A High Capacity Li-Ion Cathode: The Fe(III/VI) Super-Iron Cathode

Department of Chemistry and Institute of Basic Energy Science and Technology, George Washington University, Washington, DC 20052, USA
Energies 2010, 3(5), 960-972; https://doi.org/10.3390/en3050960
Submission received: 31 March 2010
/
Accepted: 12 April 2010
/
Published: 6 May 2010
(This article belongs to the Special Issue Lithium-ion Batteries)
Abstract
:A super-iron Li-ion cathode with a 3-fold higher reversible capacity (a storage capacity of 485 mAh/g) is presented. One of the principle constraints to vehicle electrification is that the Li-ion cathode battery chemistry is massive, and expensive. Demonstrated is a 3 electron storage lithium cathodic chemistry, and a reversible Li super-iron battery, which has a significantly higher capacity than contemporary Li-ion batteries. The super-iron Li-ion cathode consists of the hexavalent iron (Fe(VI)) salt, Na2FeO4, and is formed from inexpensive and clean materials. The charge storage mechanism is fundamentally different from those of traditional lithium ion intercalation cathodes. Instead, charge storage is based on multi-electron faradaic reduction, which considerably enhances the intrinsic charge storage capacity.
1. Introduction
Vehicle electrification provides significant advantages of fuel efficiency, which will decrease greenhouse gas emission, decrease the dependence on fossil fuel resources, and facilitate the transition to the renewable energy economy. However the rate of societal transition to electric vehicles is constrained by the low travel range and high battery cost of these vehicles. One of the principle constraints to vehicle electrification is that the Li-ion cathode battery chemistry is massive, and expensive. Demonstrated here is a transformative 3e- storage lithium cathodic chemistry, and a reversible Li super-iron battery with 3-fold higher capacity than contemporary cathodes. This super-iron Li-ion cathode has a storage capacity of 485 mAh/g, consists of the hexavlanent iron (FeVI)) salt, Na2FeO4, and is formed from inexpensive and clean materials. The charge storage mechanism is fundamentally different from those of traditional lithium ion intercalation cathodes. Instead, charge storage is based on multi-electron faradaic reduction, which considerably enhances the intrinsic charge storage capacity.
The introduction of Li-ion systems has substantially increased electrochemical energy storage capacity [1]. Yet, contemporary rechargeable lithium batteries have only one fifth the volumetric energy density of gasoline, and require five times the gas tank volume to travel the same distance [2]. The cathode comprises the main mass component of contemporary Li-ion batteries. For example, the commonly used LiCoO2 cathode and Li-Co-Mn-Ni variants have capacities of 80–150 mAh/g, but are cost limited by the relative scarcity and high price of cobalt (which is up two orders of magnitude more expensive than iron). The cobalt is associated with significant toxicity hazards.
An alternative to LiCoO2 cathode, LiMn2O4, has an even lower capacity of 100 mAh/g. A popular new cathode LiFePO4, contains divalent iron, Fe(II), and can store up to 170 mh/g [3,4]. An attraction of this cathode is the availability and low cost of the source reagents (iron is the most common metal in the earth’s crust). LiFePO4 can sustain higher power densities than equivalent cobalt or manganese cathodes. However, Li-ion LiFePO4 batteries operate at lower voltage than those with cobalt and manganese based cathodes, and also have approximately 20% lower energy storage density.
Development of 3e- Fe(VI) charge storage. An unusual class of iron salts was pioneered by our group as inorganic charge storage salts in 1999, and named super-irons due to their highly oxidized hexavalent iron state [5,6].
Key milestones in the super-iron battery development to date are of Table 1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Development | Reference |
|---|---|---|
| 1999 | introduction of super-iron charge storage & super-iron alkaline battery | [5] |
| 2000** | introduction of super-iron lithium primary (single discharge) battery | [7] |
| 2001 | demonstration of the solid state stability of the hexavalent iron | [8] |
| 1999-5 | chemical syntheses of an array of super-iron salts | [5,7,9,10,11,12,13,14,15,16] |
| 2000-4 | inexpensive, electrochemical syntheses of super-iron salts | [17,18,19,20,21,22,23,24,25,26] |
| 2003-5** | electrolyte optimization for super-iron lithium batteries | [27,28] |
| 2003 | reversibility of alkaline, nanothick (3 nm) Fe(VI) cathodes | [29] |
| 2006 | rechargeable alkaline super-iron battery | [30] |
| 2006** | reversibility of non-aqueous, nanothick (3 nm) Fe(VI) cathodes | [31] |
| 2007-8 | zirconia encapsulation–stabilization of alkali super-irons | [32,33,34,35] |
| 2009** | rechargeable super-iron lithium battery, 4 V cathode | [6] |
**=lithium super-iron battery development.
2. Results and Discussion
2.1. The challenge of facile Fe(VI) charge transfer
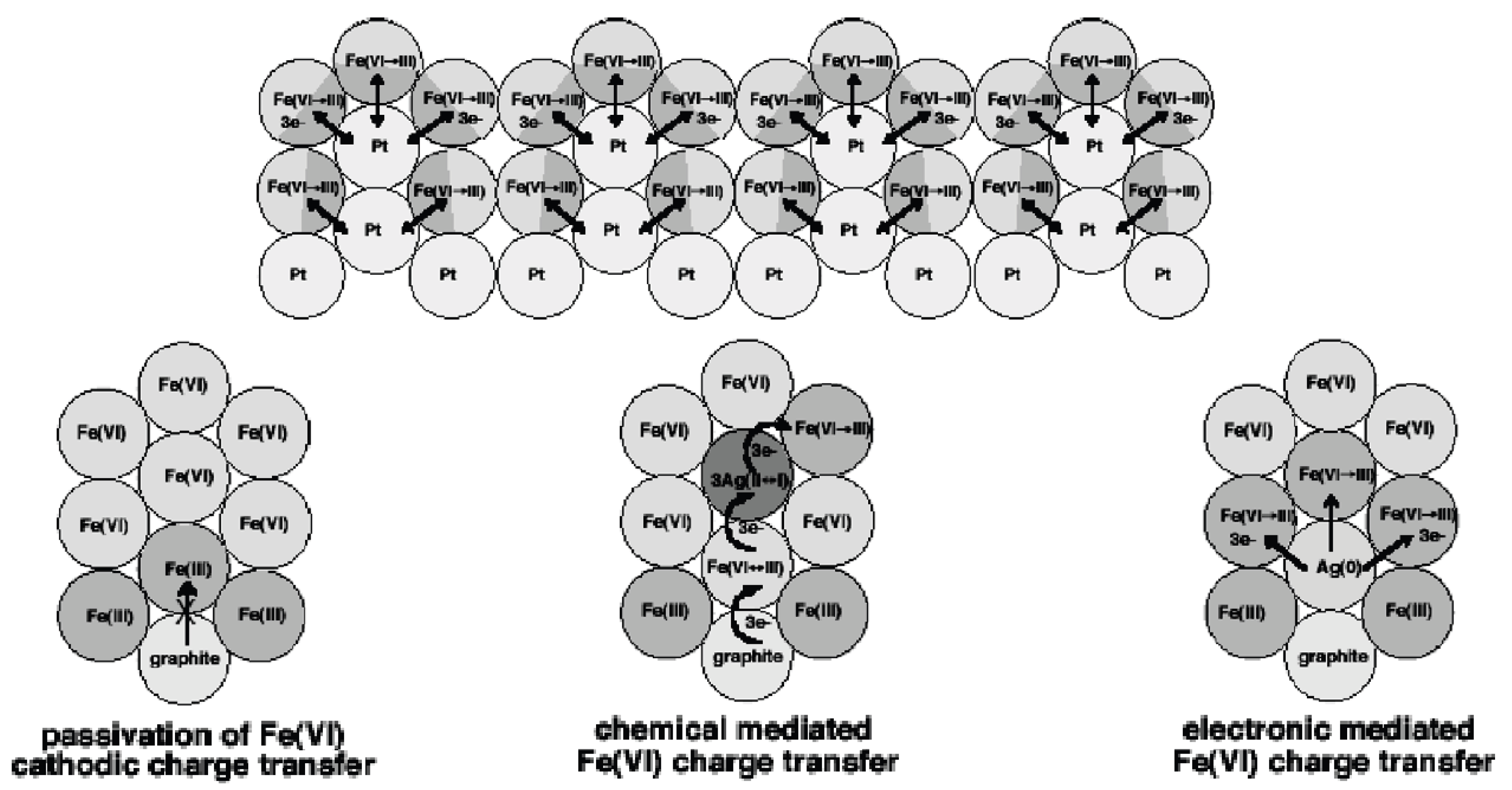
The principal limitation to facile Fe(VI) charge transfer has been passivation of the couple due to resistive buildup of low-conductivity ferric (Fe(III)) salts, as schematically represented in the lower left corner of Scheme 1 [32,36,37]. We have demonstrated that the addition of simple transition metal oxides, such as manganese or silver oxide (shown in the scheme), to form a composite with alkali or alkali earth super-irons, considerably facilitates the rate of super-iron charge transfer through chemical and mediation of charge transport mechanisms [38,39,40,41,42].
Chemical mediation acts to displace passivating Fe(III) centers into the bulk and away from the current collectors, while the electronic mediation provides alternative, more conductive pathways for charge transport. We have also demonstrated that a zirconia overlayer facilitates alkal Fe(VI) charge transfer in alkali media [32,33,34,35]. Most recently, we have also shown that an external conductive matrix, such as platinized, platinum considerably enhances reversible, non-aqueous Fe(VI) charge transfer [5].
Scheme 1.
Modes of Fe(VI) charge transfer and passivation. Bottom: middle-Chemical mediated, and right-electronic mediated, Fe(VI) charge transfer. Bottom-left: Fe(III) inhibition and passivation of charge transfer. Top: Nanofilm enhanced reversible Fe(VI) charge transfer.
Scheme 1.
Modes of Fe(VI) charge transfer and passivation. Bottom: middle-Chemical mediated, and right-electronic mediated, Fe(VI) charge transfer. Bottom-left: Fe(III) inhibition and passivation of charge transfer. Top: Nanofilm enhanced reversible Fe(VI) charge transfer.

2.2. Reversible non-aqueous 3e- Fe(VI) charge storage
The basis for improved electrochemical storage capacity using the super-iron Li battery:
- (i)
- Fe(VI) nonaqueous charge transfer is constrained by reduction, not intercalation.
- (ii)
- Fe(VI) cathodes store the charge equivalent to 3 electrons per iron center,
- (iii)
- an extended conductive matrix facilitates reversible Fe(VI) reduction,
- (iv)
- the redox Fe(VI) potential is 0.25 V larger than that of Li-Mn or Li-Co cathodes.
The Fe(VI) redox storage potential versus lithium has increased from an observed 3 V, to over 4 V, in the past 3 years. With advent of the 485 mAh/g lithium non-aqueous cathode, and with use of the conductive matrix, the reversible, non-aqueous super-iron has increased 200 fold in thickness (from 3 nm to over 600 nm).
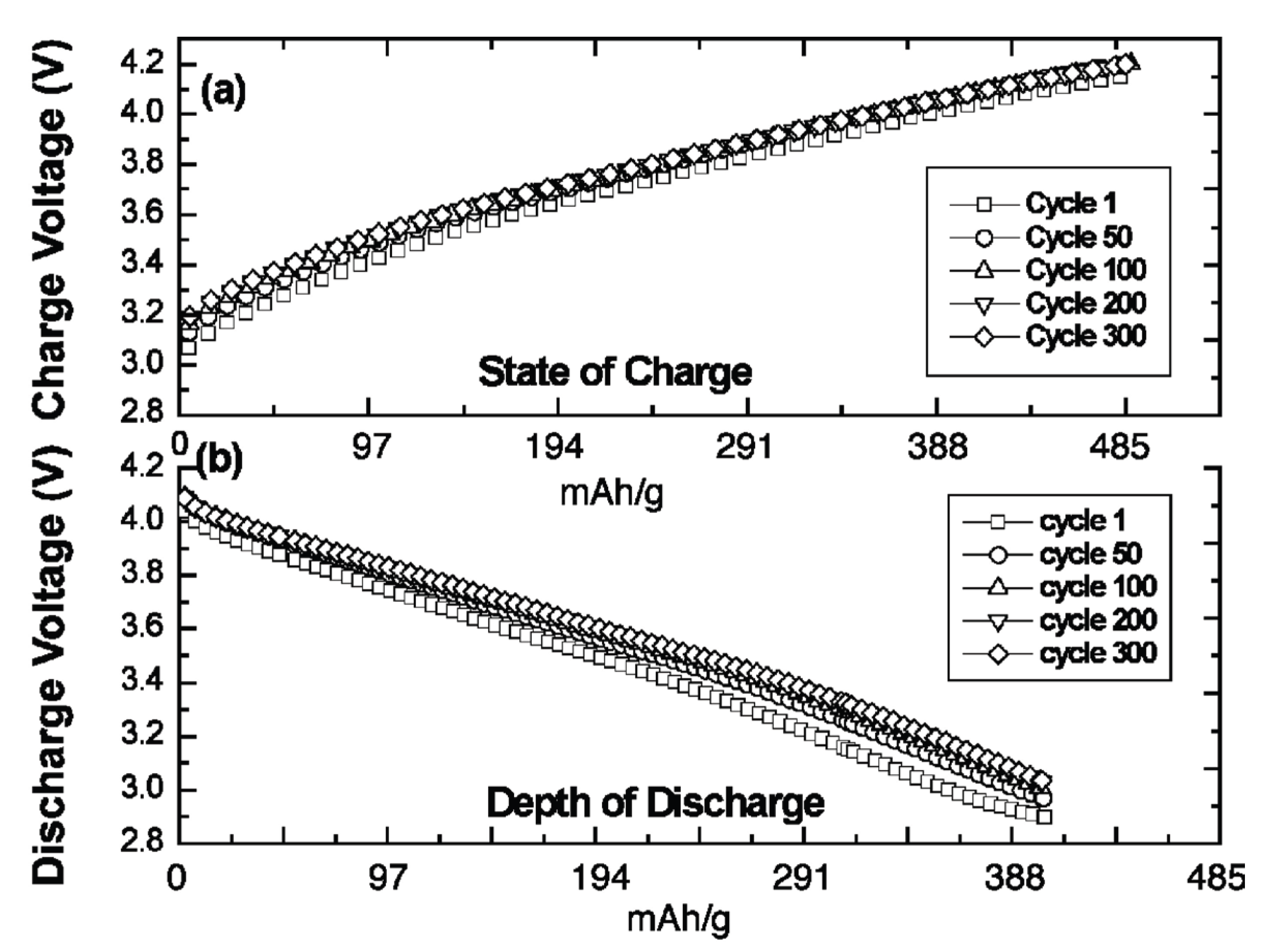
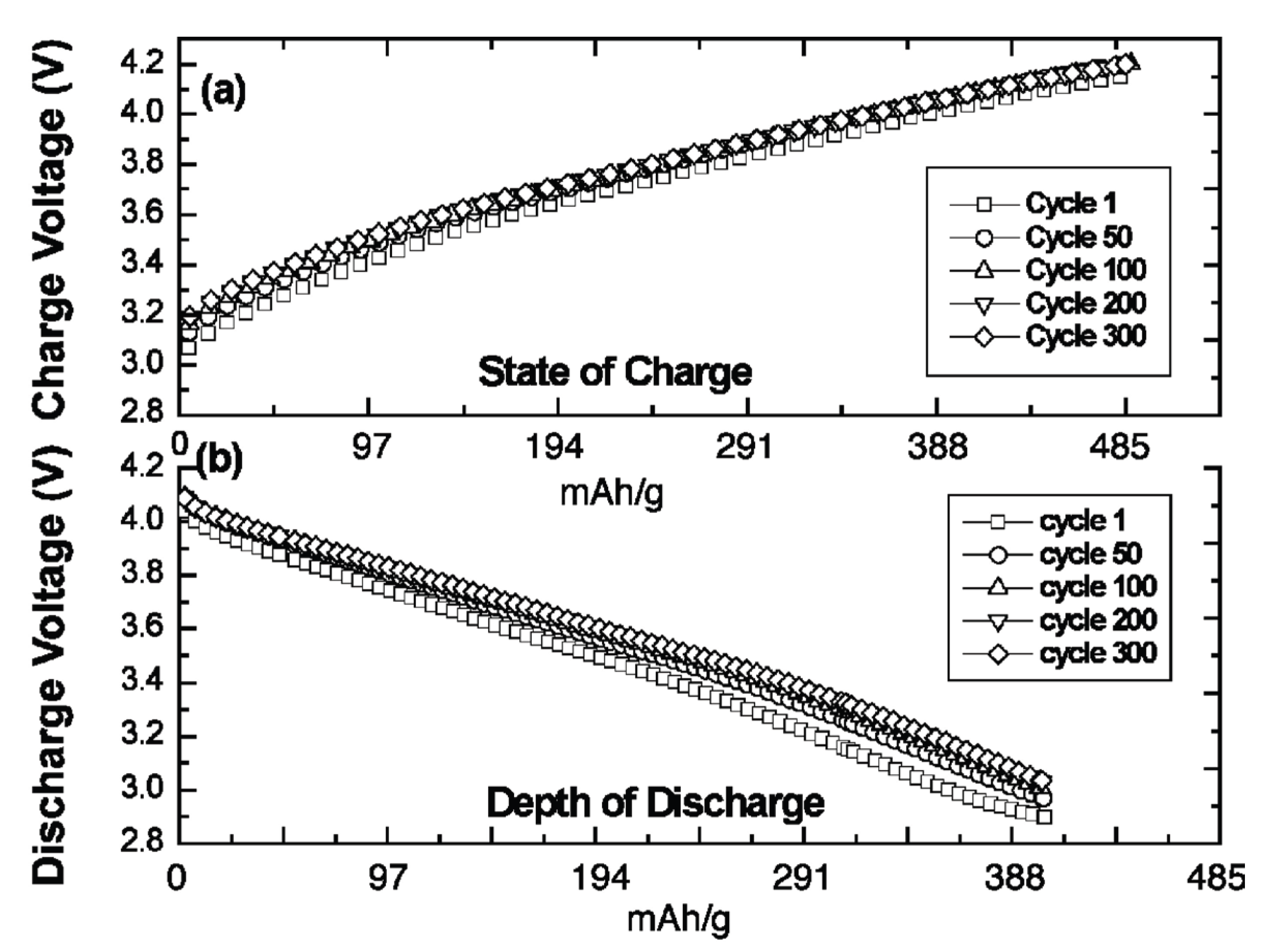
In 2009, we presented the first reversible super-iron Li batteries, cells operating at high voltage (over 4 volt) and a cathode capacity of 485 mAh/g, several fold higher than the cathodes in contemporary Li-ion batteries [6]. The capacity is based on the intrinsic three electron storage of Na2FeO4, 50 nm thick cathodes were cycled at over 90% DOD (depth of discharge), and 191 and 573 nm thick cathodes were cycled at over 80% (400 mAh/g). Charge and discharge voltage during 300 recharge cycles are shown in Figure 1.
Figure 1.
Extended galvanostatic cycling charge-transfer behavior for a super-iron Li battery containing a 191-nm thick Fe(III/VI) Na2FeO4 cathode film. (a) Cell potential during charge and (b) cell potential during discharge. Charge/discharge cycle numbers are indicated. Results are detailed in Licht, Wang, Gourdin, Enhancement of Reversible Nonaqueous Fe(III/VI) Cathodic Charge Transfer, Journal of Physical Chemistry, C, 133, 9884–9891 (2009).
Figure 1.
Extended galvanostatic cycling charge-transfer behavior for a super-iron Li battery containing a 191-nm thick Fe(III/VI) Na2FeO4 cathode film. (a) Cell potential during charge and (b) cell potential during discharge. Charge/discharge cycle numbers are indicated. Results are detailed in Licht, Wang, Gourdin, Enhancement of Reversible Nonaqueous Fe(III/VI) Cathodic Charge Transfer, Journal of Physical Chemistry, C, 133, 9884–9891 (2009).
Rather than the typical intercalation mechanism of conventional Li and Li-ion cathodes, the super-iron discharge involves reduction from Fe(VI to III), as confirmed by AA, Mossbauer and charge measurements in the electrochemical processes. To date this was demonstrated as 3 Faraday per mole of Na2FeO4 (an intrinsic 485 mAh/g capacity), K2FeO4 (an intrinsic 408 mAh/g capacity) or BaFeO4 (314 mAh/g) in accord with the discharge mechanism [6]:
MFe(VI))O4 + 3e– + 3Li+ → MO + 3/2Li2O + 1/2Fe(III)2O3, M= K2, Na2 or Ba
2.3. Preparation of Super-Iron Cathode Films
The electrochemical preparation of Fe(III/VI) super-iron thin films cathodes on an extended conductive matrix significantly facilitates such film’s nonaqueous, reversible charge transfer. Fe(VI) salts can exhibit higher cathodic capacity and environmental advantages, and the films are of relevance toward the next generation charge storage chemistry for reversible cathodes. These films were electrochemically deposited by electrochemical reduction of Na2FeO4, which retains an intrinsic 3 e– cathodic charge storage capacity of 485 mAh g–1. The influence on cathode reversibility, capacity and charge retention was probed as function of film deposition conditions (including the deposition potential and stirring rate, and the concentration of NaOH and K2FeO4, in the deposition electrolyte).
Initially to optimize deposition conditions, super-iron films were electrodeposited from various alkaline K2FeO4/NaOH solutions at an applied potentiostatic voltage of 0.11 V vs. Ag/AgCl in a voltammetric Plexiglas cell, and optimized as a function of solution composition. Subsequently, super-iron films were consistently electrodeposited from 50 mM K2FeO4 dissolved in stirred (magnetic bar), 8.0 M NaOH in a voltammetric Plexiglas cell at a galvanostatic current of 10.0 mA. The working electrode was a 1 cm2 platinum disc or a 1 cm2 platinized, platinum disc. The auxiliary electrode was a platinum rod, and the reference electrode was an Ag/AgCl/ KCl (sat) encased in a 0.1 M NaNO3 jacket. Solutions were initially de-aerated with nitrogen gas for a minimum of 5 min prior to the electro-deposition. A N2 gas atmosphere was maintained over the solution during the electrodeposition. The film electrode was carefully rinsed with 8.0 M NaOH solution, air dried for 30 minutes, and then vacuum dried for 2 days prior to use.
During the optimization of the electrodeposition process, the surface of the working electrode was rinsed at each end of the experiment by an oxidative linear potential scan in 1.0 M HCl solution. Employed voltammetric Plexiglas cells were consistently, cleaned by soaking overnight in a solution of 10–2 M nitric acid, followed by DI water rinse, and results presented are the average of three replicate measurements. For Fe(III/VI) film formation, at the optimized deposition potential employed, the electrodeposition via the 3 electron reduction of Fe(VI) to Fe(III) substantially dominates, permitting the current efficiency to be assumed as 100%. Hence, the intrinsic capacity of the super-iron films determined by integrating the current-time response curve, Q(C = coulombs = ampere seconds), provides a quantitative measure of the intrinsic charge capacity of the super-iron films and, for convenience, a relative (not absolute) measure of the film thickness. The relative comparison of film thicknesses, T, is quantitative for all compared electrodes. For example a 19 nm Fe(III/VI) films contains 69 nanaomole of Fe per cm2, and is capable of storing 20 mC of intrinsic capacity per cm2 (based on the observed 3 electrons of storage per Fe(III/VI) center). Similarly, thicker deposited 57, 191 and 573 nm Fe(III/VI) films used in this study, have an intrinsic storage capacity respectively of 60, 202 and 605 mC per cm2.
The solid state stability (stable to > 99.9% year retention of the Fe(VI) valence state), and storage time, for K2FeO4 is much greater than for Na2FeO4, and hence it has been our chemical dissolution salt of choice. In this study, the effects on Fe(III/VI) charge storage on smooth platinum were conducted by varying the concentrations of [K2FeO4], [NaOH], electrodeposition potential and magnetic stirring rate, respectively.
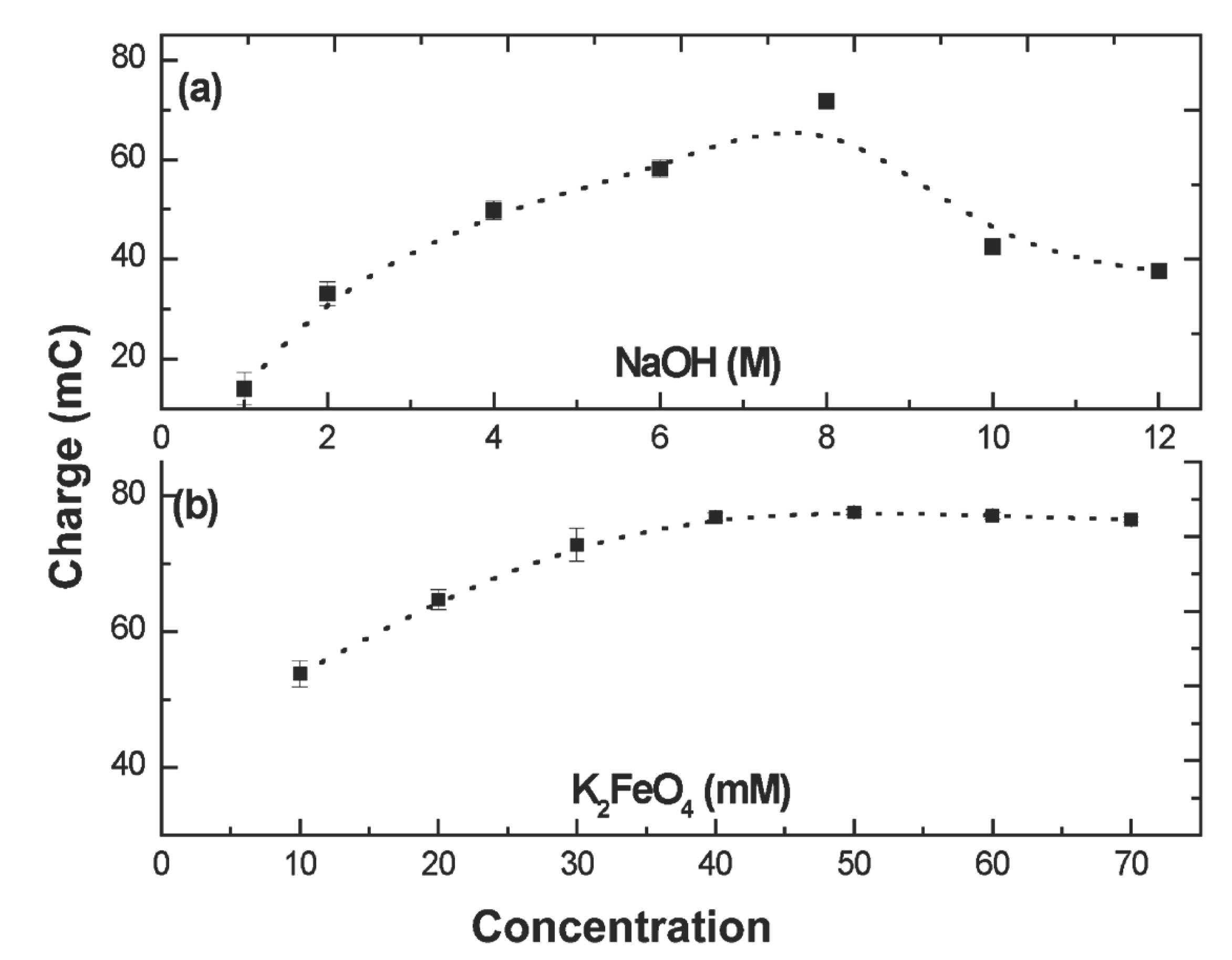
Figure 2a presents the effect of the electrolyte, NaOH, concentration on charge storage in 50 mM K2FeO4. It is seen that the Fe(III/VI) charge storage capacity increased with increasing the concentration of NaOH from 1.0 to 8.0 M, while an increase beyond 8.0 M NaOH led to a decrease of Fe(III/VI) charge storage. Increasing NaOH concentration (from 1.0 to 8.0 M) will inhibit the K2FeO4 solution phase decomposition process (Equation 7), stabilizing the MFeO4– or FeO42– species (Equations 12 and 13), and as a result, the Fe(III/VI) charge storage is increased. In competition with this is the decrease in equivalent ionic conductivity of hydroxide with increasing concentration. For the NaOH electrolyte, this decrease is significant. For example at 18°C the equivalent conduction of NOH solutions, decrease from 160 to 108 S cm2/equivalent, as concentration increases from 1 to 3 M, and the decrease is precipitous in more concentrated NaOH (decreasing to 69 S cm2/ equivalent in 5 M NaOH). Consistent with the observed decrease in charge storage at 8 M NaOH, this decrease in mobility at higher concentrations appears to dominate over the stabilization enhancement. Therefore, in order to obtain a favorable charge storage, a compromise between decomposition and ion mobility needs to be considered. Figure 2b presents the concentration effect of K2FeO4 on charge storage in 8.0 M NaOH. It was found that the storage charge increased with increasing the concentration of K2FeO4 until a plateau was formed about 45 mM of K2FeO4, which approaches the saturation point of K2FeO4 in 8.0 M NaOH. In subsequent experiments in this study, 8.0 M of NaOH and 50.0 mM of K2FeO4 were used in the following experiments, except in noted special cases. In this concentrated alkaline environment diffusion processes should be facilitated. Hence, variation of the (magnet bar) stirring rate was also probed, and generally, the higher the stirring rate, the greater the charge storage which can be obtained in the deposited Fe(III/VI) films; this is consistent with the expected compression of the diffusion layer with an increase in solution turbulence.
Figure 2.
Electrodeposition optimization of a Fe(III/VI) film. Deposition conditions—applied potential: 110 mV versus Ag/AgCl; deposition time 10 s on a smooth platinum electrode; stirring rate: maximum, without disruptive turbulence: (a) the effect of NaOH concentration on charge storage in 50 mM K2FeO4 and (b), the effect of K2FeO4 concentration on charge storage in 8.0 M NaOH.
Figure 2.
Electrodeposition optimization of a Fe(III/VI) film. Deposition conditions—applied potential: 110 mV versus Ag/AgCl; deposition time 10 s on a smooth platinum electrode; stirring rate: maximum, without disruptive turbulence: (a) the effect of NaOH concentration on charge storage in 50 mM K2FeO4 and (b), the effect of K2FeO4 concentration on charge storage in 8.0 M NaOH.

2.4. Characterization of Super-Iron Cathode Films
The 3-electron reduction of Fe(VI) can produce a variety of Fe(III) oxide and oxyhydroxide species such as (α,γ Fe2O3) and (α,γ,β,δFeOOH). Sodium, over potassium, species will dominate in the 8 M N+, 0.01 M K+ deposition solution, and a variety of cation-containing ferric salts such NaFeO2 are also possible in the film. Well-defined Fe2O3 particles gave three fundamental bands ranged from 500 cm–1, 400 cm–1 to 300 cm–1 respectively, and the bands shifted with varying in size, shape, internal structure and aggregation of particles. In our experiment, the surface morphologies of thin Fe(III) film on smooth platinum were examined by a Nicolet Nexus 670 Fourier Transform Infrared Spectrophotometer.
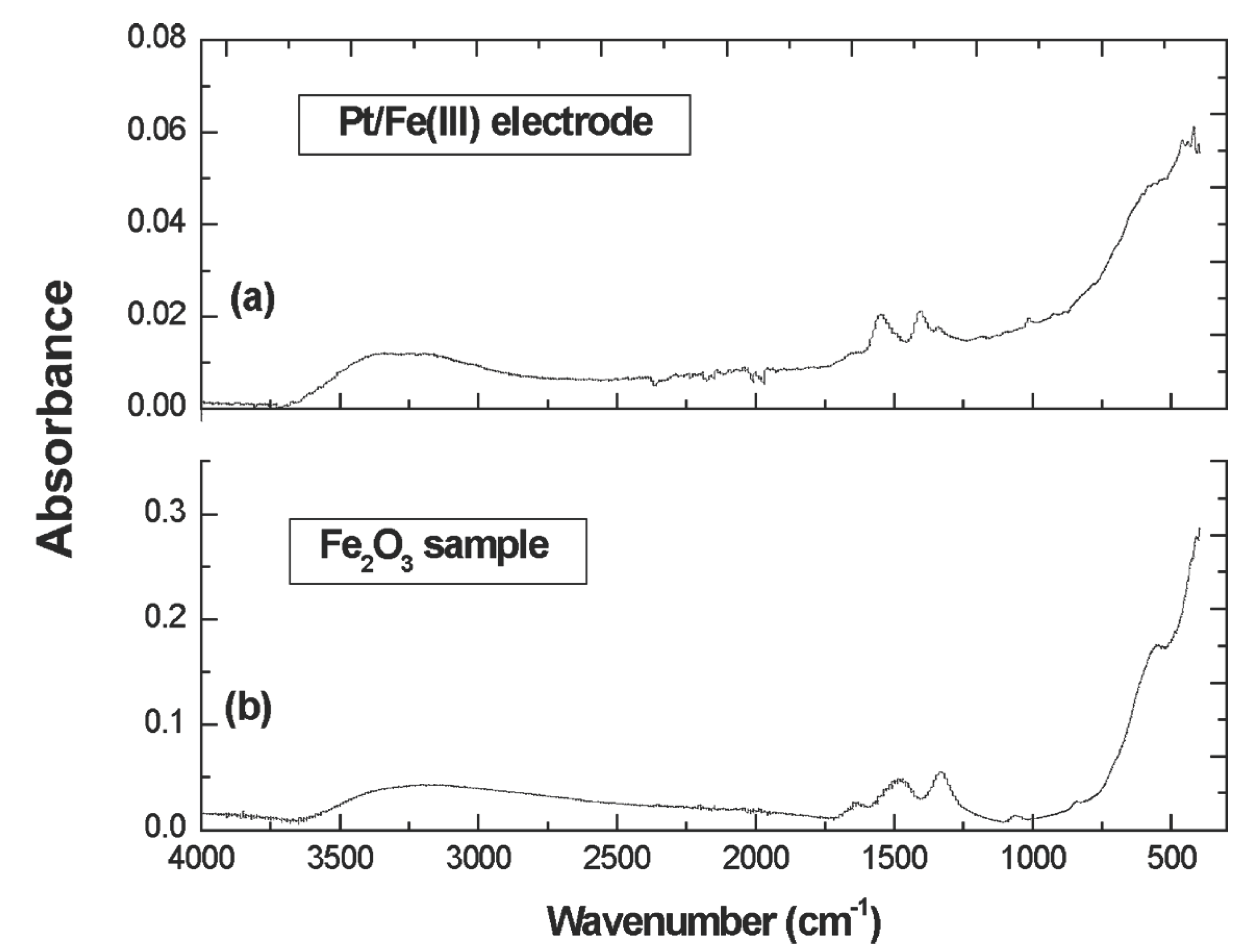
Figure 3a presents the FTIR diffuse reflectance spectra (in absorbance mode) of a 191 nm thin Fe(III) film which was freshly electrodeposited on a 1 cm2 platinum disk. A single peak at ~429 cm–1, and a shoulder near 538 cm–1 are observed (far infrared spectra less than 400 cm–1 was not examined due to the instrumental limitations). For comparison, nanocrystalline γ-Fe2O3 particles was synthesized, and the FTIR absorption spectra of this particle is showed in Figure 3b. Two adjacent peaks near 1500 cm–1 appear to be associated with residual free hydroxide.The strong similarity between our Fe(VI) electrodeposited film Fe(III) and the nanocrystalline γ-Fe2O3 particles indicates this as a principal component of the Fe(III) film. In ongoing investigations, we continue to probe the identity of the Fe(III) centers in the reduced form of the film Fe(III/VI) films.
Figure 3.
FTIR diffuse reflectance spectrum (in absorbance mode) of (a) a Pt/Fe(III) electrode and (b) a Fe2O3 sample. The electrodeposition conditions of Pt/Fe(III) are the same as above with 50 mM K2FeO4 in 8.0 M NaOH. The Fe2O3 samples are freshly synthesized.
Figure 3.
FTIR diffuse reflectance spectrum (in absorbance mode) of (a) a Pt/Fe(III) electrode and (b) a Fe2O3 sample. The electrodeposition conditions of Pt/Fe(III) are the same as above with 50 mM K2FeO4 in 8.0 M NaOH. The Fe2O3 samples are freshly synthesized.
The Fe(III/VI) films were placed in a lithium cell with 1M LiPF6 in PC: DME (1:1) electrolyte, and their quasi-reversibility characterized as a function of hydroxide and Fe(VI) concentrations in the deposition solution. Specifically, a 191 nm Fe(III/VI) on smooth platinum was galvanostically deposited (at 10 mA, for 10s) in either 10, 50 or 80 mM K2FeO4. The film was placed in a lithium cell with 1M LiPF6 in PC: DME (1:1) electrolyte, and cycled through periodic, galvanostatic charge/discharge cycles. Specifically, each cell was repeatedly subject to a 0.02 mA cm–2 charge, followed by a deep 0.01 mA cm–2 discharge.
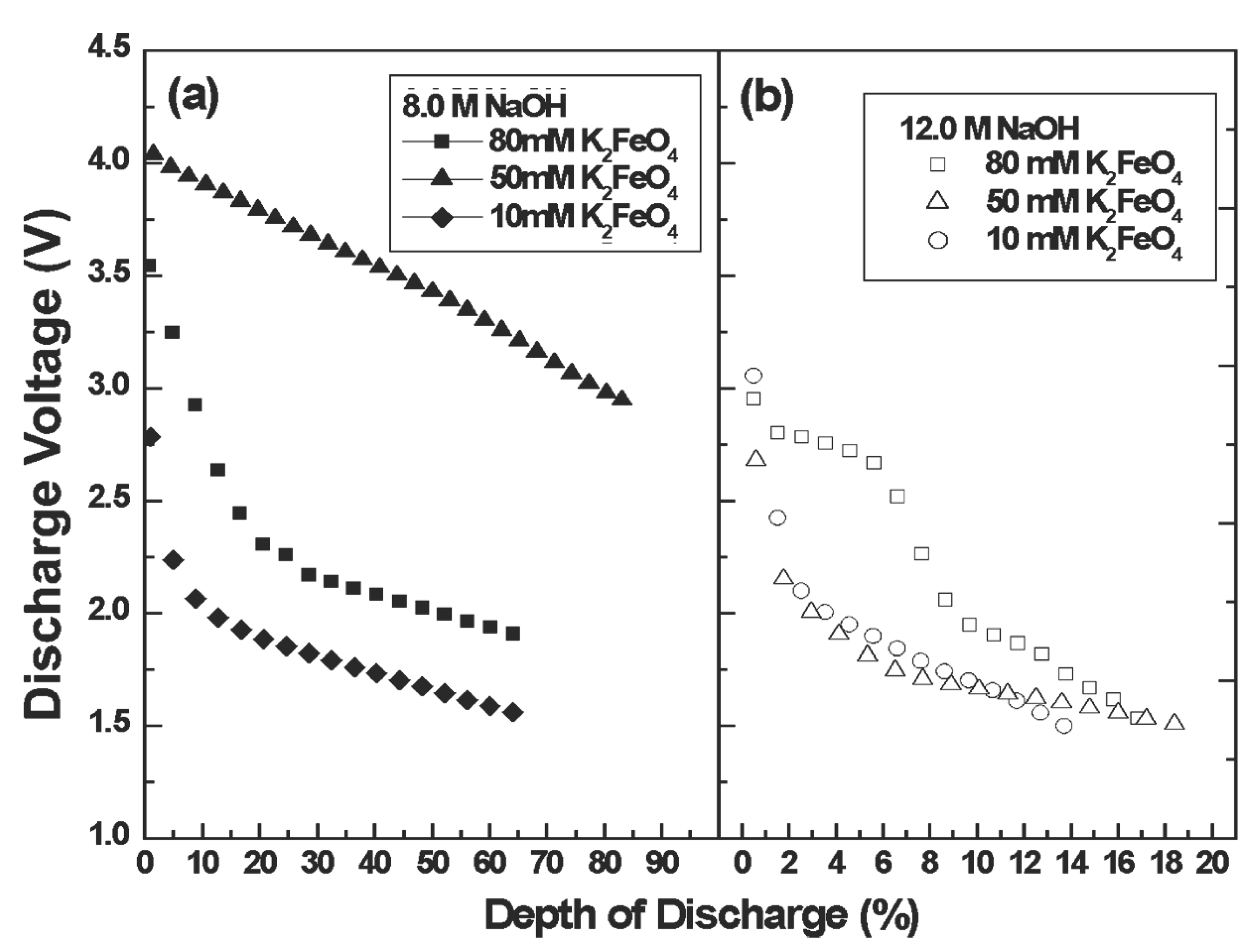
Figure 4 presents the discharge voltage during the 20th discharge cycle as a function of the intrinsic, (100 mC , 3 electron) depth of discharge of these films. Figure 4a,b, respectively presents discharge cycle behavior for films prepared from either 8.0 M (4a) or 12.0 M (4b) deposition solutions. It is evident in Figure 4 that the average discharge voltage, and the depth of discharge are significantly higher for films deposited in 8 M, rather than 12 M, NaOH. Furthermore, in the preferred 8 M NaOH deposition solution, the average discharge voltage, and the depth of discharge are significantly higher for films deposited from 50 mM, rather than 10 or 80 mM K2FeO4 solutions. It is evident that the charge storage and transfer behavior of Fe(III/VI) thin-film cells are significantly influenced by the electrochemical deposition conditions. In the 50.0 mM K2FeO4, 8.0 M of NaOH prepared film, the 20th discharge cycle commenced at 4.1 V and decayed to 3.1 V through an 80% depth of discharge, and no sharp decline of potential was observed within 20 cycles.
Figure 4.
The discharge behavior of a 191 nm Fe(VI) film on a Pt electrode with various deposition conditions at 20th cycle. Films are deposited at a constant current of 10 mA for 10 s from electrolytes containing various [K2FeO4] in 8.0 M (a) or 12.0 M NaOH (b). Subsequent nonaqueous galvanostatic cycling consists of charge at 0.02 mA cm–2 to 100% of the intrinsic 3e– Fe capacity, as limited by a maximum cut0ff voltage of 4.2 V, followed by discharge at 0.01 mA cm–2 to 90% DOD of this capacity as limited by a 1.5 V minimum voltage cutoff.
Figure 4.
The discharge behavior of a 191 nm Fe(VI) film on a Pt electrode with various deposition conditions at 20th cycle. Films are deposited at a constant current of 10 mA for 10 s from electrolytes containing various [K2FeO4] in 8.0 M (a) or 12.0 M NaOH (b). Subsequent nonaqueous galvanostatic cycling consists of charge at 0.02 mA cm–2 to 100% of the intrinsic 3e– Fe capacity, as limited by a maximum cut0ff voltage of 4.2 V, followed by discharge at 0.01 mA cm–2 to 90% DOD of this capacity as limited by a 1.5 V minimum voltage cutoff.
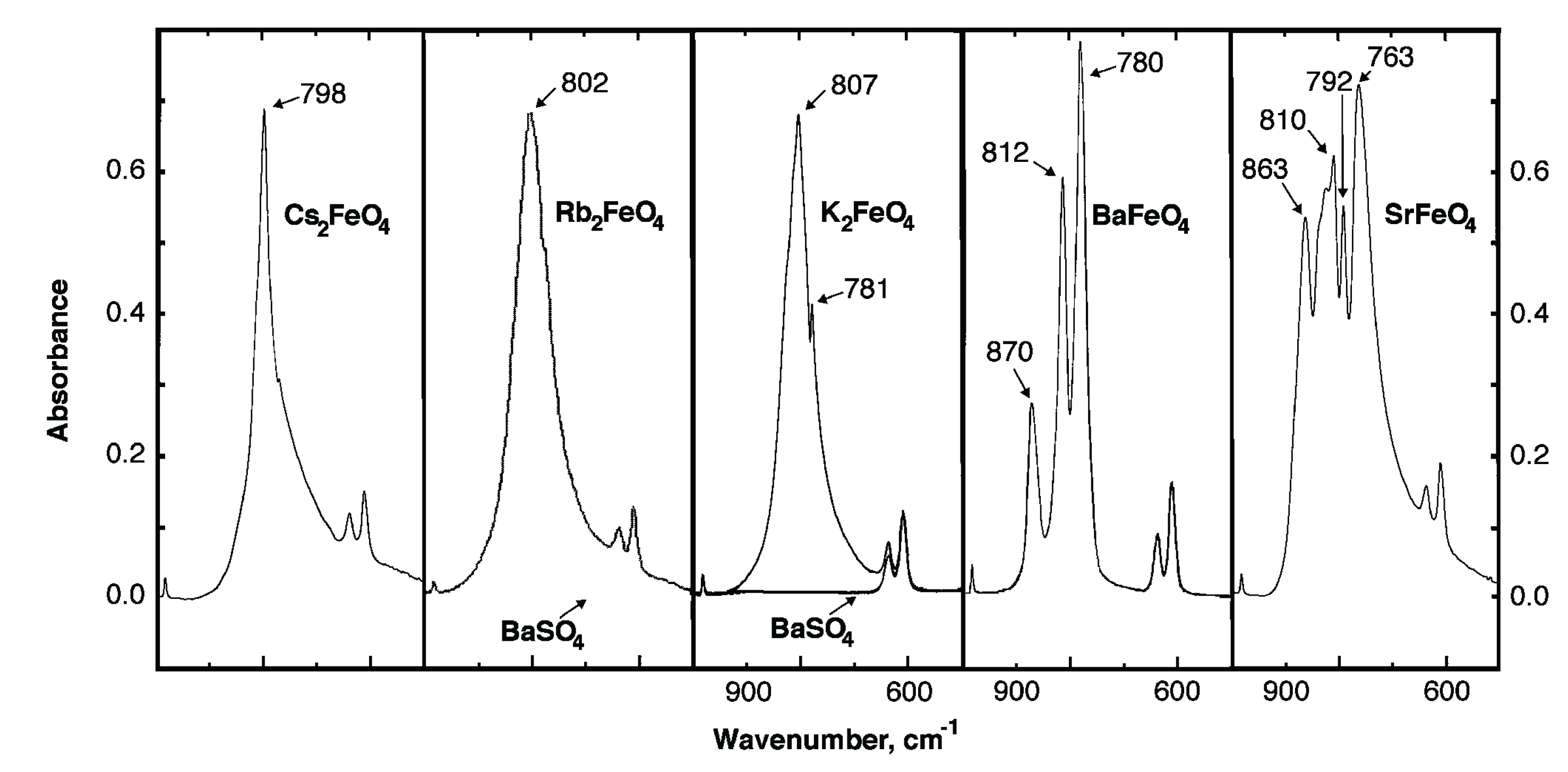
FTIR provides not only a specific "fingerprint" distinguishing the various Fe(VI) oxides, as shown in Figure 5, but importantly we have also developed it as a quantitative technique to determine the Fe(VI) salt purity through the addition of a standardized BaSO4 salt [8]. Discharge of cathode replaced, commercial alkaline button cells provides rapid screening of the redox activity of alternative salts [6,10,11,14,15,19,27,28,29,30,31,32,33,34,35].
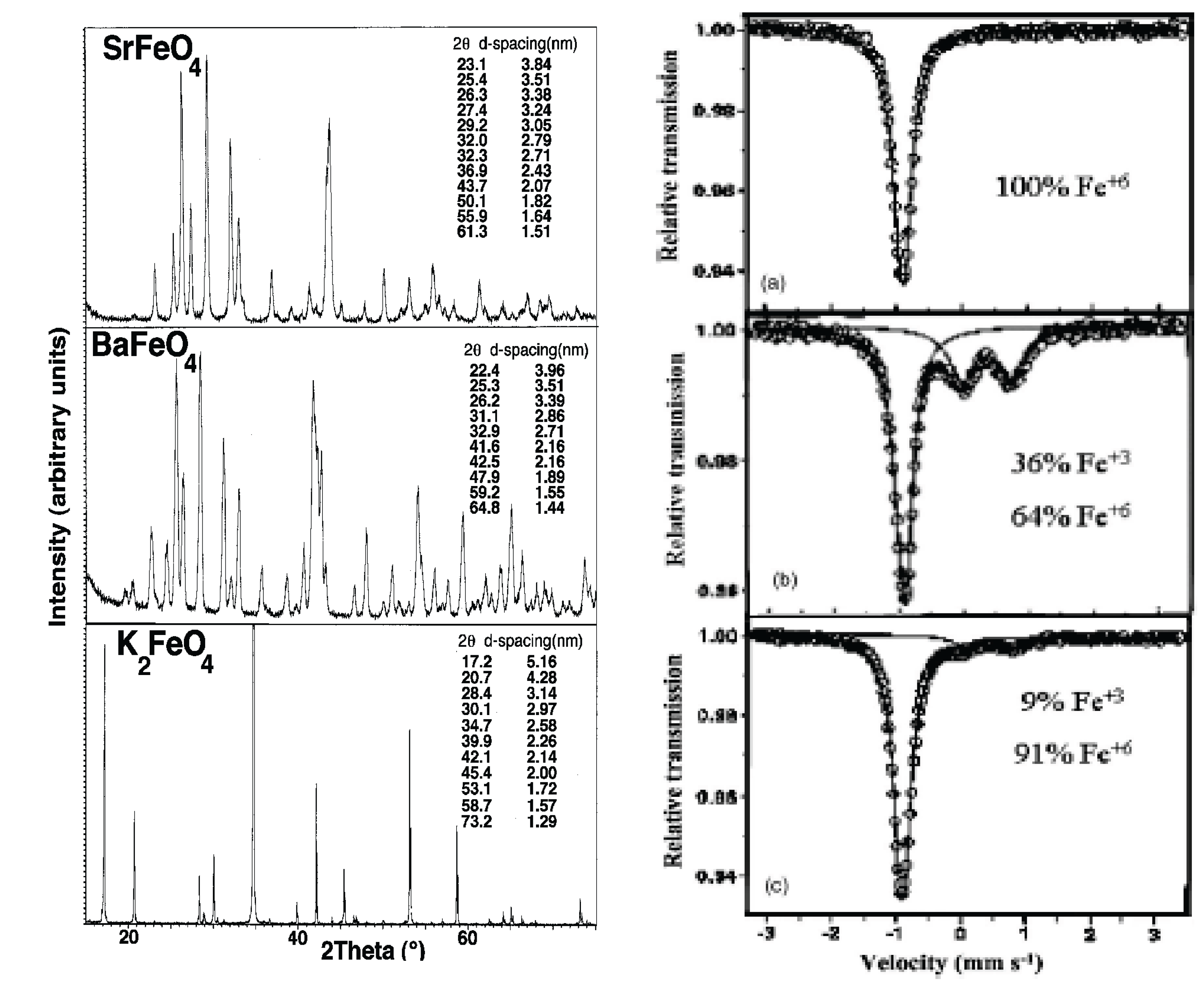
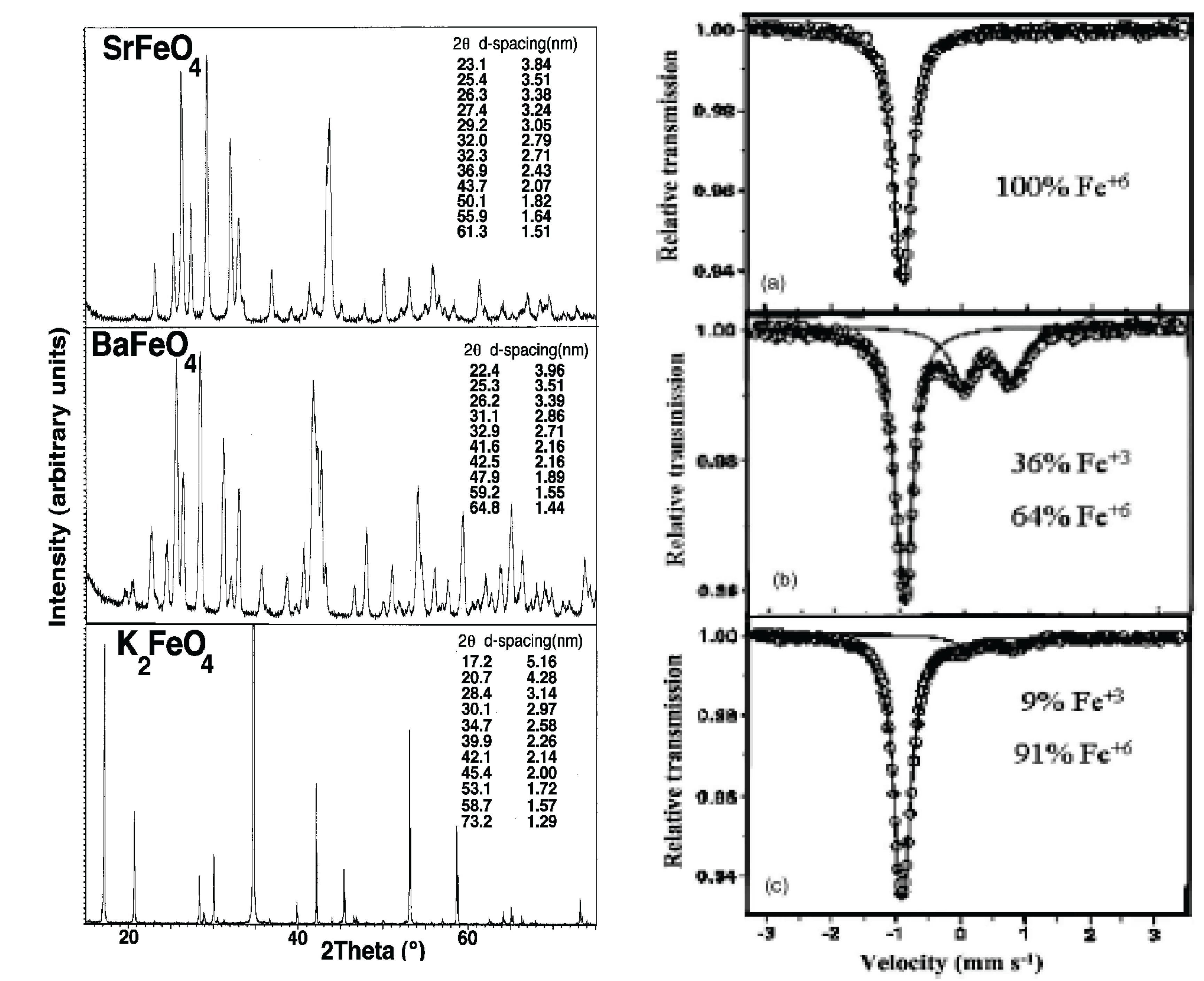
X-ray powder diffraction and Mössbauer are used to distinguish the variation of crystal structure and definitive nature of the iron state of super-iron oxides as a function of the state of charge/discharge of the cycled new cathode salts. As seen in Figure 6, we used these techniques to probe alkali and alkali earth super-iron oxides, and x-ray characterization of these salts has provided specific lattice parameters of their orthorhombic β-K2SO4 analogue structure.
As seen in Figure 1 and Figure 6(right), we have the fascinating case of a cathode which can be reversibly, reformed by faradaic charge transfer, as in aqueous cells, but with the high voltage advantage of the nonaqueous environment.
Figure 5.
IR absorption of solid K2FeO4, Rb2FeO4, Cs2FeO4, BaFeO4, and SrFeO4, mixed with a BaSO4 standard. From Licht, S.; Naschitz, V.; Rozen, D.; Halperin, N. Cathodic charge transfer and analysis of Cs2FeO4, K2FeO4 and mixed alkali Fe(VI), ferrate, super-irons. J. Electrochem. Soc. 2004, 151, A1147–A1151.
Figure 5.
IR absorption of solid K2FeO4, Rb2FeO4, Cs2FeO4, BaFeO4, and SrFeO4, mixed with a BaSO4 standard. From Licht, S.; Naschitz, V.; Rozen, D.; Halperin, N. Cathodic charge transfer and analysis of Cs2FeO4, K2FeO4 and mixed alkali Fe(VI), ferrate, super-irons. J. Electrochem. Soc. 2004, 151, A1147–A1151.

Figure 6.
Left: X-ray analysis of Fe(VI) compounds, evidence of their orthorhombic β-K2SO4 analogue structure, from "Recent advances in the synthesis and analysis of Fe(VI) cathodes." Licht, et al., J. Solid State Electrochem. 2008, 12, 1523. Right: Mössbauer spectra of K2FeO4: (a) pristine electrode, (b) polarized to 1.5V vs. Li/Li+, and (c) after one complete lithiation cycle. From "Study of Various ("super iron") MeFeO4 compounds in Li salt solutions as cathode materials for Li batteries." J. Electrochem Soc. 2006, 153, A32.
Figure 6.
Left: X-ray analysis of Fe(VI) compounds, evidence of their orthorhombic β-K2SO4 analogue structure, from "Recent advances in the synthesis and analysis of Fe(VI) cathodes." Licht, et al., J. Solid State Electrochem. 2008, 12, 1523. Right: Mössbauer spectra of K2FeO4: (a) pristine electrode, (b) polarized to 1.5V vs. Li/Li+, and (c) after one complete lithiation cycle. From "Study of Various ("super iron") MeFeO4 compounds in Li salt solutions as cathode materials for Li batteries." J. Electrochem Soc. 2006, 153, A32.
3. Conclusions
Vehicle electrification provides significant advantages of fuel efficiency, which will decrease greenhouse gas emission, decrease the dependence on fossil fuel resources, and facilitate the transition to the renewable energy economy. Principle constraints to vehicle electrification include that the Li-ion cathode battery chemistry is massive and costly. Limited battery capacity (vehicle range) and cost are hurdles to implementation. This is exemplified in a recent evaluation of Li-ion batteries for use in electrified vehicles, which concludes that after yield economies of scale, the “most significant cost component at the cell-level is the cathode active material”, and which is three times more expensive than the anode material [43].
The super-iron cathode addresses the cathode challenge with a transformative 3e– Fe(VI) storage Li-ion chemistry, with a capacity several fold higher than observed in conventional lithium-ion cathodes. Continued research will further advance these systems. For example, new nm-architectures need to be explored which preserve the high recharge voltage efficiency observed for the thin layer Fe(III/VI) films. As super-iron films are increased in thickness by a factor of two orders of magnitude, from a thickness of several nm [29] to a thickness approaching 1 µm [6], impedance losses increase, and impair the recharge voltage of efficiency. The thicker films retain a high coulombic efficiency, but exhibit a charge/discharge potential variation similar to super-capacitor, rather than battery, behavior. The super-iron lithium-ion battery is a Li-ion chemistry in which the cathode does not weigh down the battery, with a transformative potential in terms of range increase of electric vehicles.
Acknowledgements
S. Licht is grateful to the US Department of Energy and the George Washington University Institute of Basic Energy Science and Technology for partial support of this research. Yufei Wang, Gerald Gourdin and Lan Yang participated in the results reported in Figure 1 and Reference 6.
References and Notes
- Advances in Lithium-ion Batteries; van Schalkwijk, W.A.; Scrosati, B. (Eds.) Springer: Berlin, Germany, 2002.
- Licht, S.; Wu, H.; Yu, X.; Wang, Y. Renewable highest capacity VB2/air energy storage. Chem. Comm. 2008, 28, 3257–3259. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, D.; Yang, F. Developments of lithium-ion batteries & challenges of LiFePO4 as one promising cathode material. J. Mater. Sci. 2009, 44, 2435–2443. [Google Scholar] [CrossRef]
- Padhi, A.K.; Nanjundaswamy, K.S.; Goodenough, J.B. Phospho-olivines as positive-electrode materials for rechargeable lithium-ion batteries. J. Electrochem. Soc. 1997, 144, 1188–1194. [Google Scholar] [CrossRef]
- Licht, S.; Wang, B.; Ghosh, S. Energetic iron (VI) chemistry: The super-iron battery. Science 1999, 285, 1039–1042. [Google Scholar] [CrossRef] [PubMed]
- Licht, S.; Wang, Y.; Gourdin, G. Enhancement of reversible nonaqueous Fe(III/VI) cathodic charge transfer. J. Phys. Chem. C 2009, 113, 9884–9891. [Google Scholar]
- Licht, S.; Wang, B. Non aqueous Iron(VI) chemistry: The lithium super-iron battery. Electrochem. Solid State Lett. 2000, 3, A 209–A212. [Google Scholar]
- Licht, S.; Naschitz, V.; Halperin, L.; Halperin, L.; Lin, L.; Chen, J.; Ghosh, S.; Lui, B. Analysis of Ferrate(VI) compounds & super-Iron battery cathodes, FTIR, XRD, UV/Vis, ICP, electrochemical & chemical characterization. J. Power Sources 2001, 99, 7–14. [Google Scholar] [CrossRef]
- Licht, S.; Wang, B.; Ghosh, S.; Jun, Li.; Naschitz, V. Insoluble Fe(VI) compounds: Effects on the super-iron battery. Electrochem. Comm. 1999, 1, 522–526. [Google Scholar] [CrossRef]
- Licht, S.; Naschitz, V.; Ghosh, S.; Liu, B.; Halperin, N.; Halperin, L.; Rozen, D. Chemical synthesis of battery grade super-iron barium and potassium Fe(VI) ferrate compounds. J. Power Sources 2001, 99, 7–14. [Google Scholar] [CrossRef]
- Licht, S.; Naschitz, V.; Ghosh, S.; Lin, L.; Lui, B. SrFeO4: synthesis, Fe(VI) characterization and the strontium super-iron battery. Electrochem. Comm. 2001, 3, 340–345. [Google Scholar] [CrossRef]
- Yang, W.; Wang, J.; Pan, T.; Xu, J.; Xhang, J.; Cao, C. Studies on electrochem. characteristics of K2Sr(FeO4)2 electrode. Electrochem. Comm. 2002, 4, 710–715. [Google Scholar] [CrossRef]
- Licht, S.; Naschitz, V.; Wang, B. Rapid chemical synthesis of barium ferrate, BaFe(VI)O4. J. Power Sources 2002, 109, 67–70. [Google Scholar] [CrossRef]
- Licht, S.; Naschitz, V.; Rozen, D.; Halperin, N. Cathodic charge transfer and analysis of Cs2FeO4, K2FeO4 and mixed alkali Fe(VI), ferrate, super-irons. J. Electrochem. Soc. 2004, 151, A1147–A1151. [Google Scholar] [CrossRef]
- Licht, S.; Yang, L.; Wang, B. Synthesis and analysis of Ag2FeO4 Fe(VI) ferrate super-iron cathodes. Electrochem. Comm. 2005, 7, 931–936. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, J.; Shao, H.; Tang, Z.; Zhang, J. Preliminary investigation on the physiochemical properties of calcium ferrate. Electrochem. Comm. 2007, 9, 371–377. [Google Scholar] [CrossRef]
- Licht, S. Electrolytic production of solid Fe(VI) salts, PCT application No. LI00588. Patent No. WO0,121,856, 2000. [Google Scholar]
- Lapique, F.; Valentin, G. Direct electrochemical preparation of solid potassium ferrate. Electrochem. Comm. 2002, 4, 764–766. [Google Scholar]
- Licht, S.; Tel-Vered, R.; Halperin, L. Direct electrochemical preparation of solid Fe(VI) compounds and super-iron battery compounds. Electrochem. Comm. 2002, 4, 933–937. [Google Scholar] [CrossRef]
- Lee, J.; Tryk, D.; Fujishima, A.; Park, S. Electrochemical generation of ferrate in acidic media at boron-doped diamond electrodes. Chem. Comm. 2002, 5, 486–487. [Google Scholar] [CrossRef] [PubMed]
- De Koninck, M.; Brousse, T.; Belanger, D. The electrochemical generation of ferrate at pressed iron powder electrode: Effect of different parameters. Electrochim. Acta 2003, 48, 1425–1433. [Google Scholar] [CrossRef]
- Licht, S.; Tel-Vered, R.; Halperin, L. Towards efficient electrochemical synthesis of Fe(VI) ferrate, and super-iron battery compounds. J. Electrochem. Soc. 2004, 151, A31–A39. [Google Scholar] [CrossRef]
- Zhang, F.C.; Liu, Z.; Wu, F.; Lin, L.; Qi, F. Electrochemical generation of ferrate on SnO2-Sb2O3/Ti electrodes in strong concentration basic condition. Electrochem. Comm. 2005, 6, 1104–1109. [Google Scholar] [CrossRef]
- Cañizares, P.; Arcís, M.; Sáez, C.; Rodrigo, M.A. Electrochemical synthesis of ferrate using boron doped diamond anodes. Electrochem. Comm. 2007, 9, 2286–2290. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, J.; Mao, W.; He, W.; Chen, Q.; Zhang, J. The effects of ultrasound on the direct electrosynthesis of solid K2FeO4 and the anodic behaviors of Fe in 14 M KOH solution. J. Solid State Electrochem 2008, 11, 413–420. [Google Scholar] [CrossRef]
- Yu, X.; Licht, S. Advances in electrochemical Fe(VI) synthesis and analysis. J. Appl. Electrochem. 2008, 38, 731–742. [Google Scholar] [CrossRef]
- Licht, S.; Rozen, D.; Tel-Vered, R.; Halperin, L. Enhancement of nonaqueous Fe(VI) super-iron primary cathodic charge transfer. J. Electrochem. Soc. 2003, 150, A1671–A1675. [Google Scholar] [CrossRef]
- Koltypin, M.; Licht, S.; Tel-Vered, R.; Nashitz, V.; Aurbach, D. The Study of Licht, S.: Various ("super iron") MeFeO4 (F6+-super iron) compounds in Li salt solutions. J. Power Sources 2005, 146, 723–726. [Google Scholar] [CrossRef]
- Licht, S.; Tel-Vered, R. Rechargeable Fe(III/VI) super-iron cathodes. Chem. Comm. 2004, 6, 628–629. [Google Scholar] [CrossRef] [PubMed]
- Licht, S.; DeAlwis, C. Conductive matrix mediated charge transfer in Fe(III/VI): Three electron storage, reversible super-iron thin film cathodes. J. Phys. Chem. B 2006, 110, 12394–123404. [Google Scholar] [CrossRef] [PubMed]
- Koltypin, M.; Licht, S.; Nowik, I.; Levi, E.; Gofer, Y.; Aurbach, D. The study of various ("super-iron") MeFeO4 compounds in Li salt solutions as potential cathode materials for Li batteries. J. Electrochem. Soc 2006, 153, A32–A41. [Google Scholar] [CrossRef]
- Licht, S.; Yu, X.; Dhong, Z. Cathodic chemistry of high performance Zr coated alkaline materials. Chem. Comm. 2006, 2006, 4341–4343. [Google Scholar] [CrossRef]
- Licht, S.; Yu, X.; Qu, D. A novel alkaline redox couple: Chemistry of the Fe6+/B2– super-iron boride battery. Chem. Comm. 2007, 2007, 2753–2755. [Google Scholar] [CrossRef]
- Licht, S.; Yu, X.; Wang, Y. Stabilized alkaline Fe(VI) charge transfer: Zirconia coating stabilized super-iron alkaline batteries. J. Electrochem. Soc. 2008, 155, A1–A7. [Google Scholar] [CrossRef]
- Licht, S.; Yu, X.; Wang, Y.; Wu, H. The super-iron boride battery. J.Electrochem. Soc. 2008, 155, A297–A303. [Google Scholar] [CrossRef]
- Ghosh, S.; Wen, W.; Urian, R.C.; Heath, C.; Srinivasamurthi, V.; Reiff, W.M.; Mukerjee, S.; Naschitz, V.; Licht, S. The reversible behavior of K2FeO4. Electrochem. Solid State Lett. 2003, 6, A260–A264. [Google Scholar] [CrossRef]
- Yu, X.; Licht, S. Recent advances in synthesis and analysis of Fe(VI) cathodes: solution phase and solid-state Fe(VI) syntheses, reversible thin-film Fe(VI) synthesis, coating-stabilized Fe(VI) synthesis. J. Solid State Electrochem. 2008, 12, 1523–1540. [Google Scholar] [CrossRef]
- Licht, S.; Ghosh, S.; Naschitz, V.; Halperin, N.; Halperin, L. Fe(VI) catalyzed manganese redox chemistry: Permanganate and super-iron batteries. J. Phys. Chem. B 2001, 105, 11933–11936. [Google Scholar] [CrossRef]
- Licht, S.; Naschitz, V.; Ghosh, S. Silver mediation of Fe(VI) charge transfer: Activation of the K2FeO4 super-iron cathode. J. Phys. Chem. B 2002, 106, 5947–5955. [Google Scholar] [CrossRef]
- Yu, X.; Licht, S. Advances in Fe(VI) charge storage: Part II. Reversible alkaline super-iron batteries and nonaqueous super-iron batteries. J. Power Sources 2007, 171, 1010–1022. [Google Scholar] [CrossRef]
- Licht, S.; Ghosh, S. High power BaFe(VI)O4 /MnO2 composite cathode alkaline super-iron batteries. J. Power Sources 2002, 109, 465–468. [Google Scholar] [CrossRef]
- Licht, S.; Naschitz, V.; Ghosh, S. Hydroxide activated AgMnO4 alkaline cathodes, alone, and in combination with Fe(VI) super-iron, BaFeO4. Electrochem. Solid State Lett. 2001, 4, A209–A212. [Google Scholar] [CrossRef]
- Anderson, D.L. An evaluation of current and future costs for lithium-ion batteries for use in electrified vehicle powertrains. Master Thesis; Duke University: Durham, NC, USA, May 2009. Available online: http://dukespace.lib.duke.edu/dspace/bitstream/10161/1007/1/Li-Ion%20Battery%20Costs%20-%20Anderson%20-%20MP%20Final.pdf (accessed on 25 April 2010).
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Licht, S. A High Capacity Li-Ion Cathode: The Fe(III/VI) Super-Iron Cathode. Energies 2010, 3, 960-972. https://doi.org/10.3390/en3050960
AMA Style
Licht S. A High Capacity Li-Ion Cathode: The Fe(III/VI) Super-Iron Cathode. Energies. 2010; 3(5):960-972. https://doi.org/10.3390/en3050960
Chicago/Turabian StyleLicht, Stuart. 2010. "A High Capacity Li-Ion Cathode: The Fe(III/VI) Super-Iron Cathode" Energies 3, no. 5: 960-972. https://doi.org/10.3390/en3050960