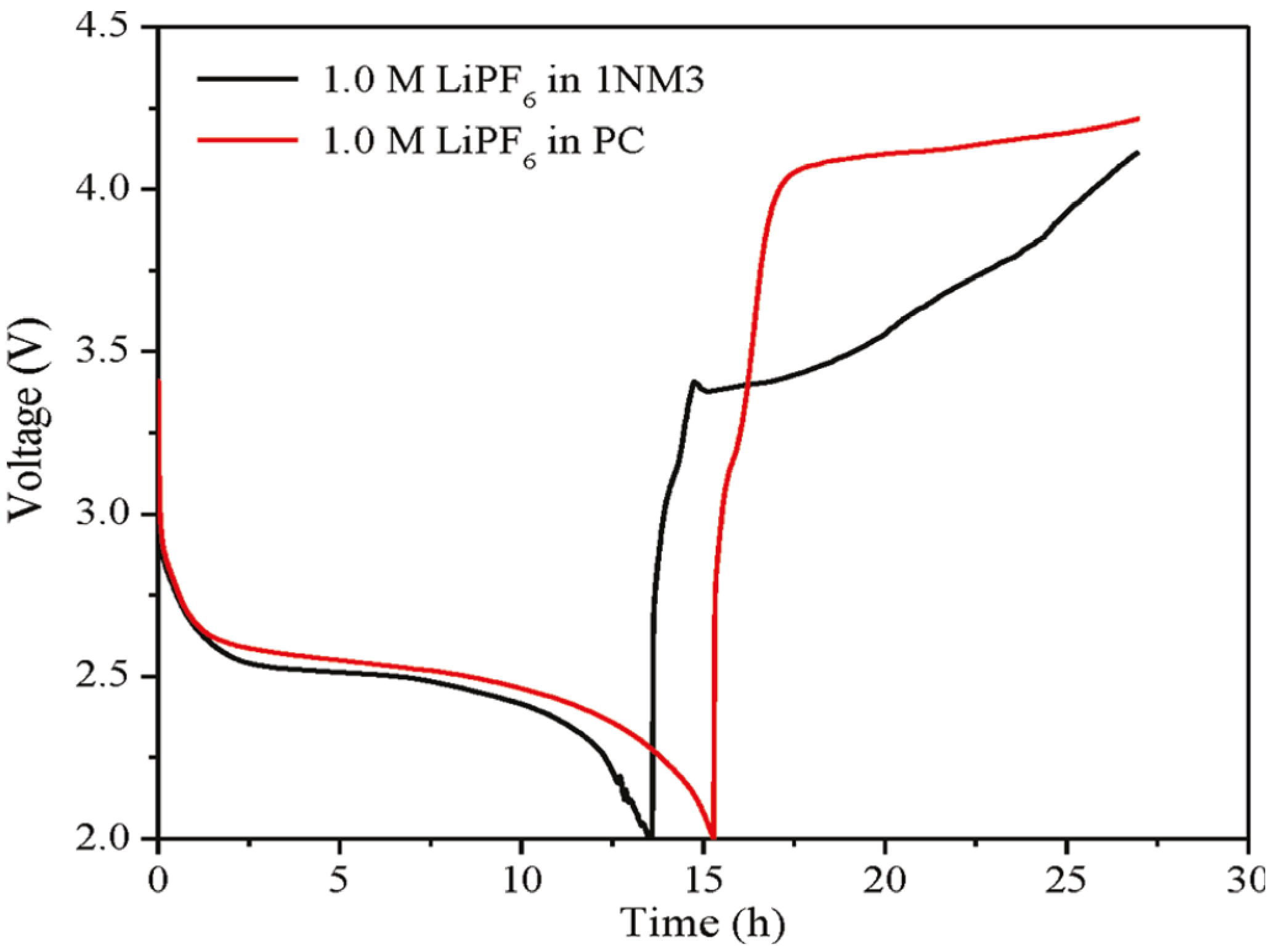
Figure 2b. When 1NM3 is used in place of PC in a Li-air cell, the XPS data collected after a single discharge show that only lithium oxides are formed; moreover, they are partially decomposed upon charging (
Figure 1c). The increased stability of the 1NM3 solvent results in a significantly lower overpotential for the Li-air cell, as shown in
Figure 3. The finding that the product from charging in the 1NM3 is oxygen needs confirmation, but the lower overpotential could be due the oxidation of lithium oxides being easier than the oxidation of the carbonate that is formed in the PC-electrolyte cells. Clearly, the electrolyte solvent stability plays a key role in the performance of Li-air batteries and will be a factor in improving their efficiencies. While the stability during discharge found for the 1NM3 electrolyte is an encouraging result for Li-air cell development, electrolytes that provide stability during charge are also needed to aid in reversibility of the reactions. Note that the reversibility and cycling stability of 1NM3-based Li-air cells were still not satisfactory, which might be attributed to the failure caused by decomposition of the lithium oxides on charge, since some of the lithium oxides might require a higher potential for decomposition.
Figure 3.
First charge and discharge cycles of a Li-air cell with PC and 1NM3. Reprinted with permission [
27]; Copyright 2011, American Chemical Society.
Figure 3.
First charge and discharge cycles of a Li-air cell with PC and 1NM3. Reprinted with permission [
27]; Copyright 2011, American Chemical Society.
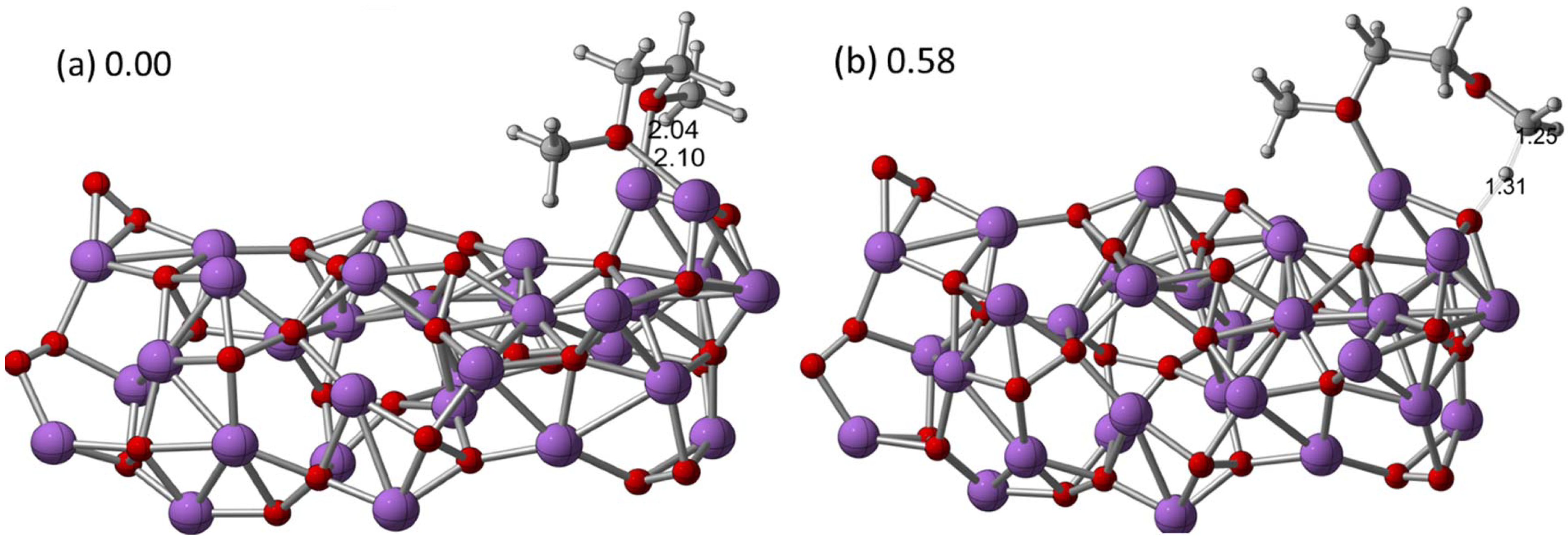
The reactions of the electrolyte at the lithium oxide/peroxide surfaces (the discharge products in a Li-air cell) could also be responsible for the degradation of the solvent, in addition to the O
2− that has been postulated to be present in the electrolyte. Recent DFT calculation results on the interactions of dimethoxyethane (DME) with Li
2O
2 clusters indeed demonstrated a high possibility of such reactions [
22]. In this work, small Li
2O
2 cluster models of up to 16 Li
2O
2 units were used to represent possible surface sites on the Li
2O
2 discharge products to investigate the reactions with ether, as shown in
Figure 4. The computations indicated that hydrogen abstraction by an unpaired spin on a Li–O–Li site (if present on the surface) may be favorable for decomposition of DME, which likely leads to the formation of reactive radicals and, subsequently, in the presence of oxygen, can lead to oxidized species such as aldehydes and carboxylates as well as LiOH on the surface of the Li
2O
2. In contrast, the decomposition initiated by the proton abstraction from the secondary position of DME by the singlet cluster (O–O site) results in an endothermic product, and subsequent reactions require the presence of oxygen or superoxide to be exothermic. Thus, pathways involving proton abstraction are less likely than those involving hydrogen abstraction. These results suggest that, if Li–O–Li sites with spin exist on a Li
2O
2 surface, they could be detrimental to Li-air cells by providing energetically favorable chemical mechanisms for DME decomposition at the Li
2O
2-electrolyte interface during discharge or charge. It is reasonable to expect that the hydrogen abstraction mechanism would also likely occur for other longer chain ether solvents as well, such as tetraethylene glycol dimethyl ether (TEGDME), one of the most widely used solvents in Li-air cells recently.
Figure 4.
Optimized structures for the (
a) (Li
2O
2)16-dimethoxyethane (DME) cluster (quintet) and (
b) transition state structure for the abstraction of a primary hydrogen from the DME at the B3LYP/6-31G (d) level of theory. Selected bond distances (Å) and relative energies (eV) were also shown. Reprinted with permission [
22]; Copyright 2013, American Chemical Society.
Figure 4.
Optimized structures for the (
a) (Li
2O
2)16-dimethoxyethane (DME) cluster (quintet) and (
b) transition state structure for the abstraction of a primary hydrogen from the DME at the B3LYP/6-31G (d) level of theory. Selected bond distances (Å) and relative energies (eV) were also shown. Reprinted with permission [
22]; Copyright 2013, American Chemical Society.
Ryan
et al. [
26] experimentally investigated the stability of ether-based electrolytes in a Li-air cell by a combined electrochemical and spectroscopic (FT-IR) measurement. In particular, they attempted to identify electrolyte decomposition species that may remain soluble in the electrolyte, if any. Through well-controlled experiments, they drew the conclusion that the electrochemical stability of the ether-based solvents may not be as great as previously considered. They found that ethers, for instance, 1NM3 or TEGDME, are consumed as part of the charging process in working Li-air cells, even under moderate voltages in the absence of O
2, which is consistent with the theoretical calculations discussed above. In another study on the stability of TEGDME electrolyte using
in-situ synchrotron X-ray diffraction (XRD), Ryan
et al. [
20] found the reversible formation of Li
2O
2 upon discharge and charge, as shown in
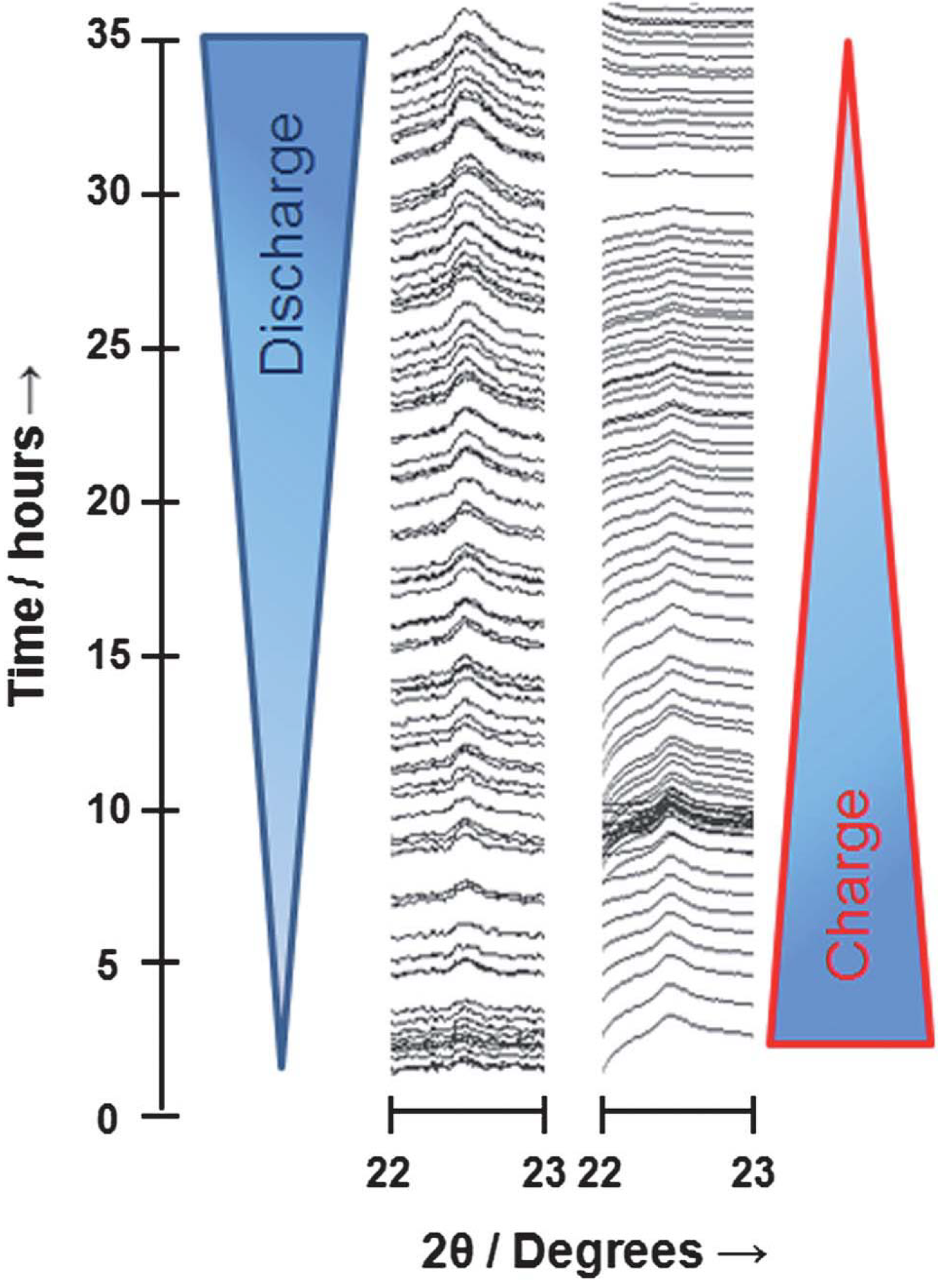
Figure 5. Through the Rietveld refinement of the discharge data on the Li
2O
2 (100), (101), and (110) reflections, they also reported a general trend of increasing grain size of Li
2O
2 from 10 nm to 120 nm as the discharge progresses. However, if an identical cell is allowed to charge first to force some of the electrolyte to decompose prior to the discharge, Ryan
et al. [
20] observed the reversible formation of LiOH rather than Li
2O
2 on the following cycle test. This surprising result may indicate that electrolyte decomposition can completely alter the nature of the stable discharge products in the Li-air cell.
Figure 5.
Three-dimensional line plot for the (110) reflection of Li
2O
2 which shows complete reversibility on discharge and subsequent charge. Reprinted with permission [
20]; Copyright 2013, Royal Society of Chemistry.
Figure 5.
Three-dimensional line plot for the (110) reflection of Li
2O
2 which shows complete reversibility on discharge and subsequent charge. Reprinted with permission [
20]; Copyright 2013, Royal Society of Chemistry.
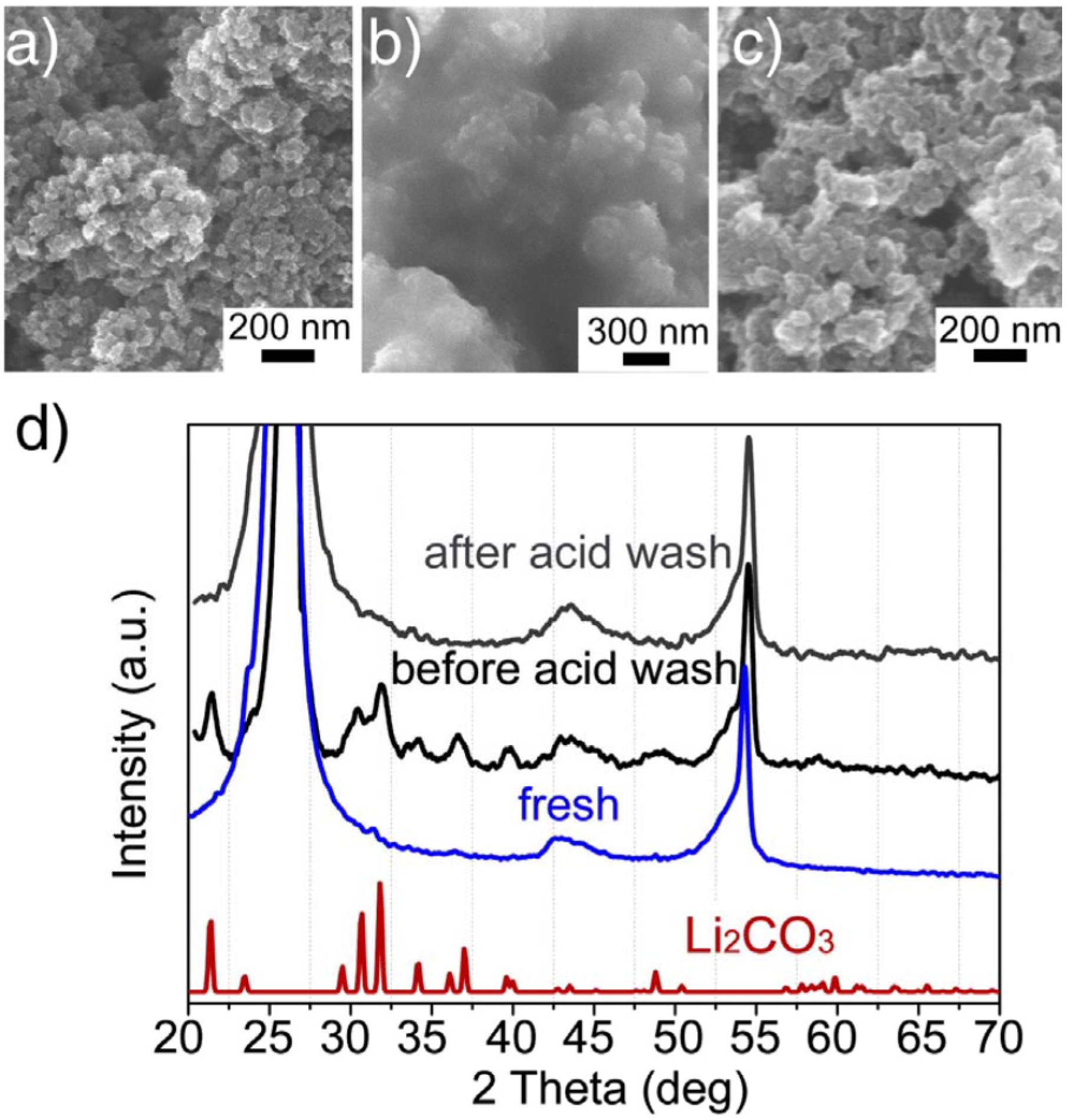
As a consequence of the electrolyte decomposition, the byproducts (insoluble lithium precipitates) from these side reactions will accumulate on the cathode, which not only blocks the oxygen diffusion, but also increases the cathode impedance. Whether the failed cathode was permanently damaged or recoverable, a repairable cathode suggests that the battery life could be dramatically extended if a more stable electrolyte were used. In an attempt to address this issue, Shui
et al. [
15] investigated the cathode’s failure and recoverability of the Li-air cell and inferred that passivation by insoluble lithium precipitates from the electrolyte decomposition was responsible for the cathode failure. The spent cathode could nearly return to its original performance upon removal of the precipitates (mainly lithium carbonate) by acidic treatment, which is attributed to the recovery of the porous structure of the cathode, as confirmed by scanning electron microscopy (SEM) images (
Figure 6). This study indicated that the lifespan of the Li-air battery could be extended, in principle, if the passivation layer were removed in a timely manner.
Shui
et al. [
14,
18] also investigated the distribution of insoluble lithium precipitates in the separator region of non-aqueous Li-air batteries, using microfocused synchrotron X-ray diffraction (µ-XRD). They reported that, unexpectedly, a significantly higher concentration of precipitates was found in the separator region than in the cathode. The µ-XRD results showed that these precipitates are mainly crystallized Li
2CO
3, which grew on the separator fiber surface, as revealed in the SEM images in
Figure 7. Under severe electrolyte decomposition, such precipitate formation could lead to the blockage of the pores in the middle layer of the separator, thereby constricting the electrolyte-mediated ion transport. Moreover, these precipitates in the separator are electrochemically non-decomposable, since the separator is insulated from the electrochemical reactions, resulting in higher observed accumulation at the separator.
Figure 6.
Scanning electron microscopy (SEM) images of the cathode surface of: (
a) a fresh cathode, (
b) a used cathode from a failed cell after multi cycles, (
c) the used cathode after being washed in 0.5 M H
2SO
4 for 10 min; and (
d) X-ray diffraction (XRD) patterns of cathodes shown by (
a), (
b) and (
c) with reference Li
2CO
3. Reprinted with permission [
15]; Copyright 2013, Elsevier.
Figure 6.
Scanning electron microscopy (SEM) images of the cathode surface of: (
a) a fresh cathode, (
b) a used cathode from a failed cell after multi cycles, (
c) the used cathode after being washed in 0.5 M H
2SO
4 for 10 min; and (
d) X-ray diffraction (XRD) patterns of cathodes shown by (
a), (
b) and (
c) with reference Li
2CO
3. Reprinted with permission [
15]; Copyright 2013, Elsevier.
Figure 7.
SEM images of (
a) fresh separator and (
b–
i) separator slices from a failed battery sampled at equal distances from the cathode to the anode. The scale bar is 2 µm. Reprinted with permission [
14]; Copyright 2012, Wiley-VCH Verlag GmbH& Co. KGaA, Weinheim.
Figure 7.
SEM images of (
a) fresh separator and (
b–
i) separator slices from a failed battery sampled at equal distances from the cathode to the anode. The scale bar is 2 µm. Reprinted with permission [
14]; Copyright 2012, Wiley-VCH Verlag GmbH& Co. KGaA, Weinheim.
Besides the stability of solvents, another source of detrimental electrolyte decomposition in Li-air batteries could be from side reactions due to the decomposition of lithium salt used in the electrolytes. The stability of lithium salt, especially in the presence of reduced oxygen species (O
2−) and trace amount of H
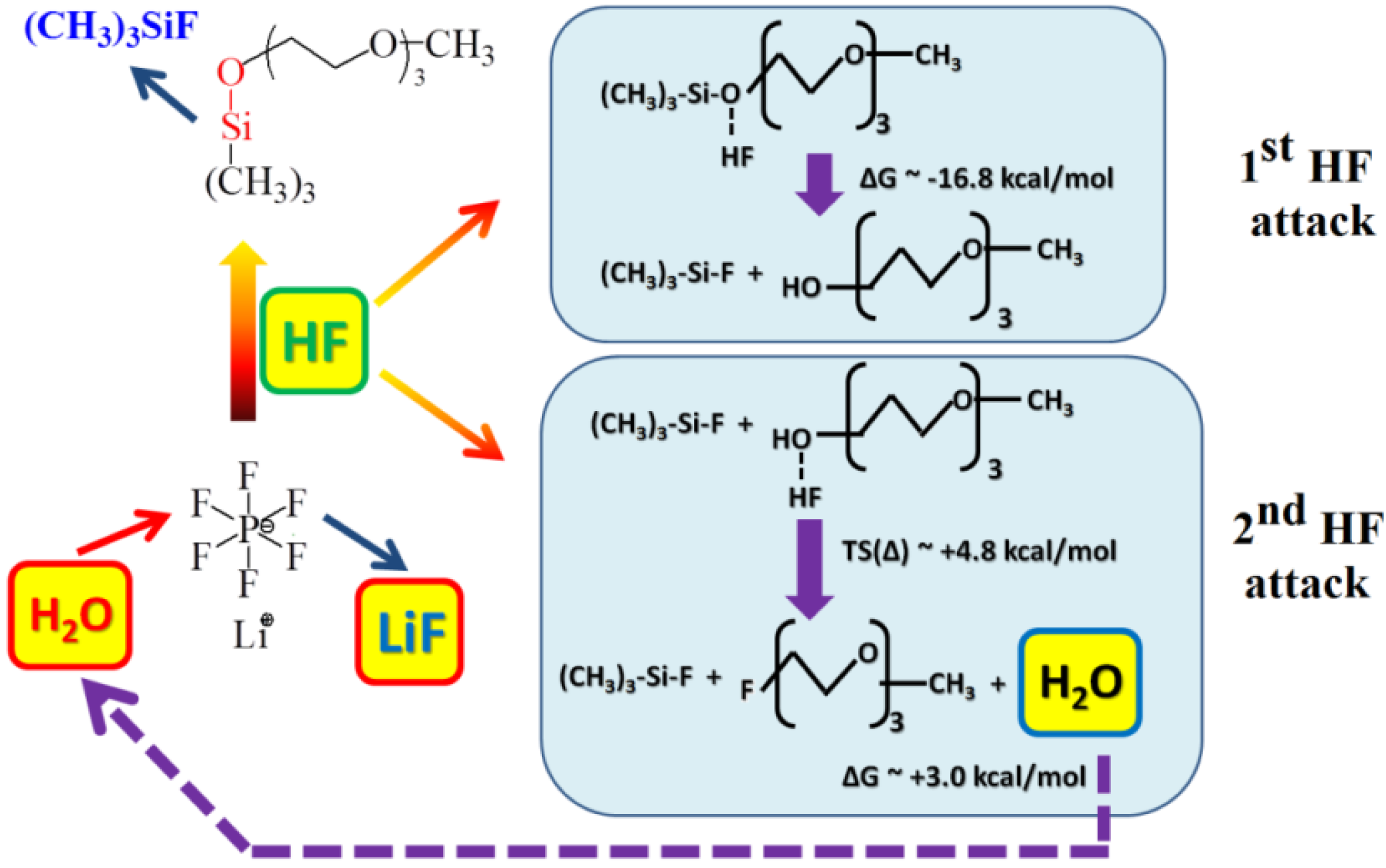
2O, may significantly affect the cyclability and capacity of Li-air cells. Moreover, these side reactions, if they are not electrochemical reactions, will not be evident on the voltage/capacity profiles and, therefore, will be difficult to investigate. Applying a combined experimental and computational approach, Du
et al. [
32] provided evidence that the stability of the electrolyte used in the Li-air cell strongly depends on the compatibility of the lithium salt with the solvent. In the case of the LiPF
6-1NM3 electrolyte, the decomposition of LiPF
6 occurs in the cell, as evidenced by
in-situ XRD, FT-IR, and XPS analyses. This decomposition forms HF and triggers the decomposition of the 1NM3 solvent.
Figure 8 shows a possible mechanism for the decomposition of the 1NM3-LiPF
6 electrolyte observed experimentally, which was further confirmed by
ab initio molecular dynamics simulations of the bulk 1NM3-LiPF
6 electrolyte. These reactions lead to degradation of the electrolyte and cause the poor cyclability of the cell. The same reactions are not observed when lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) and LiCF
3SO
3 are used as the lithium salt in 1NM3 solvent, or when LiPF
6 is used in TEGDME solvent, suggesting that the stability of the electrolyte in Li-air cells depends on the compatibility of lithium salt with solvent.
Figure 8.
Schematic reaction mechanism for decomposition of LiPF
6-1NM3 electrolyte in the Li-O
2 cell. Reproduced from [
32] with permission from the PCCP Owner Societies.
Figure 8.
Schematic reaction mechanism for decomposition of LiPF
6-1NM3 electrolyte in the Li-O
2 cell. Reproduced from [
32] with permission from the PCCP Owner Societies.
4. Structure and Magnetic Properties of Li2O2 and Relevance to Lithium-Air Battery
Better understanding of the electronic and structural properties of lithium oxides (Li
xO
y) could help us elucidate the charge and discharge chemistries involved in the Li-air battery. Unfortunately, the Li-O
2 phase diagram remains elusive and far from complete. In general, the most commonly known stoichiometric Li
xO
y compounds with a thermodynamically stable structure in condensed phase are Li
2O (lithia) and Li
2O
2 [
25]. Similar to Li
2O, crystalline bulk Li
2O
2 is known from theoretical predictions to be a semiconductor with an electronic band gap of ~4.9 eV [
69,
70,
71]. Such high band gap fails to explain the electronic conductivity and charge transfer required for the redox chemistry in an operating Li-air cell cathode. To address this issue, Hummelshoj
et al. [
69,
72] recently proposed a theory based on metallicity in bulk Li
2O
2 induced by Li vacancies, whose formation energy is predicted to be ~3.0 eV and depends on the vacancy concentration. The theory is supported by the finding that the Li
2O
2 observed as the discharge product of a Li-air cell is generally in the form of nanoparticles [
71,
73].
The mechanism of the formation and growth of Li
2O
2 products in Li-air batteries during discharge is currently not well understood, nor is the mechanism of the decomposition of Li
2O
2 during charge. Therefore, computational studies are needed to investigate the structural and electronic properties of small Li
2O
2 clusters, considering that computations on large clusters or particles would be much more complicated and costly. The information gathered from theoretical computations could provide insight into the nucleation of Li
2O
2 during battery discharge as well as the properties of larger nanoparticles. In a recent article, Lau and co-workers [
24] reported on a DFT study of the structure of (Li
2O
2)
n clusters (
n = 1–4). Many possible structures of the clusters and the corresponding electronic states were considered in their calculations, as shown in
Table 2. High-level G4 theory calculations showed, surprisingly, that the triplet state is significantly stabilized relative to the singlet in these clusters, especially for clusters larger than the dimer. The DFT calculations also showed that a cluster with
n = 16 has a high spin state, which can be characterized by O–O pairs protruding from the surface but still chemically bonded to the Li of the clusters. The distinct O–O pairs found on the high spin stoichiometric Li
2O
2 clusters suggest the existence of superoxide-like surface structures, considering the short O–O distances and a localized unpaired spin on the surface compared to a peroxide pair as found in a Li
2O
2 bulk crystal, which has longer O–O distance and no spin. These superoxide-like surface structures, if they indeed exist experimentally, could have important implications for the electrochemistry of formation and decomposition of Li
2O
2 in Li-air batteries, including surface electronic conductivity and electrolyte surface reactions.
Table 2.
Energies of triplet state relative to singlet state (in eV) of (Li
2O
2)
n Clusters,
n = 1–4, at various levels of theory; also given in the Table is the energy of the quintet state relative to the singlet for the (Li
2O
2)
16 cluster. Reprinted with permission [
24]; Copyright 2012, American Chemical Society.
Table 2.
Energies of triplet state relative to singlet state (in eV) of (Li2O2)n Clusters, n = 1–4, at various levels of theory; also given in the Table is the energy of the quintet state relative to the singlet for the (Li2O2)16 cluster. Reprinted with permission [24]; Copyright 2012, American Chemical Society.
| N | State a | B3LYP/6-31G(2df)//B3LYP/6-31G(2df) | PBE/PW//PBE/PW b | G4 theory |
|---|
| Ee | G298 | Ee | Ee | G298 |
|---|
| 1 | singlet (1) | 0.00 | 0.0 | 0.0 | 0.0 | 0.0 |
| triplet (1) | 0.96 | 0.86 | 0.88 | 1.27 | 1.17 |
| 2 | singlet (4) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| triplet (4) | −0.04 | −0.21 | −0.02 | 0.55 | 0.36 |
| 3 | singlet (11) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| triplet (11) | −0.48 | −0.72 | −0.49 | −0.08 | −0.32 |
| 4 | singlet (22) | 0.0 | 0.0 | 0.0 | 0.0 c | 0.0 c |
| 16 | triplet (22) | −0.45 | −0.64 | −0.55 | −0.17 c | −0.35 c |
| singlet (1) | 0.0 d | 0.0 d | 0.0 |
| quintet (1) | −1.09d | −1.39 d | −1.03 |
Inspired by the above computational study, Lu
et al. [
13] recently investigated the magnetic properties of the Li
2O
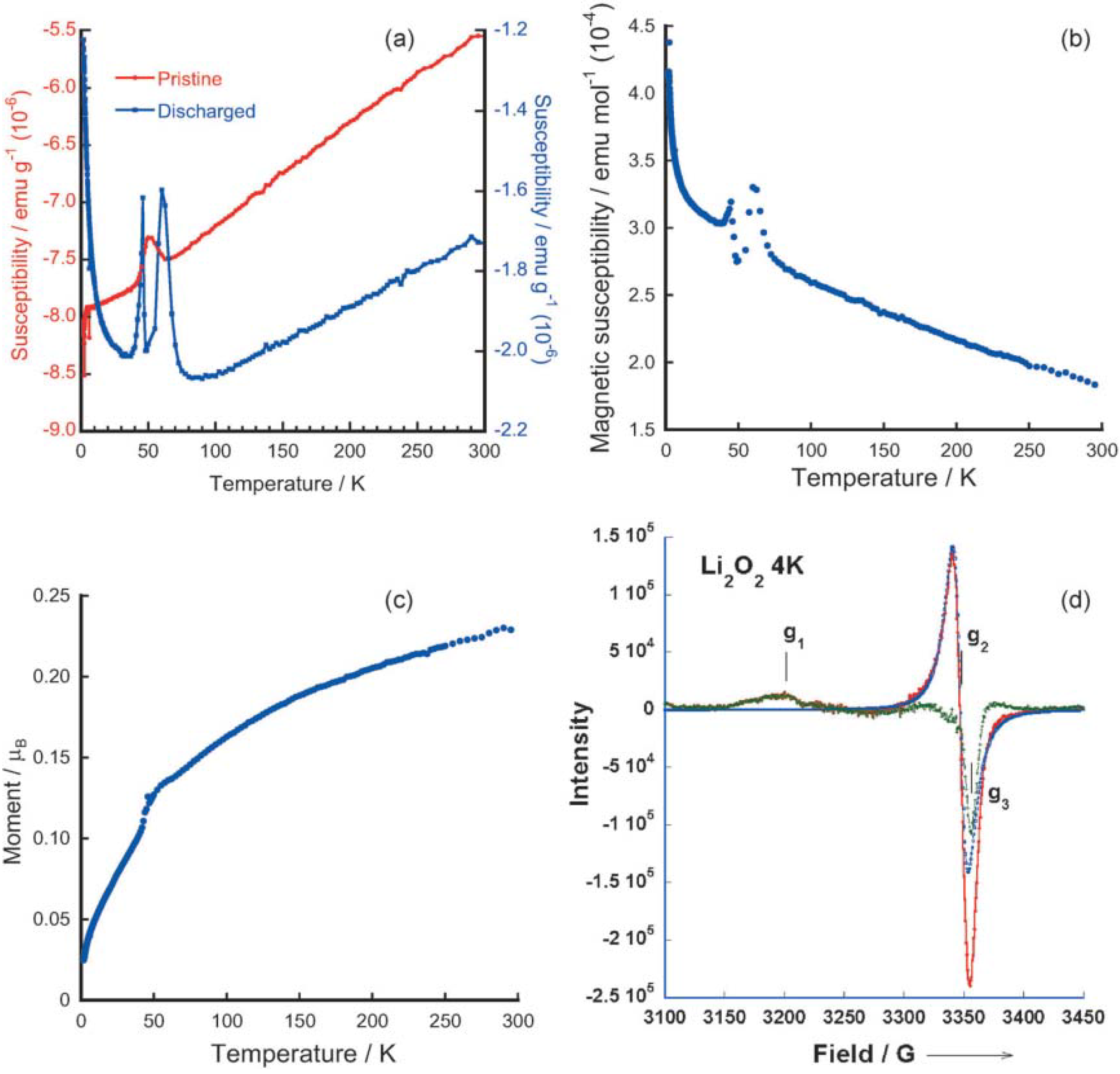
2 discharge products formed in an ether (1NM3)-based electrolyte, which lead to a better understanding on preparing successful Li-air batteries. Using electron paramagnetic resonance (EPR) and superconducting quantum interference device (SQUID) magnetometry, they found evidence for paramagnetism in the Li
2O
2 discharge product, as shown in
Figure 9. The EPR spectra, by comparison to the EPR spectra of LiO
2 from the literature, also provided direct evidence that the spin in the discharge Li
2O
2 product is caused by a superoxide-type structure. This finding confirmed the DFT calculations that superoxide-type surface oxygen groups with unpaired electrons exist on some stoichiometric Li
2O
2 nanoparticle surfaces. In addition, the number of spins predicted by using DFT is approximately consistent with the magnetic measurements of the discharged Li
2O
2 product.
Figure 9.
Magnetic susceptibility data from superconducting quantum interference device (SQUID) measurements for: (
a) pristine carbon cathode before cycling and discharged cathode with 1NM3-LiCF
3SO
3 electrolyte; (
b) difference between pristine carbon cathode and initially discharged cathode; (
c) magnetic moment data from SQUID measurement for Li
2O
2 bulk powder; and (
d) electron paramagnetic resonance (EPR) spectrum of Li
2O
2 bulk powder at 4 K. The red trace is the experimental result. The blue trace is the calculated one line Lorentzian spectrum to fit the central peak at g
2 value; and the green trace is the difference between red and blue traces to reveal peaks at g
1 and g
3. Reprinted with permission [
13]; Copyright 2013, Wiley-VCH Verlag GmbH& Co. KGaA, Weinheim.
Figure 9.
Magnetic susceptibility data from superconducting quantum interference device (SQUID) measurements for: (
a) pristine carbon cathode before cycling and discharged cathode with 1NM3-LiCF
3SO
3 electrolyte; (
b) difference between pristine carbon cathode and initially discharged cathode; (
c) magnetic moment data from SQUID measurement for Li
2O
2 bulk powder; and (
d) electron paramagnetic resonance (EPR) spectrum of Li
2O
2 bulk powder at 4 K. The red trace is the experimental result. The blue trace is the calculated one line Lorentzian spectrum to fit the central peak at g
2 value; and the green trace is the difference between red and blue traces to reveal peaks at g
1 and g
3. Reprinted with permission [
13]; Copyright 2013, Wiley-VCH Verlag GmbH& Co. KGaA, Weinheim.
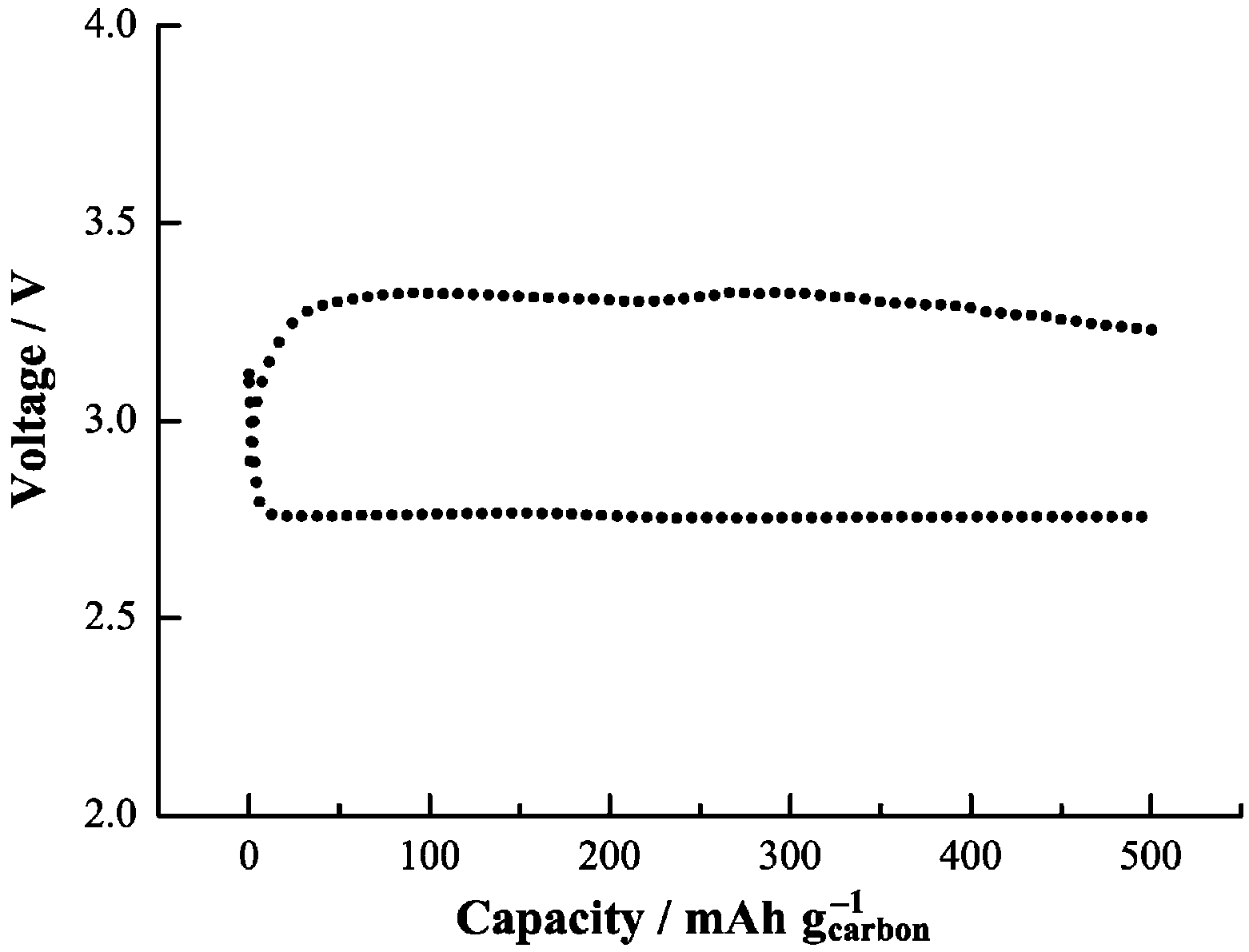
The presence of magnetism in the Li
2O
2 discharge product could play an important role in the charge and discharge chemistries of Li-air batteries, since the superoxide-type surface oxygen groups with spin could enable the electronic conductivity mechanism that is required for the reversible formation and decomposition of Li
2O
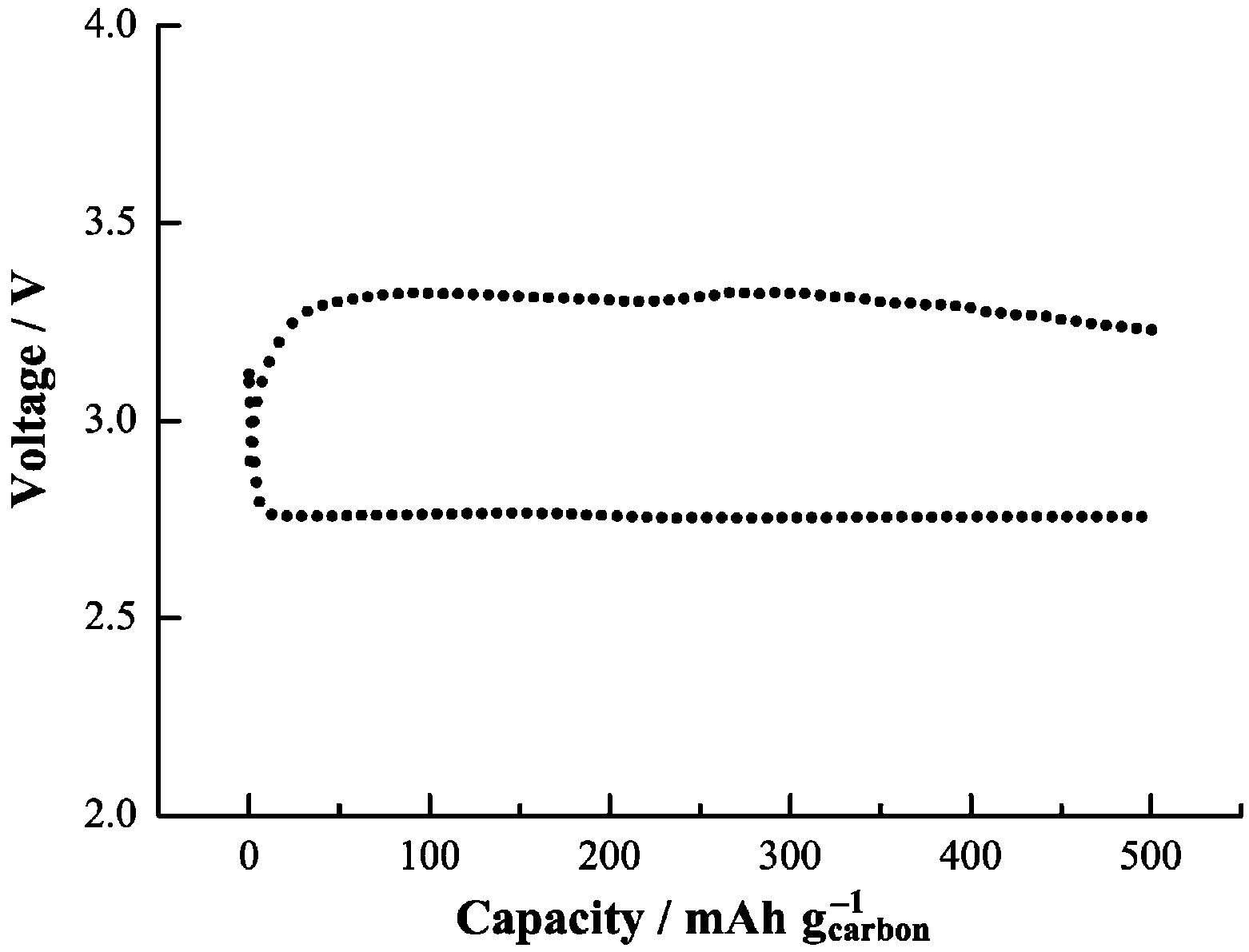
2. As consequence, Li-air cells with such discharge products achieved a low charge potential (3.3 V), as shown in
Figure 10. The presence of spin, as found in the experiments and as suggested to be present on the Li
2O
2 surface in the calculations, provides a possible mechanism for the electronic conductivity for the reversible electrochemical formation and decomposition of Li
2O
2 [
13]. The calculated DFT density of states from the “superoxide” species for the high spin state of a Li
2O
2 surface indicates the presence of a finite density of states in the band gap. Analysis of the density of states indicates that these states come from the surface superoxide species, which could give rise to surface electrical conductivity. Thus, electrochemical decomposition and formation of Li
2O
2 could be enabled by this surface conductivity, as well as effective connectivity through grain boundaries between nanoparticles and interfaces with the carbon.
Figure 10.
Voltage profile during first discharge-charge of Li-O
2 cell based on the 1NM3-LiCF
3SO
3 electrolyte. Reprinted with permission [
13]; Copyright 2013, Wiley-VCH Verlag GmbH& Co. KGaA, Weinheim.
Figure 10.
Voltage profile during first discharge-charge of Li-O
2 cell based on the 1NM3-LiCF
3SO
3 electrolyte. Reprinted with permission [
13]; Copyright 2013, Wiley-VCH Verlag GmbH& Co. KGaA, Weinheim.
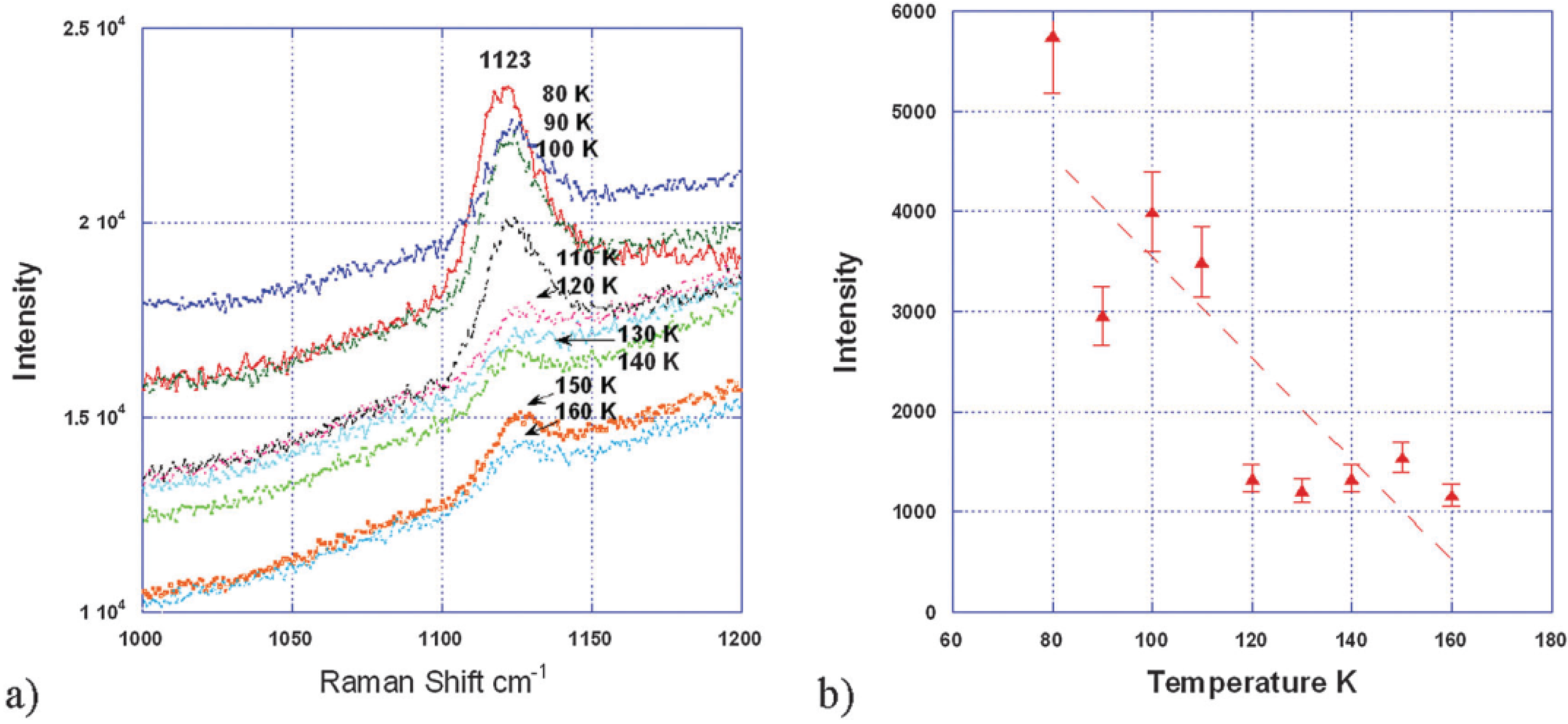
Recent work by Yang and co-workers [
33] further confirmed the presence of a LiO
2-like species in the Li
2O
2 discharge product by the presence of a peak at 1125 cm
−1 in the Raman spectra (
Figure 11), in addition to the peaks expected for Li
2O
2.
Figure 11.
(
a) Raman spectra of the discharged activated carbon (AC) surface between 160 K and 80 K showing that the superoxide peak ~1125 cm
−1 increases with decreasing temperature; (
b) superoxide peak intensity estimated from its peak height between 160 K and 80 K. The dash line is a guide for the eyes. Reproduced from [
33] with permission from the PCCP Owner Societies.
Figure 11.
(
a) Raman spectra of the discharged activated carbon (AC) surface between 160 K and 80 K showing that the superoxide peak ~1125 cm
−1 increases with decreasing temperature; (
b) superoxide peak intensity estimated from its peak height between 160 K and 80 K. The dash line is a guide for the eyes. Reproduced from [
33] with permission from the PCCP Owner Societies.
In this work, they applied a petroleum coke-based activated carbon (AC) as cathode material and a TEGDME-LiCF
3SO
3 electrolyte to investigate the discharge/charge behavior of a Li-air cell. Electrochemical measurement surprisingly showed two distinct voltage plateaus during charging, at 3.2–3.5 V and 4.2–4.3 V
vs. Li
+/Li, where the lower plateau corresponds to a form of Li
2O
2 with superoxide-like properties characterized by a low temperature magnetic phase transition (49.7 K) and a high O–O stretching frequency (1125 cm
−1). Yang
et al. [
33] also observed that the magnetic phase transition and the high O–O stretching frequency disappear when the cell is charged to above 3.7 V. This finding suggests that a superoxide surface species with a low-temperature phase transition results in a lower charge potential, and control of the forms of Li
2O
2 produced on discharge is important for improving the efficiency of Li-air batteries.
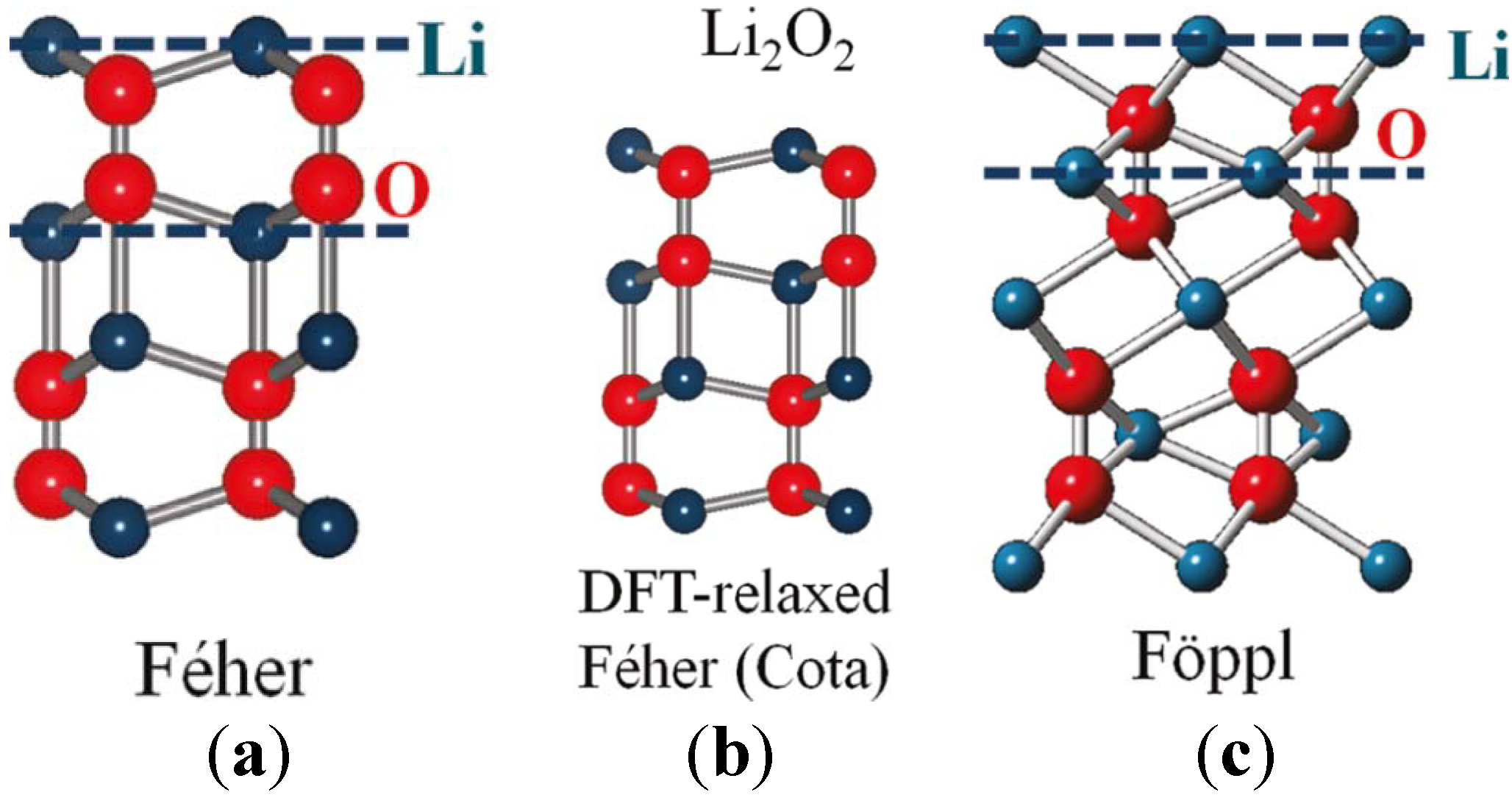
In addition to the electronic and magnetic properties, the crystalline structure of Li
2O
2 is also of vital importance to the study of Li-air batteries. In the 1950s, two disparate crystal structures were proposed for Li
2O
2 from XRD studies, the Féher [
74] and Fӧppl [
75] structures, which have different lithium sublattices, as shown in
Figure 12. Along the
c-lattice direction, Féher’s original structure consists of lithium and oxygen atoms nominally sharing each plane, whereas Fӧppl’s revised structure positions of the lithium sites between adjacent oxygen planes. Although these two structures share similar nearest-neighbor Li–O distances (1.91 Å in Féher’s
vs. 1.98 Å in Fӧppl’s), the O–O bond distances are dramatically different (1.28 and 1.55 Å, respectively). To assess these competing Li
2O
2 structures, Chan
et al. [
28] recently employed a combination of high-energy XRD and nonresonant inelastic X-ray scattering (NIXS) spectra with first-principle calculations using the Bethe-Salpeter Equation. Their data indicated that Fӧppl’s structure is more appropriate for Li
2O
2. The measured and computed spectra and data presented in their work are very useful as benchmarks for future characterization of Li
2O
2 during the discharge of Li-air cells. Also note that NIXS using hard X-rays is a suitable analytical technique for providing bulk sensitive information on Li-air battery discharge products with element specificity [
23]. Also known as X-ray Raman scattering, NIXS can be considered as an alternative approach toward obtaining soft X-ray absorption spectroscopy (XAS)-like information using hard X-rays, despite crystalline or amorphous components being present.
Figure 12.
(
a) Féher’s; (
b) density functional theory (DFT)-relaxed Féher’s according to Cota; and (
c) Fӧppl’s proposed crystal structures for Li
2O
2. Red (larger) spheres represent oxygen atoms and blue (smaller) spheres represent lithium atoms. Blue (dashed) horizontal lines indicate Li planes. We can see that Li atoms are roughly in-plane with the O atoms in the Féher structure while Li planes cut through the O–O bonds in the Fӧppl structure. Reprinted with permission [
28]; Copyright 2011, American Chemical Society.
Figure 12.
(
a) Féher’s; (
b) density functional theory (DFT)-relaxed Féher’s according to Cota; and (
c) Fӧppl’s proposed crystal structures for Li
2O
2. Red (larger) spheres represent oxygen atoms and blue (smaller) spheres represent lithium atoms. Blue (dashed) horizontal lines indicate Li planes. We can see that Li atoms are roughly in-plane with the O atoms in the Féher structure while Li planes cut through the O–O bonds in the Fӧppl structure. Reprinted with permission [
28]; Copyright 2011, American Chemical Society.
Using DFT together with classical statistical mechanical analyses, Lau and Curtiss [
25] investigated the thermodynamic stability of bulk crystalline LiO
2, Li
2O, and Li
2O
2 as a function of the oxygen environment, which is relevant to the basic bulk Li-O
2 electrochemical couples observed in the Li-air battery. Their calculated results demonstrated that Li
2O
2(s) and superoxide [LiO
2(s)] are likely to be stable only under O
2-rich conditions with high oxygen partial pressures, whereas Li
2O is most stable at ambient conditions.
5. Air-Breathing Cathode
Cathode materials represent a major technology challenge in rechargeable Li-air battery development. The specific capacity and power capability of Li-air cells strongly depend on the materials and microstructures of the air-breathing cathode, which contribute to most of its voltage drop [
3,
6,
76,
77]. A high performance cathode should be able to provide: (a) good electronic and ionic conductivity; (b) fast oxygen diffusion; and (c) stable electrode integrity. It should also have an efficient catalyst supported by a high surface area substrate, similar to that in a proton exchange membrane fuel cell. Since the first demonstration of a lithium-oxygen system using pyrolyzed cobalt phthalocyanine over carbon as the cathode [
63], many new cathode materials have been reported, particularly in the area of new catalysts for improving the battery efficiency [
8,
40,
42,
44,
62].
In 2006, Bruce
et al. [
2] demonstrated high-energy-density Li-air cells using a porous carbon cathode with electrolytic manganese dioxide. Since that time, MnO
2-based compounds have been widely investigated as the cathode catalyst to improve the performance of the air-breathing cathode. In their study, they found that different catalytic reactivity for the oxygen evolution reaction could be achieved for MnO
2 with different crystal structures and morphologies, among which the use of α-MnO
2 nanowires was found to deliver capacities of 3000 mA h/g of carbon [
78]. Several manganese dioxide materials have been investigated as electrocatalysts for non-aqueous Li-air cells, which were prepared by various approaches with different crystalline structures and morphologies [
9,
10,
17]. One is MnO
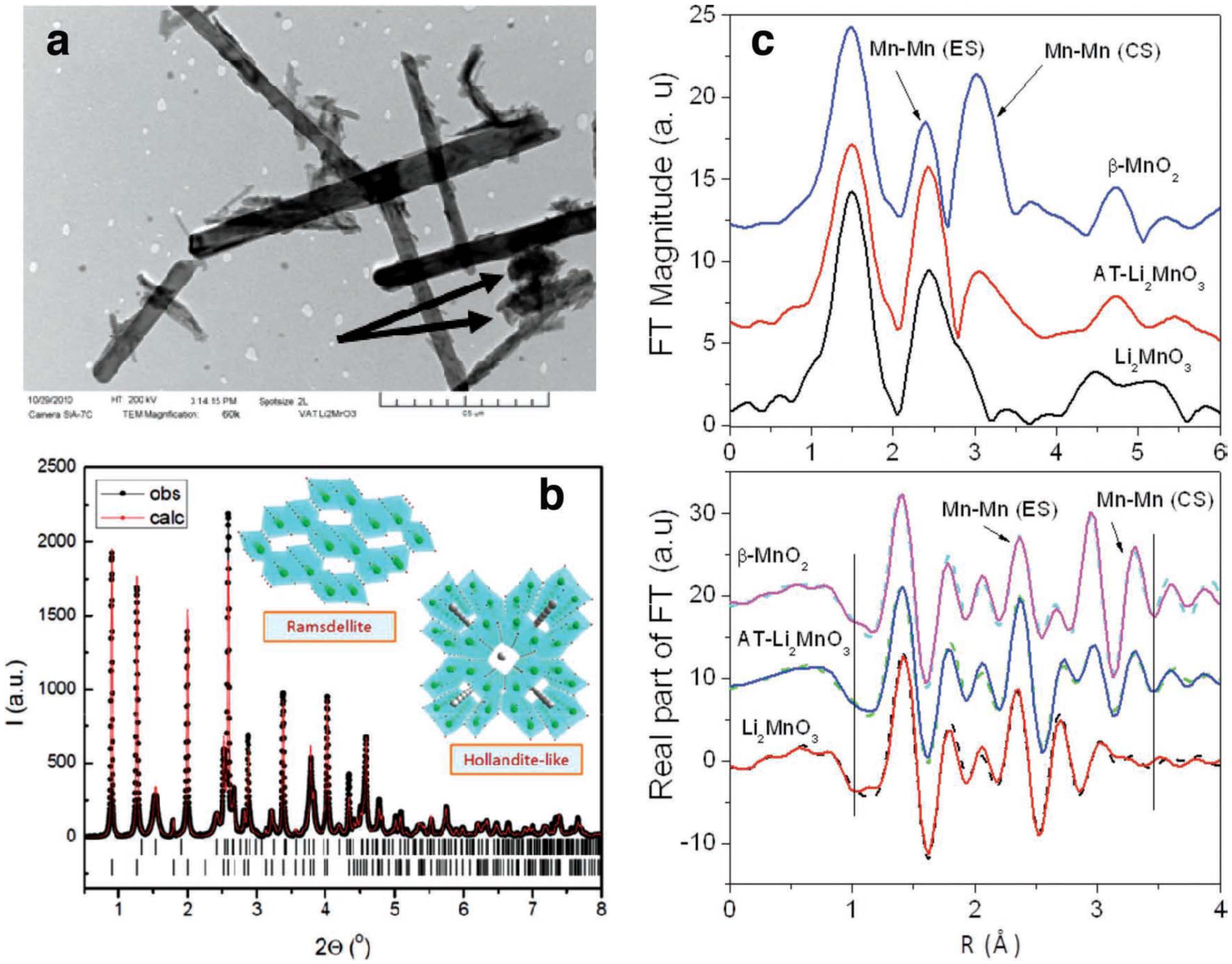
2 with a “tunnel” structure reported by Trahey
et al. [
10], meaning there are parallel, one-dimensional lines of open space through the structures. In their work, they produced nanostructured MnO
2 with dual electrode/electrocatalyst functionality that has an affinity to react with Li
2O by treating Li
2MnO
3 (Li
2O·MnO
2) with highly concentrated acid. Both XRD and XAS measurements showed that the acid-treated Li
2MnO
3 electrodes consist of a composite α-MnO
2/ramsdellite-MnO
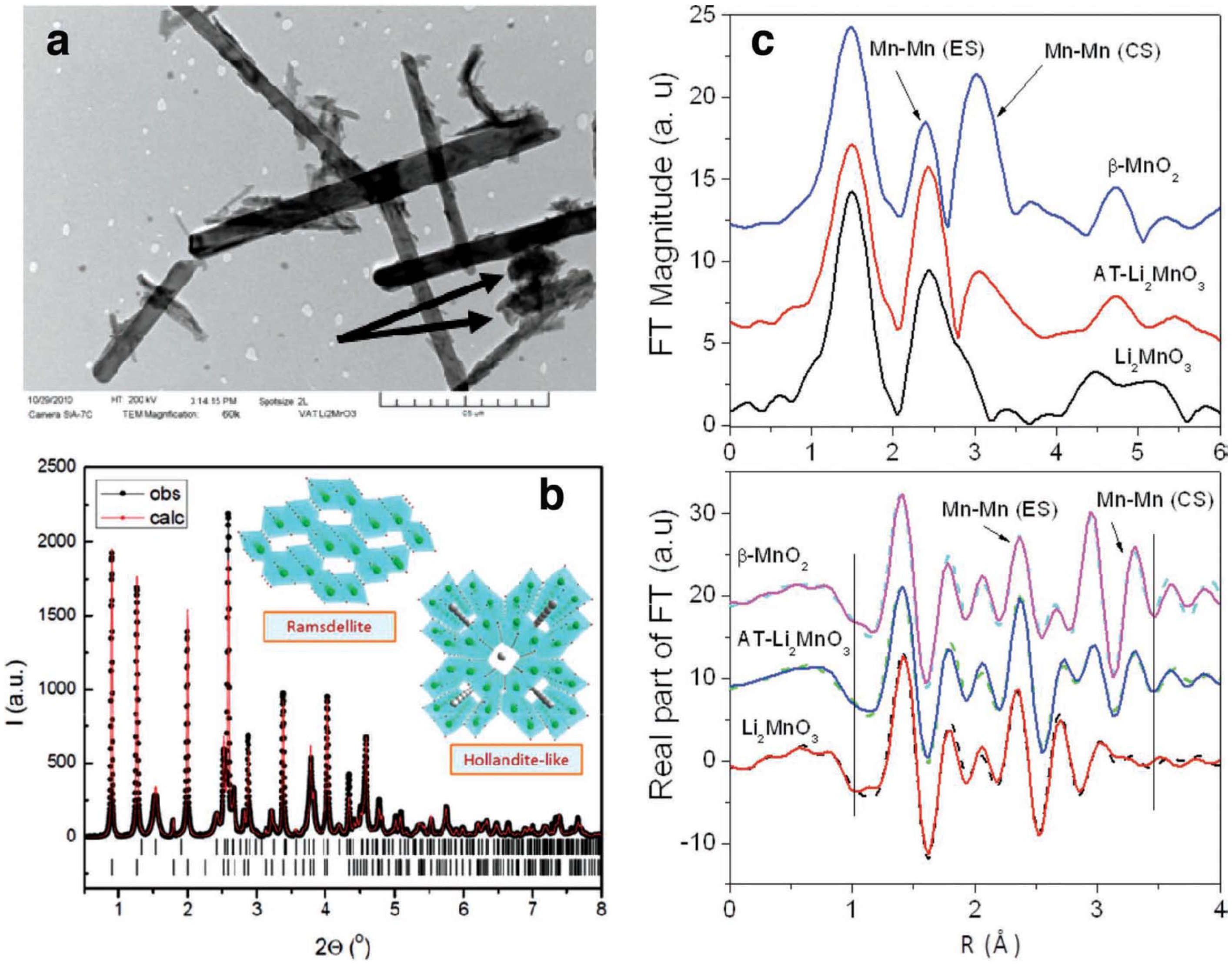
2 product with a one-to-one ratio between corner-shared and edge-shared oxygen octahedral around Mn ions. This finding confirms the presence of tunnels in the parent structure, as shown in
Figure 13. Transmission electron microscopy (TEM) (
Figure 13a) revealed the existence of nanorod-shaped particles. The presence of such MnO
2 catalyst on the cathode reduces the charging potential of the cell. As determined by XPS, the discharge products (solids) formed using PC as the electrolyte appeared to be lithium carbonate and lithium oxide, and lithium oxide disappeared upon charge. These results suggest that the α-MnO
2/ramsdellite-MnO
2 catalyst is playing a key role in the reversibility of the lithium oxide products, even though evidence suggests that PC is partially decomposing during the discharge/charge cycles.
Figure 13.
(
a) Transmission electron microscopy (TEM) image of α-MnO
2 needles and R-MnO
2 crystallites (arrowed); (
b) observed and calculated X-ray powder diffraction of the acid-treated Li
2MnO
3 product, with schematic structures of the α-MnO
2 and ramsdellite-MnO
2 components; the short vertical lines indicate the peak positions of the two phases; (
c, top) phase uncorrected Fourier transform (FT) magnitude of Mn K-edge extended X-ray absorption fine structure (EXAFS) (
k3 weighted) for acid-treated Li
2MnO
3 and Li
2MnO
3 and β-MnO
2 standards. The peaks at ~2.4 Å and ~3.1 Å arise due to edge sharing (ES) and corner sharing (CS) Mn–Mn correlations from MnO
6 octahedral linkages, respectively; and (
c, bottom) R-space fits (solid line) of the real part of the FT of Mn EXAFS data along with the experimental data (dashed lines). The vertical lines show the fit range covered. Reprinted with permission [
10]; Copyright 2013, Wiley-VCH Verlag GmbH& Co. KGaA, Weinheim.
Figure 13.
(
a) Transmission electron microscopy (TEM) image of α-MnO
2 needles and R-MnO
2 crystallites (arrowed); (
b) observed and calculated X-ray powder diffraction of the acid-treated Li
2MnO
3 product, with schematic structures of the α-MnO
2 and ramsdellite-MnO
2 components; the short vertical lines indicate the peak positions of the two phases; (
c, top) phase uncorrected Fourier transform (FT) magnitude of Mn K-edge extended X-ray absorption fine structure (EXAFS) (
k3 weighted) for acid-treated Li
2MnO
3 and Li
2MnO
3 and β-MnO
2 standards. The peaks at ~2.4 Å and ~3.1 Å arise due to edge sharing (ES) and corner sharing (CS) Mn–Mn correlations from MnO
6 octahedral linkages, respectively; and (
c, bottom) R-space fits (solid line) of the real part of the FT of Mn EXAFS data along with the experimental data (dashed lines). The vertical lines show the fit range covered. Reprinted with permission [
10]; Copyright 2013, Wiley-VCH Verlag GmbH& Co. KGaA, Weinheim.
Since MnO
2 nanoparticles can exhibit various crystallographic structures (e.g., α, β, δ, γ, and λ forms) and morphologies, controlled synthesis of nanostructured MnO
2 with high crystalline purity and uniform morphology is critical to precisely tailored properties and better performance in Li-air batteries. To address this issue, Truong
et al. [
9] produced high-quality nanosheet-based δ-MnO
2 microflowers, α-MnO
2 nanowires, and α-MnO
2 nanotubes in large quantity through microwave-assisted hydrothermal reduction of potassium permanganate in the presence of hydrochloric acid, as shown in
Figure 14. They found that the chemical reaction determines the formation of δ-MnO
2 microflowers in the earlier stage of the process, while the two-step Ostwald ripening process dominates the crystalline and morphological transition from the δ-MnO
2 nanosheets to α-MnO
2 nanowires and nanotubes in the late stage of the reactions. In terms of the electrocatalytic activity for these MnO
2 nanoparticles, the single-crystal α-MnO
2 nanotubes exhibit much better performance to catalyze the electrochemical processes involved in Li-air batteries, leading to a significant improvement in specific capacity and cycle life.
Figure 14.
SEM images of MnO
2 products obtained at different reaction times: (
A) 2; (
B) 5; (
C) 15; (
D) 30; (
E) 120; and (
F) 360 min. The temperature of the reaction solutions and the atmosphere pressure above the reaction solutions were 150 °C and 70 psi, respectively. The concentrations of KMnO
4 and HCl in the reaction solution were 0.05 and 0.2 M, respectively, before the reaction was initiated. The insets are the high-magnification SEM images of the corresponding structures. The scale bars in (
F) also apply to (
A)–(
E). Reprinted with permission [
9]; Copyright 2013, American Chemical Society.
Figure 14.
SEM images of MnO
2 products obtained at different reaction times: (
A) 2; (
B) 5; (
C) 15; (
D) 30; (
E) 120; and (
F) 360 min. The temperature of the reaction solutions and the atmosphere pressure above the reaction solutions were 150 °C and 70 psi, respectively. The concentrations of KMnO
4 and HCl in the reaction solution were 0.05 and 0.2 M, respectively, before the reaction was initiated. The insets are the high-magnification SEM images of the corresponding structures. The scale bars in (
F) also apply to (
A)–(
E). Reprinted with permission [
9]; Copyright 2013, American Chemical Society.
The high surface area and porous structure of the carbon cathode are critical for the high electrochemical performance of Li-air batteries. In general, a larger surface area provides more surface to uniformly disperse catalytic particles and more active sites to aid the electrochemical reactions, while a porous structure with an appropriate pore size provides the space to store the discharge products. However, a problem has been encountered in previous studies [
2,
16,
55,
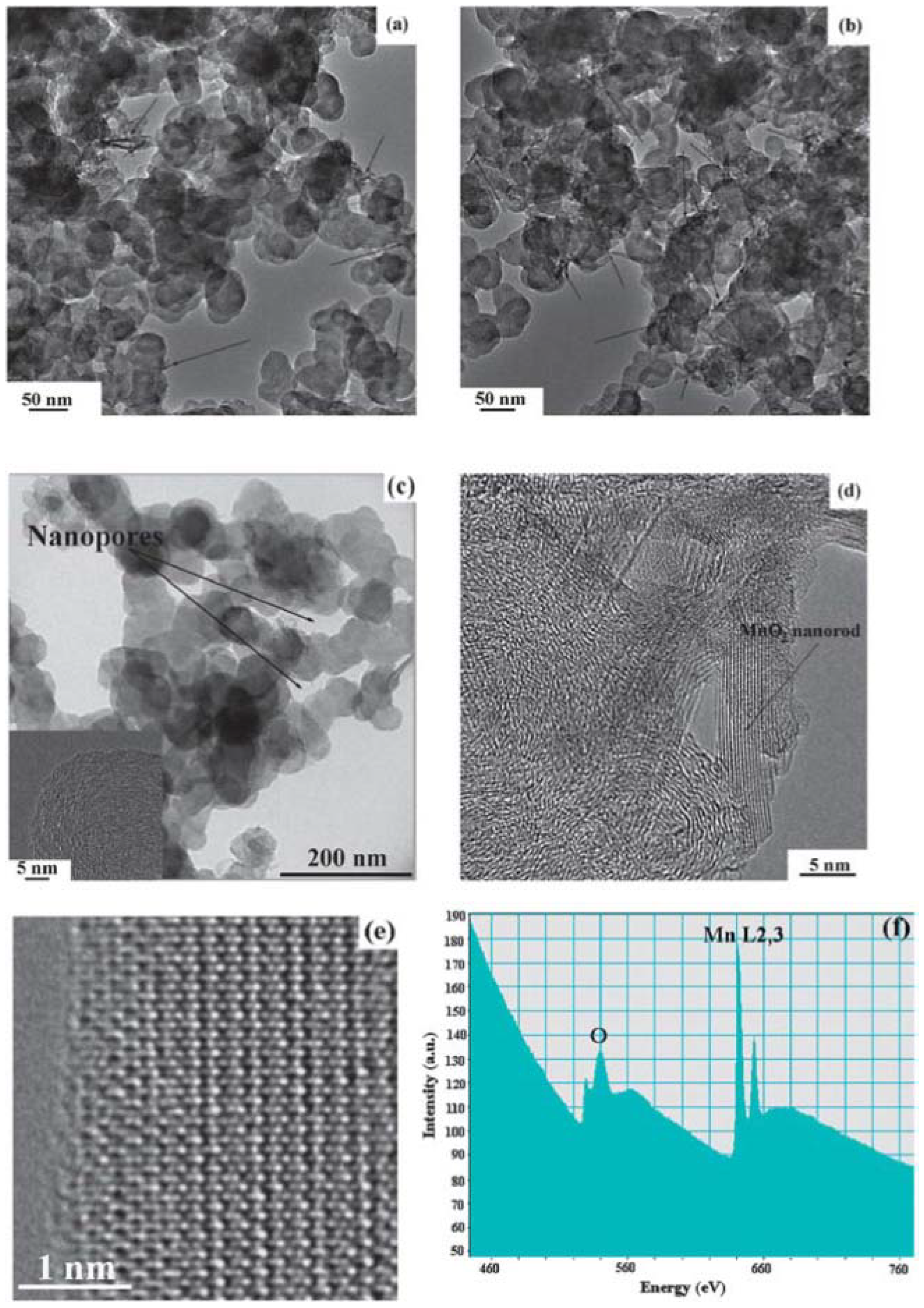
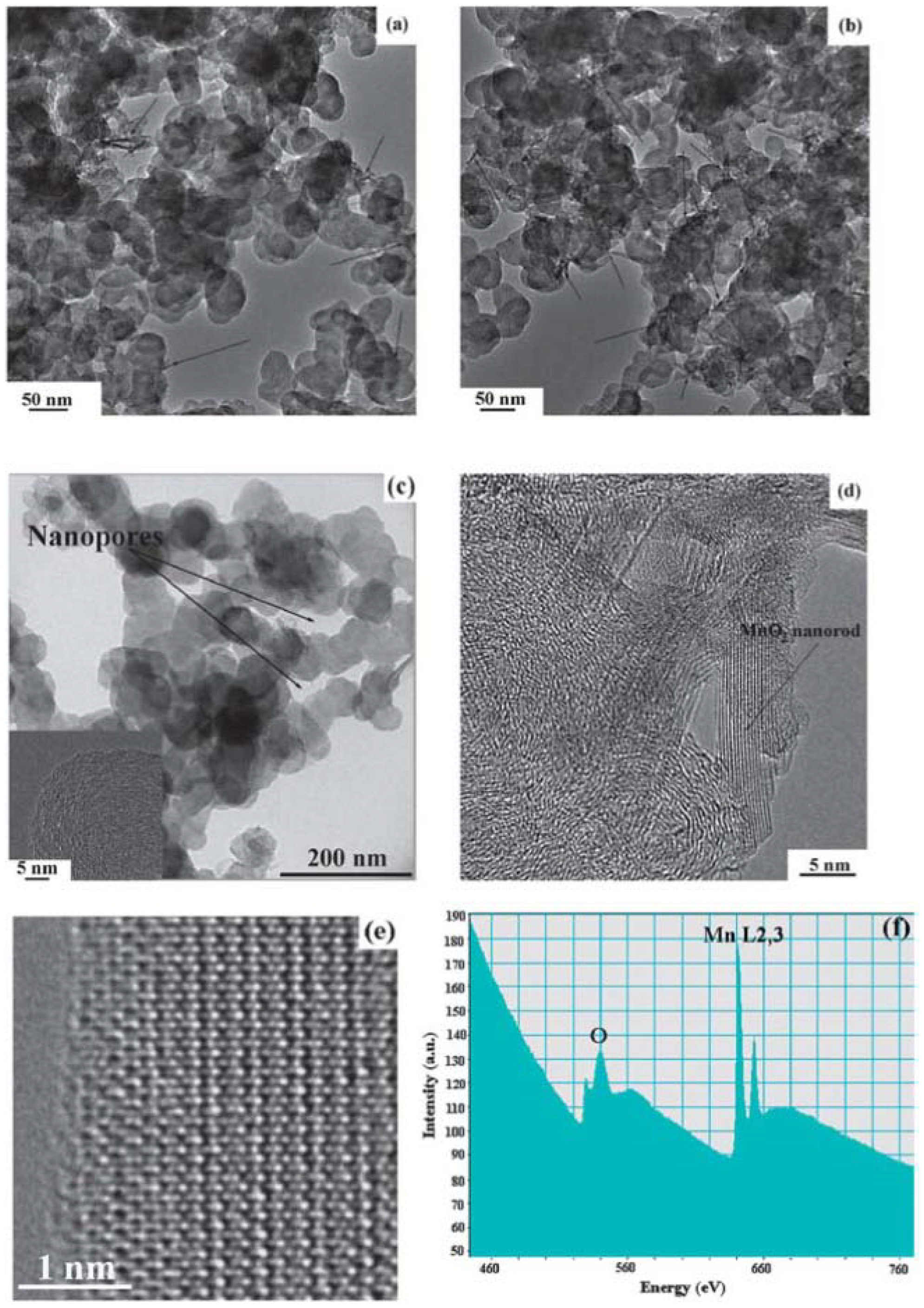
78], because the catalysts are pre-synthesized and then loaded onto carbon support by blending with carbon powder. This process introduces not only the potential of destroying the porous structure of the carbon support, if high-energy ball milling is applied, but also the non-uniform dispersion of the catalysts on the carbon support. Aiming to develop a new method to uniformly disperse the catalyst onto the carbon support while still preserving the original porous structure of carbon during the synthesis, Qin
et al. [
17] developed an
in-situ wet-chemistry approach to incorporate MnO
2 nanoparticles into porous carbon, which avoids any aggressive post-treatment of the carbon materials and thus preserves the original structure of the carbon matrix. The TEM images in
Figure 15 clearly indicate that most of the as-prepared MnO
2 particles are uniformly dispersed onto the carbon matrix with rod-like shape. The SEM images of the samples before and after being loaded with MnO
2 catalysts confirm that the porous structure of carbon was well preserved during the synthesis process. These as-prepared MnO
2/C composites with porous structures and high specific surface area provide more active sites for enabling the oxygen reduction and oxygen evolution reactions, and, therefore, lead to significant enhancement of cell performance. The as-prepared MnO
2/C composite cathode achieved a low charge overpotential (3.5 V) in a cell tested with a TEGDME-based electrolyte.
Figure 15.
TEM images of (
a) as-prepared MnO
2/C composite (Sample 1); (
b) as-prepared MnO
2/C composite (Sample 2); and (
c) original Super P Li (SPL) carbon; (
d) and (
e) high-resolution transmission electron microscopy (HRTEM) image of Sample 1; and (
f) electron energy loss spectra (EELS) of Sample 1. Reproduced from [
17] with permission from The Royal Society of Chemistry.
Figure 15.
TEM images of (
a) as-prepared MnO
2/C composite (Sample 1); (
b) as-prepared MnO
2/C composite (Sample 2); and (
c) original Super P Li (SPL) carbon; (
d) and (
e) high-resolution transmission electron microscopy (HRTEM) image of Sample 1; and (
f) electron energy loss spectra (EELS) of Sample 1. Reproduced from [
17] with permission from The Royal Society of Chemistry.
Recently, Lei
et al. [
30] also employed atomic layer deposition (ALD) for dispersing Pd catalysts with well-controlled particle size uniformly onto the carbon support. The ALD technique is capable of depositing highly uniform and conformal coatings on surfaces with complex topographies and infiltrating mesoporous materials [
79,
80,
81]. This capability is particularly attractive for the synthesis of heterogeneous catalysts requiring highly dispersed catalytic species on high-surface-area mesoporous supports. The good dispersion of the active particles on the support during ALD decreases the metal loading while still achieving the same catalytic activity as the catalysts with higher metal loading prepared by other methods. This feature is especially important with noble metal materials, where the excess use of the raw materials should be avoided. Lei
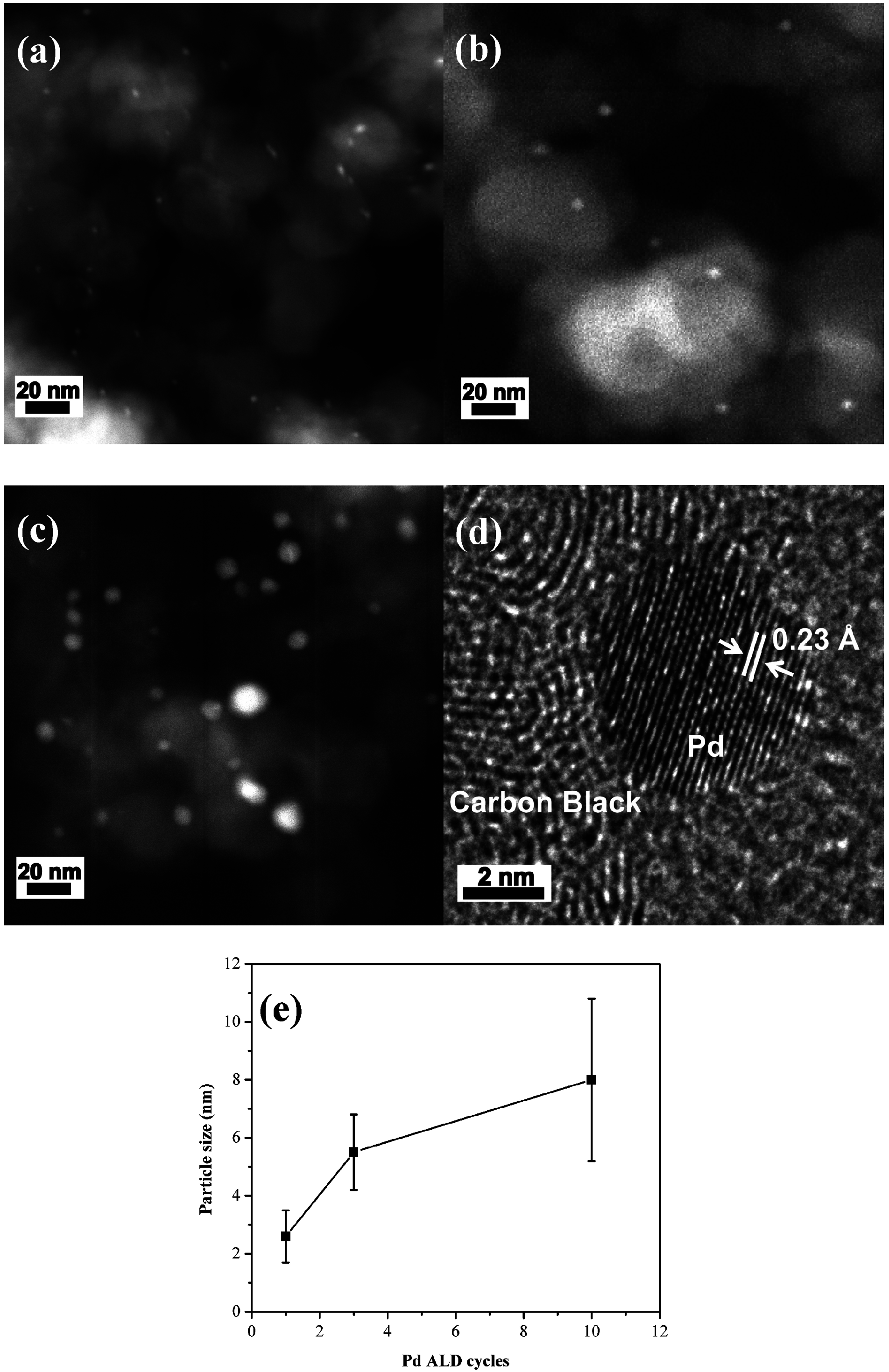
et al. [
30] demonstrated that the Pd nanoparticles can be uniformly distributed on the porous carbon surface with average particle size controlled by the number of ALD Pd cycles to be in the range of 2–8 nm, as shown in scanning transmission electron microscopy (STEM) images (
Figure 16). Such Pd/C composites with porous structures and high specific surface area provide more active sites to absorb O
2 molecules and, therefore, enhance the catalytic activity for the oxygen reduction reaction, as evident by a significantly higher discharge capacity achieved in the Li-air cell.
Figure 16.
Scanning transmission electron microscopy (STEM) images of (
a) 1c Pd/C; (
b) 3c Pd/C; (
c) 10c Pd/C; (
d) HRTEM of a Pd nanoparticle ~5.5 nm in diameter prepared by atomic layer deposition (ALD) supported over carbon; (
e) Pd particle size as a function of ALD cycles. Reprinted with permission [
30]; Copyright 2013, American Chemical Society.
Figure 16.
Scanning transmission electron microscopy (STEM) images of (
a) 1c Pd/C; (
b) 3c Pd/C; (
c) 10c Pd/C; (
d) HRTEM of a Pd nanoparticle ~5.5 nm in diameter prepared by atomic layer deposition (ALD) supported over carbon; (
e) Pd particle size as a function of ALD cycles. Reprinted with permission [
30]; Copyright 2013, American Chemical Society.
In addition, several Fe-based electrocatalysts were successfully incorporated to carbon supports and tested in the Li-air cells as the active cathode materials. For instance, Trahey
et al. [
16] designed electrocatalysts by either electrochemical activation of Li
5FeO
4 (5Li
2O·Fe
2O
3) or acid activation of Li
2MnO
3·LiFeO
2 ([Li
2O·MnO
2]·[Li
2O·Fe
2O
3]) to remove Li
2O, which delivers high discharge capacity. Lu
et al. [
34] synthesized a uniformly dispersed core-shelled Fe/Fe
3O
4 nanocomposite on porous carbon via a wet-chemistry approach, which was tested as a cathode material in the Li-air battery, showing an enhanced catalytic effect toward the electrochemical reactions, in particular, the oxygen reduction reaction. The XRD and XPS data demonstrated that Li
2O
2 participated in the reversible reactions in a Li-O
2 cell with TEGDME-based electrolyte. Shui
et al. [
19] produced a Fe/N/C composite via pyrolysis of an organometallic precursor at high temperature and evaluated its catalytic performance in connection with the rechargeable Li-air battery. Compared with the well-studied α-MnO
2/XC-72 composite and its unmodified carbon-only counterpart, this chemically modified carbon exhibited lower charge-discharge overpotential and improved the battery lifespan. A gas analysis at the end of discharge-charge cycling found no CO
2 formation for the cathode made with Fe/N/C catalyst, despite the charge potential exceeding 4.0 V. According to the authors, the catalytic selectivity of the Fe/N/C composite promotes the decomposition of Li
2O
2 over that of the electrolyte and, therefore, leads to an enhanced battery lifespan under controlled cycling.
6. Oxygen Crossover Effect on Lithium Anode
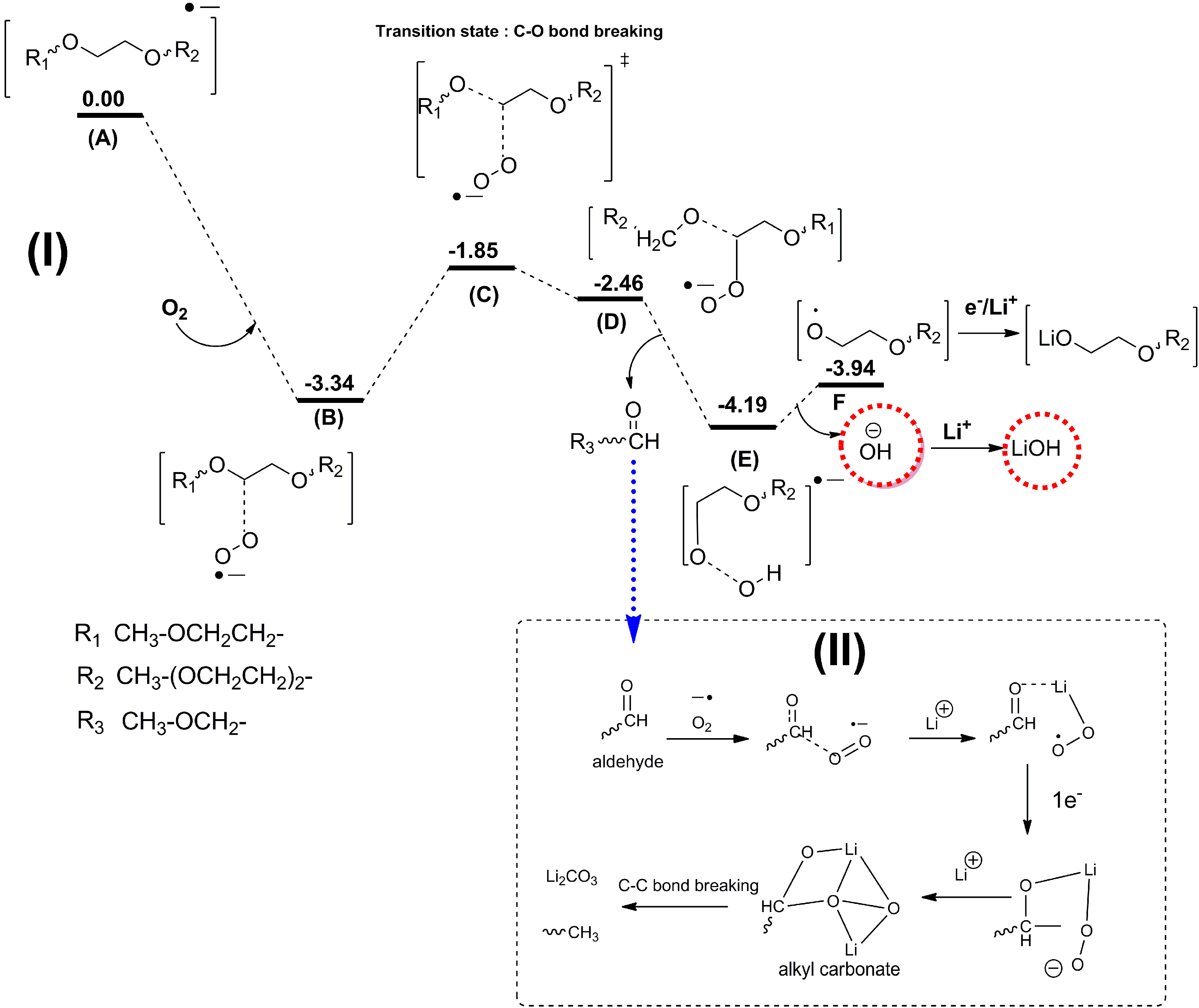
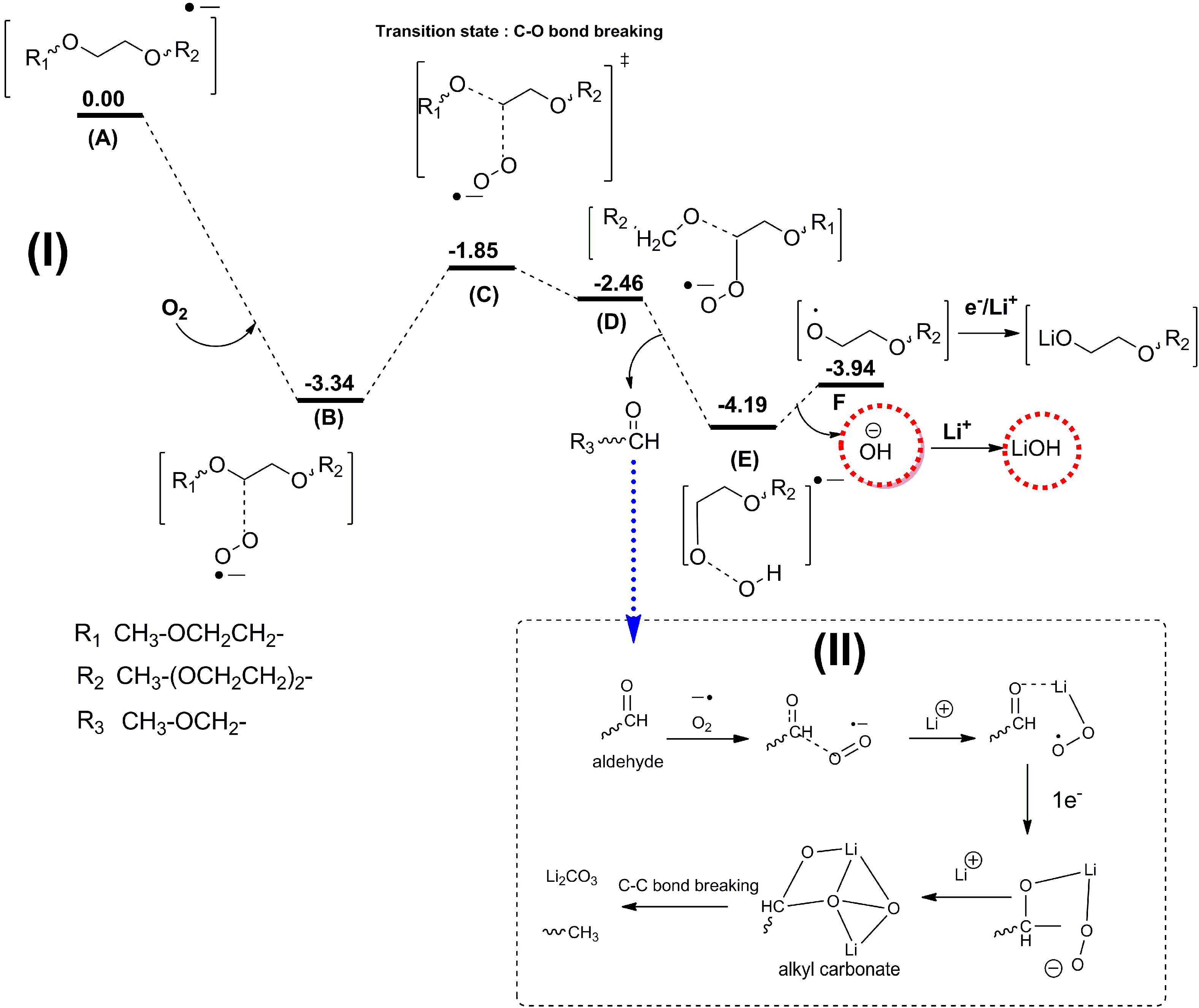
As discussed above, the current major focus of studies on Li-air batteries has been the roles that electrolyte decomposition and the catalytic air cathode play in the cell performance. It is well established that the electrolyte decomposition mainly arises from chemical or electrochemical reactions at the electrolyte-cathode interface and electrolyte-Li2O2 interface. However, another source of detrimental electrolyte decomposition in Li-air batteries, which has not been investigated in much detail yet, could be electrolyte decomposition at the lithium anode. In Li-ion batteries, reactions at the Li anode are a problem (dendrite formation), which is much more severe for Li-O2 batteries considering the presence of the oxygen environment. Because the separators currently used in Li-air cells cannot prevent O2 diffusion to the anode, oxygen crossover can be a significant problem, leading to fast decay of the Li anode.
Recently, Assary
et al. [
12] performed experimental and computational studies focused on the possible reactions that may occur at the lithium anode under conditions used in a TEGDME-based Li-O
2 cell, where oxygen crossover occurs. DFT calculation indicated that the presence of O
2 from an oxygen crossover effect from the cathode to the anode results in favorable subsequent reaction pathways, since the binding of O
2 with the TEGDME anion radical is exothermic by 3.3 eV. Due to the larger electron affinity of TEGDME, the electron is transferred to O
2 upon binding of O
2 to the reduced species. The subsequent cleavage of the C–O bond of the ether by these O
2 anion radicals is no apparent barrier due to the large exothermicity of the first step (A → B), as shown in
Figure 17. This reaction pathway can lead to the formation of various alkoxy radicals, the hydroxide ion, and aldehydes. The hydroxide ion can then bind to a Li cation in solution to form LiOH and, as the concentration of this species increases, it will result in the formation of insoluble LiOH on the anode. Further reduction and reaction with O
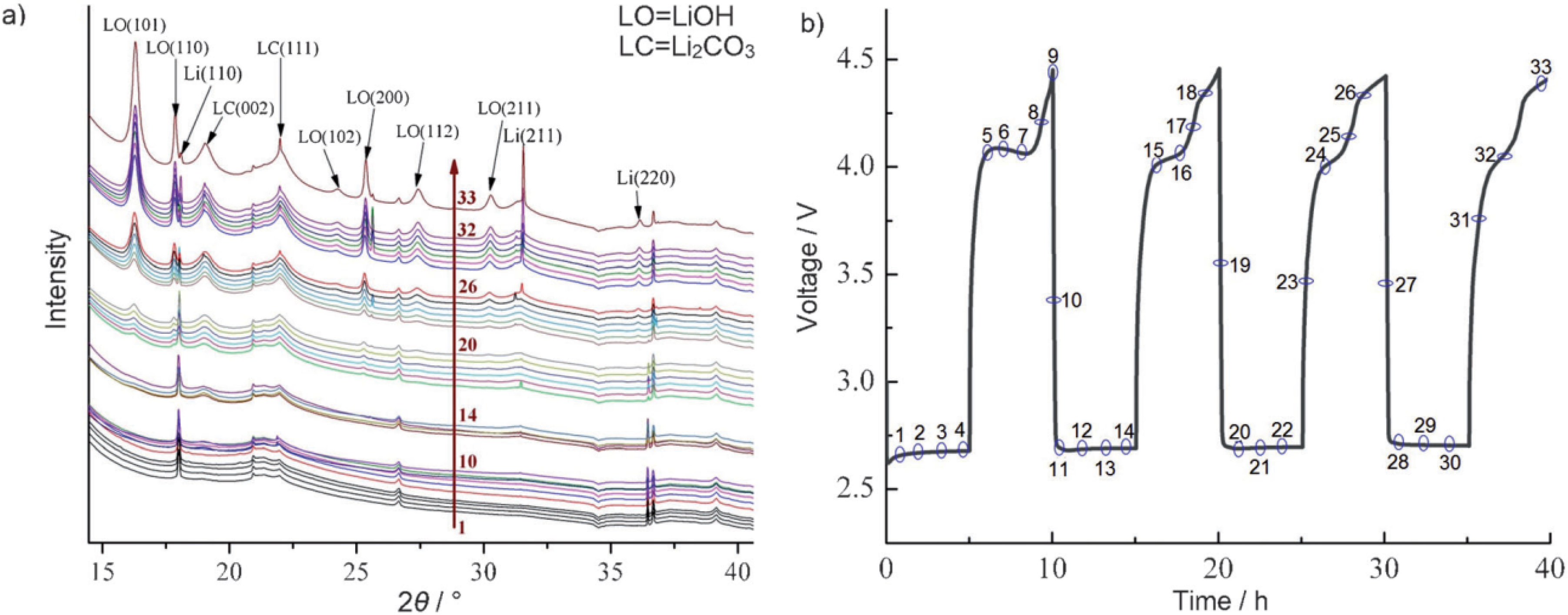
2 of the other fragments (alkoxy radicals and aldehydes) can lead to the formation of lithium alkylcarbonates or lithium carbonates, which may end up on the Li anode as well. In order to investigate potential products that are formed at the anode in a Li-air battery, they have carried out a detailed characterization study of the anode during various stages of discharge and charge using
in-situ XRD measurements. The
in-situ XRD data shown in
Figure 18 clearly indicate the formation of LiOH and Li
2CO
3 on cycling, which confirm the theoretical prediction as discussed above. This study provides evidence that controlling reactions of electrolytes at the lithium anode through suitable membranes or passivation films is essential for achieving good performance of Li-air cells.
Figure 17.
Proposed mechanism and energetics for the formation of LiOH from an ether-based solvent (TEGDME) at the anode upon reduction and oxygen crossover. Also shown in the box is a mechanism for the further formation of lithium alkyl carbonates and carbonates from resulting aldehydes. All energetics are changes in enthalpies (eV) in solution (
ε = 21) at 298 K computed at the B3LYP/6-31+G(d) level of theory (see supporting information for details of computation in [
12]). Reprinted with permission [
12]; Copyright 2013, Wiley-VCH Verlag GmbH& Co. KGaA, Weinheim.
Figure 17.
Proposed mechanism and energetics for the formation of LiOH from an ether-based solvent (TEGDME) at the anode upon reduction and oxygen crossover. Also shown in the box is a mechanism for the further formation of lithium alkyl carbonates and carbonates from resulting aldehydes. All energetics are changes in enthalpies (eV) in solution (
ε = 21) at 298 K computed at the B3LYP/6-31+G(d) level of theory (see supporting information for details of computation in [
12]). Reprinted with permission [
12]; Copyright 2013, Wiley-VCH Verlag GmbH& Co. KGaA, Weinheim.
Figure 18.
(
a)
In situ XRD patterns of Li anode and LiOH formation during the operations conditions; and (
b) corresponding voltage
vs. time profile. Numbers on XRD data correspond to those on voltage profile. Reprinted with permission [
12]; Copyright 2013, Wiley-VCH Verlag GmbH& Co. KGaA, Weinheim.
Figure 18.
(
a)
In situ XRD patterns of Li anode and LiOH formation during the operations conditions; and (
b) corresponding voltage
vs. time profile. Numbers on XRD data correspond to those on voltage profile. Reprinted with permission [
12]; Copyright 2013, Wiley-VCH Verlag GmbH& Co. KGaA, Weinheim.
To eliminate the O
2 crossover to the anode responsible for limiting the reversibility of Li-air cells, it is critical to develop thin, active membranes that can be embedded within passive porous polymeric membranes, which serve as the supports. These composite membranes should meet the following criteria for successful implementation in a rechargeable Li-air cell: (1) block diffusion of oxygen from the air cathode to the lithium anode; (2) allow the transport of lithium ions to support current flow; and (3) exhibit excellent mechanical flexibility and stability to be compatible with the mechanical flexibility of the supporting polymer membranes and battery design/processing. Engineering the active membranes with a nanometer-scale thickness could potentially meet these criteria. As an example, nanoribbons of single-crystal silicon with thickness of 100 nm exhibit buckled geometries and mechanical flexibilities on a rubber substrate, as shown in
Figure 19 [
82].
Figure 19.
Structure nanoribbons of single crystal silicon with a thickness of 100 nm (scale bar 100 µm). Reprinted with permission [
82]; Copyright 2006, Nature Publishing Group.
Figure 19.
Structure nanoribbons of single crystal silicon with a thickness of 100 nm (scale bar 100 µm). Reprinted with permission [
82]; Copyright 2006, Nature Publishing Group.
Truong
et al. [
11] fabricated single-crystal silicon membranes with high lithium conductivity and tested them in Li-air batteries. Their results demonstrated that the single-crystal Si membranes are capable of conducting Li
+ ions through their lattices to support current density as high as 1 mA/cm
2. However, compared with the best NASICON-type lithium ion conducting membranes (LICMs), the Li
+ conductivity of the single-crystal Si membranes is still too low (3–4 orders lower). Their ionic conductivity might be improved by doping the surface lattices of single-crystal Si membranes, lowering the interfacial energy barriers for Li
+ insertion/extraction from the Si lattices, or partially lithiating the single-crystal Si membranes to increase the concentration of Li
+ ions in the membranes and thereby reduce their diffusive resistance. Direct contact of the single-crystal Si membranes with Li metal should be avoided due to the severe electrochemical reactions, although the Si membranes are stable in many electrolytes, including neutral/acidic aqueous electrolytes and aprotic organic electrolytes. More detail about Li-ion conducting membranes for the Li-air battery application can be found in a recent review article by Sun [
29].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}