Application of Sulfated Tin (IV) Oxide Solid Superacid Catalyst to Partial Coupling Reaction of α-Pinene to Produce Less Viscous High-Density Fuel

Abstract
:1. Introduction
2. Materials and Methods
2.1. Catalyst Preparation
2.2. Catalyst Characterization
2.3. Catalytic Tests
3. Results and Discussion
3.1. Catalyst Characteristics
3.1.1. Catalyst Surface Morphology
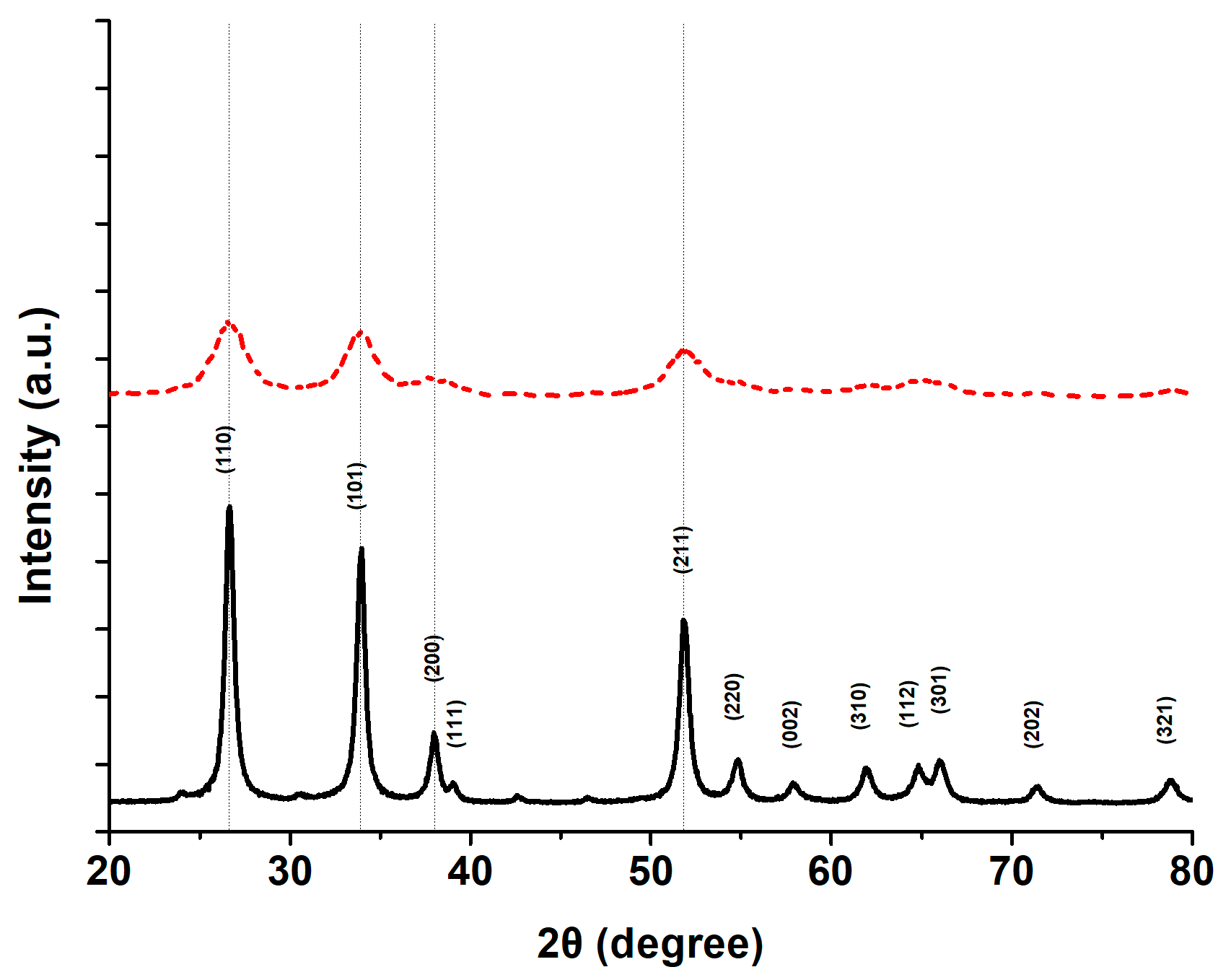
3.1.2. Catalyst Crystal Structure
3.1.3. Catalyst Thermostability
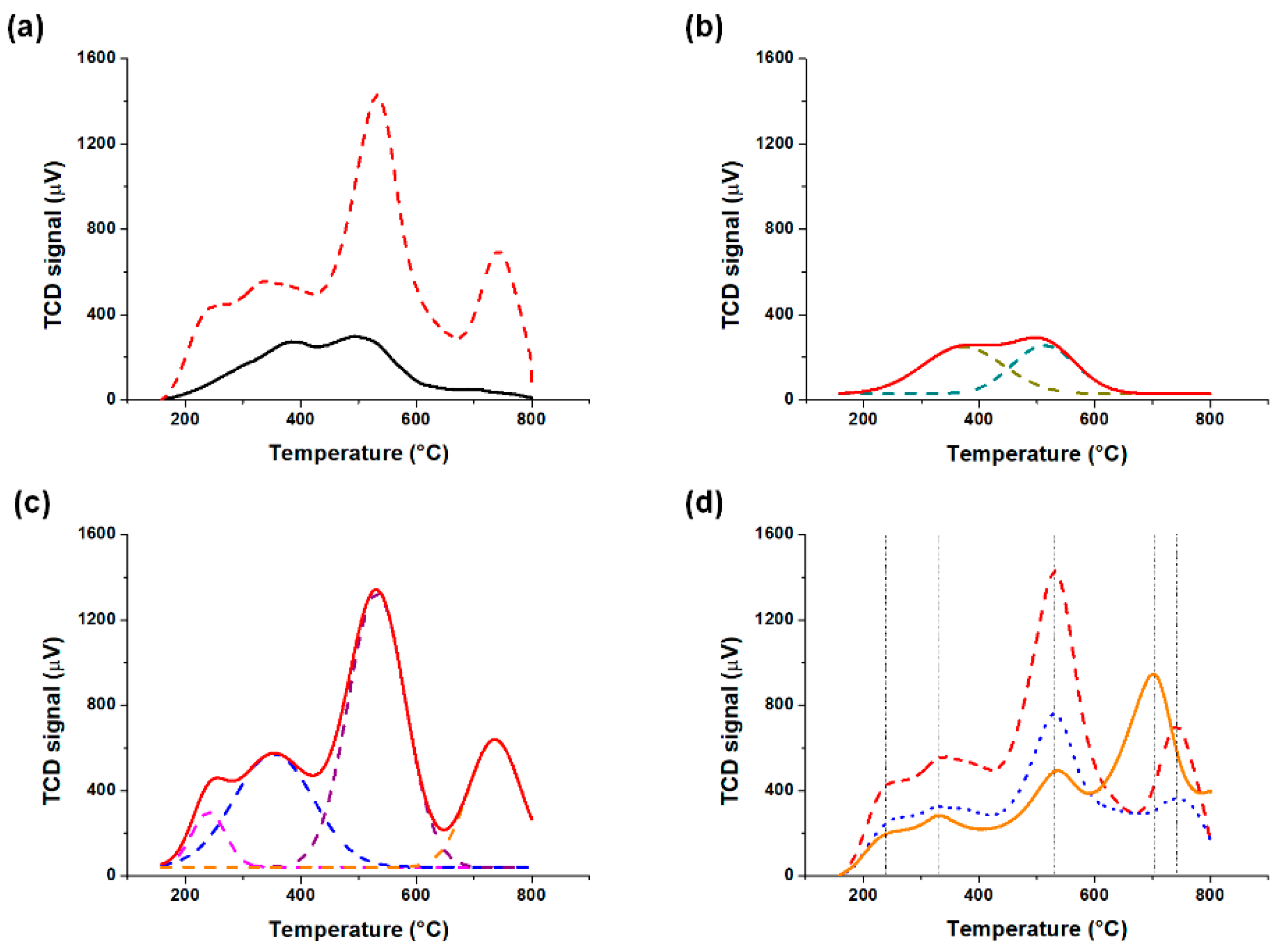
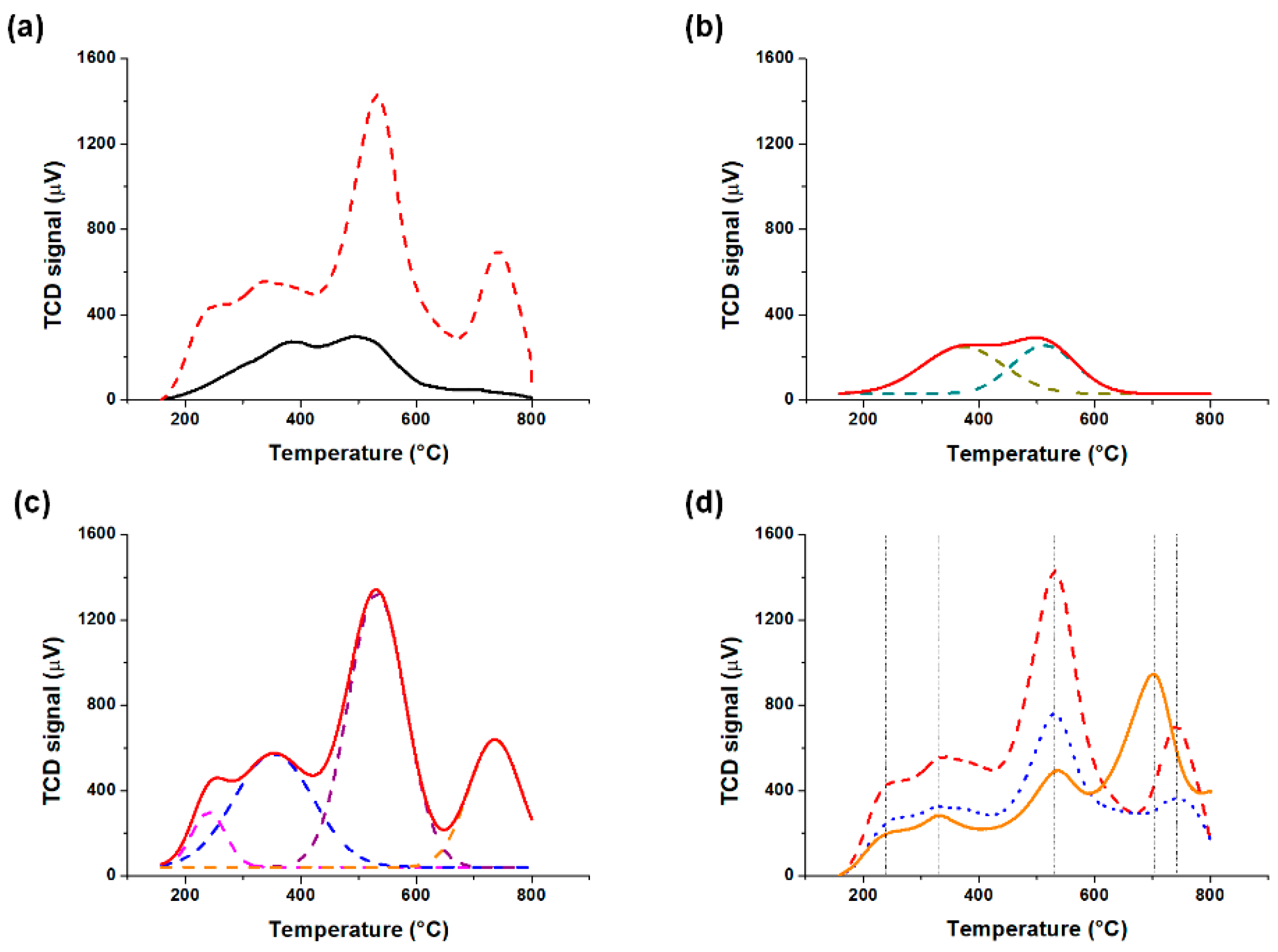
3.1.4. Catalyst Acidity
3.2. Catalytic Tests and Reaction Mechanism
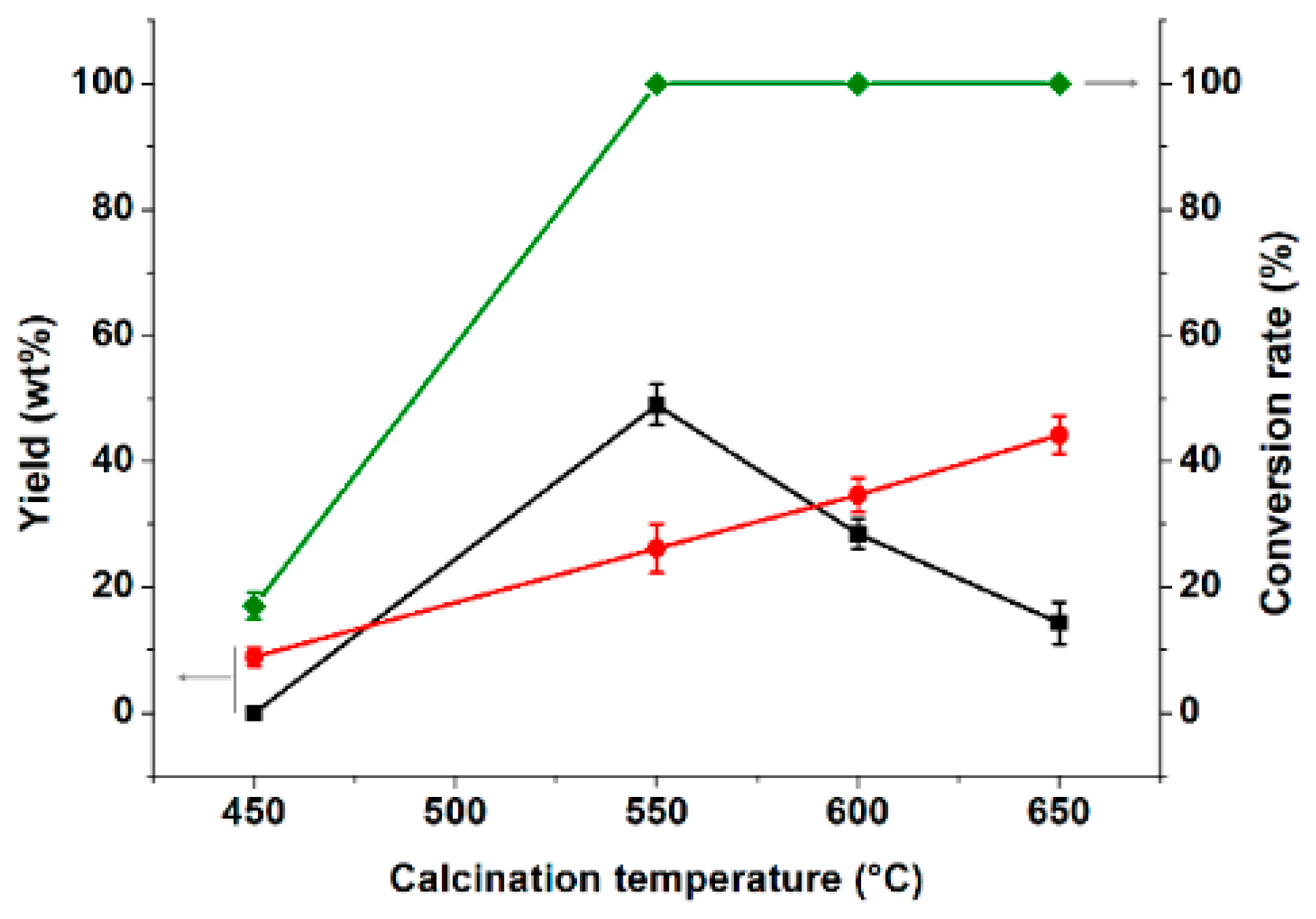
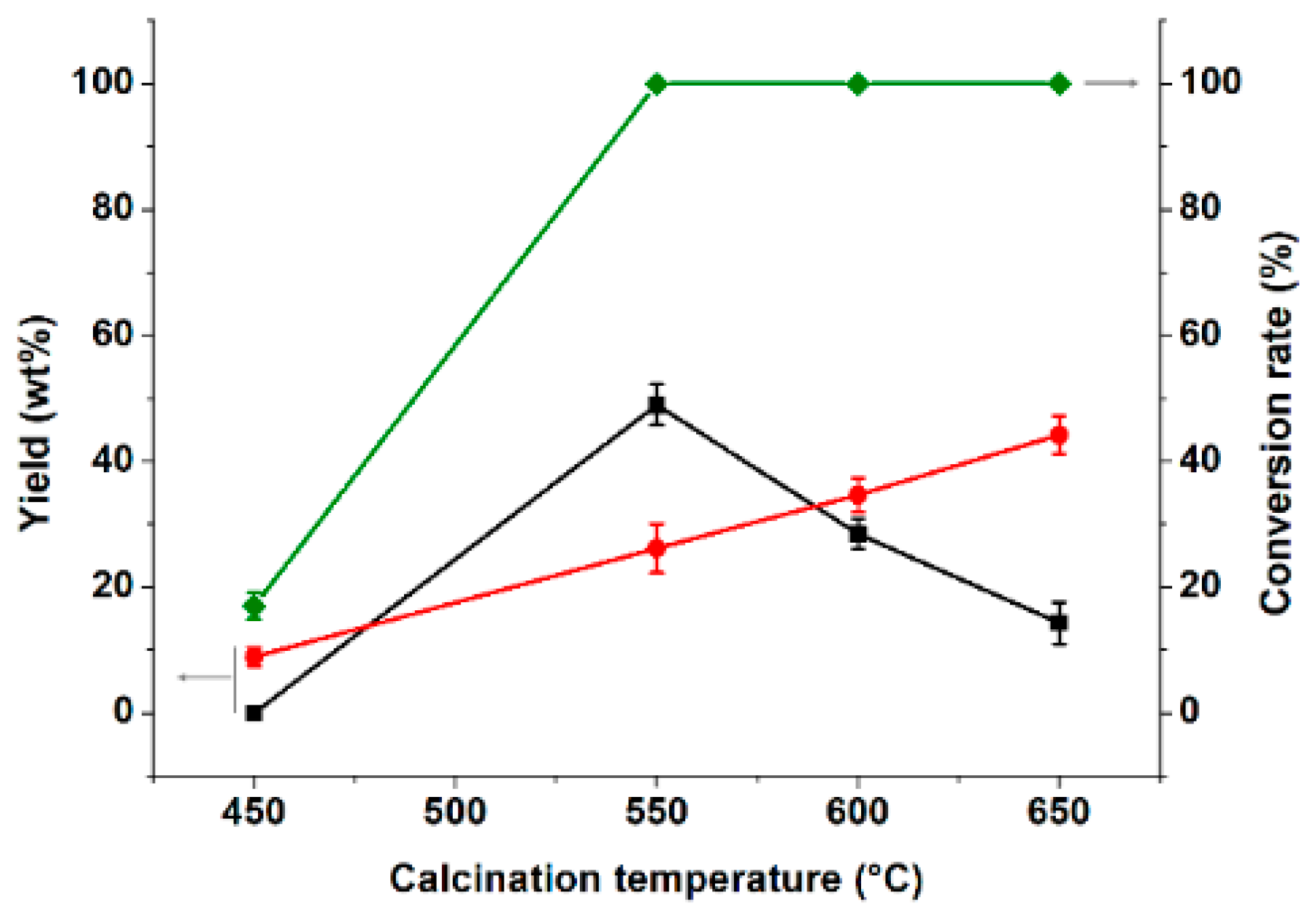
3.2.1. Effect of Catalyst Calcination Temperature on Partial Coupling Reaction of α-Pinene
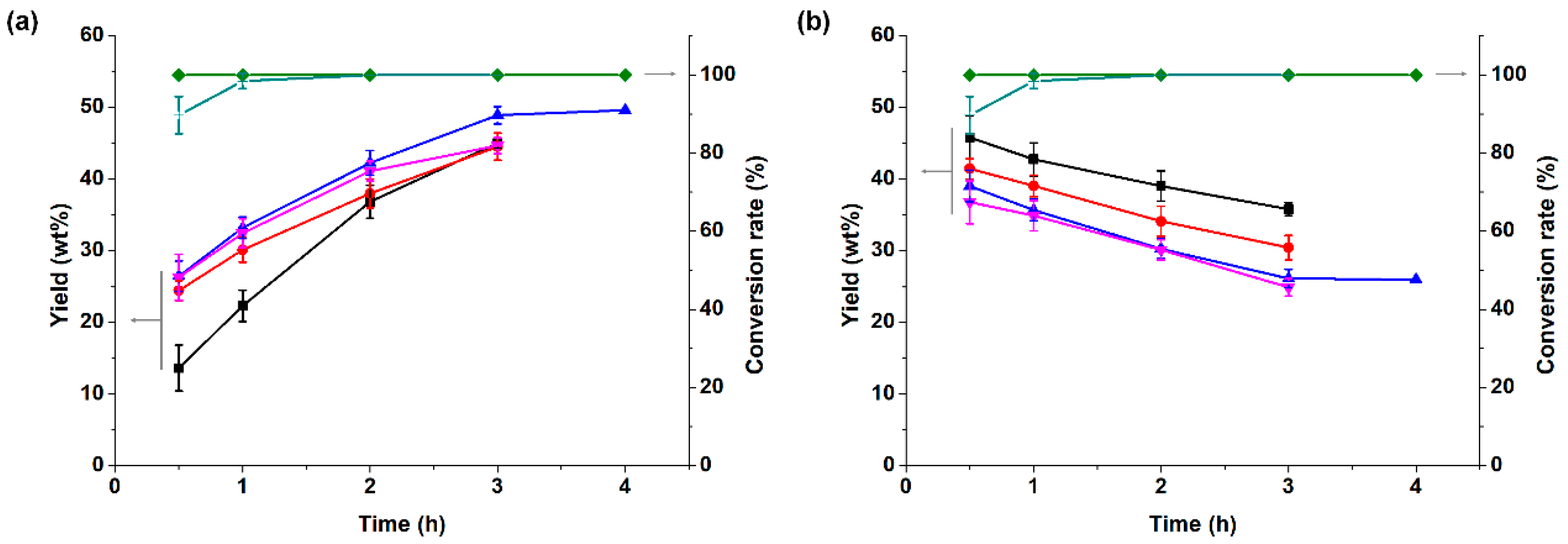
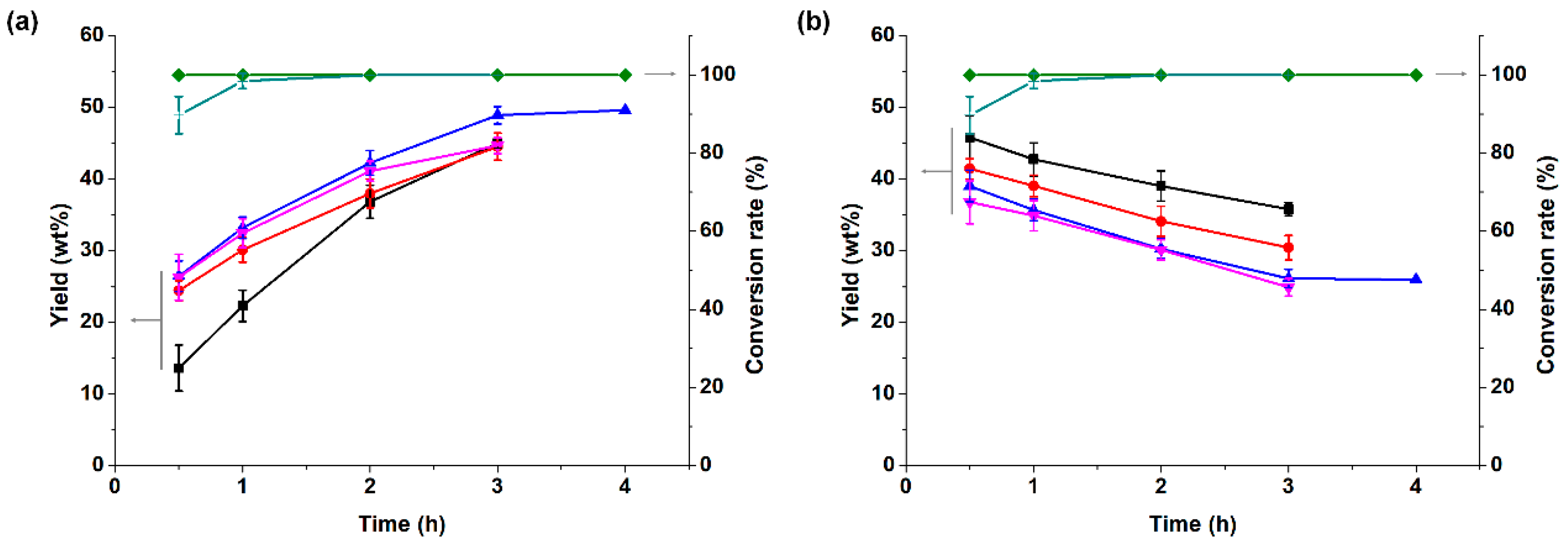
3.2.2. Effect of Reaction Time and Temperature on Partial Coupling Reaction of α-Pinene
3.2.3. Mechanism of Isomerization of α-Pinene over Sulfated Tin Oxide
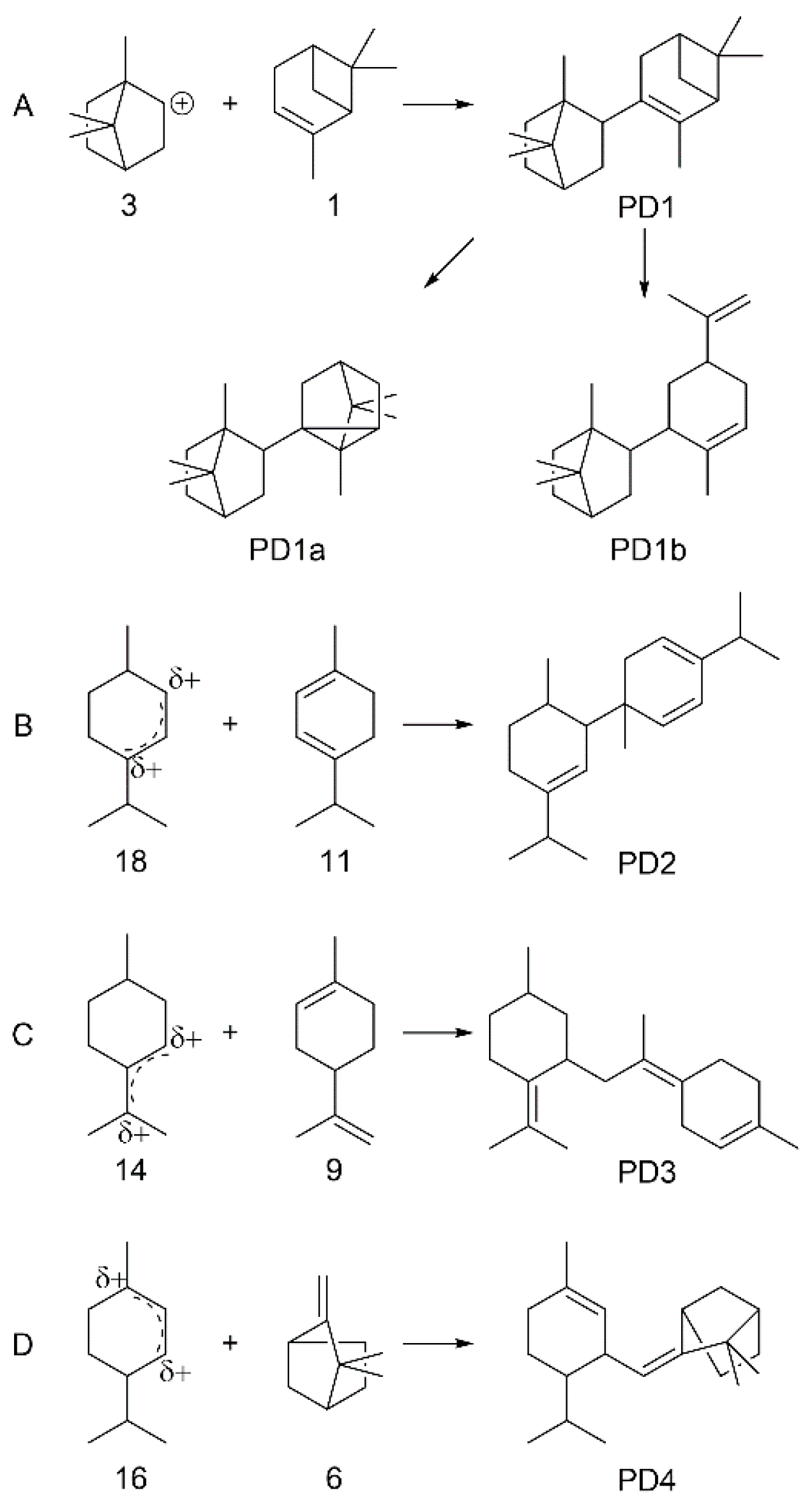
3.2.4. Mechanism of Coupling of α-Pinene over Sulfated Tin Oxide
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gscheidmeier, M.; Fleig, H. Turpentines. In Ullmann’s Encyclopedia of Industrial Chemistry; BASF Aktiengesellschaft: Ludwigshafen, Germany, 2012. [Google Scholar]
- Yumrutaş, R.; Alma, M.H.; Özcan, H.; Kaşka, Ö. Investigation of Purified Sulfate Turpentine on Engine Performance and Exhaust Emission. Fuel 2008, 87, 252–259. [Google Scholar] [CrossRef]
- Arpa, O.; Yumrutas, R.; Alma, M. Effects of Turpentine and Gasoline-Like Fuel Obtained from Waste Lubrication Oil on Engine Performance and Exhaust Emission. Energy 2010, 35, 3603–3613. [Google Scholar] [CrossRef]
- Anand, B.P.; Saravanan, C.; Srinivasan, C.A. Performance and Exhaust Emission of Turpentine Oil Powered Direct Injection Diesel Engine. Renew. Energy 2010, 35, 1179–1184. [Google Scholar] [CrossRef]
- Dubey, P.; Gupta, R. Influences of Dual Bio-Fuel (Jatropha Biodiesel and Turpentine Oil) on Single Cylinder Variable Compression Ratio Diesel Engine. Renew. Energy 2018, 115, 1294–1302. [Google Scholar]
- Harvey, B.G.; Wright, M.E.; Quintana, R.L. High-Density Renewable Fuels Based on the Selective Dimerization of Pinenes. Energy Fuels 2009, 24, 267–273. [Google Scholar] [CrossRef]
- Meylemans, H.A.; Quintana, R.L.; Harvey, B.G. Efficient Conversion of Pure and Mixed Terpene Feedstocks to High Density Fuels. Fuel 2012, 97, 560–568. [Google Scholar]
- Jung, J.K.; Lee, Y.; Choi, J.-W.; Jae, J.; Ha, J.-M.; Suh, D.J.; Choi, J.; Lee, K.-Y. Production of High-Energy-Density Fuels by Catalytic β-Pinene Dimerization: Effects of the Catalyst Surface Acidity and Pore Width on Selective Dimer Production. Energy Convers. Manag. 2016, 116, 72–79. [Google Scholar] [CrossRef]
- Meylemans, H.A.; Baldwin, L.C.; Harvey, B.G. Low-Temperature Properties of Renewable High-Density Fuel Blends. Energy Fuels 2013, 27, 883–888. [Google Scholar] [CrossRef]
- Cho, S.-M.; Kim, J.-H.; Kim, S.-H.; Park, S.-Y.; Kim, J.-C.; Choi, I.-G. A Comparative Study on the Fuel Properties of Biodiesel from Woody Essential Oil Depending on Terpene Composition. Fuel 2018, 218, 375–384. [Google Scholar] [CrossRef]
- Zou, J.J.; Chang, N.; Zhang, X.; Wang, L. Isomerization and Dimerization of Pinene using Al-Incorporated MCM-41 Mesoporous Materials. ChemCatChem 2012, 4, 1289–1297. [Google Scholar] [CrossRef]
- Nie, G.; Zou, J.-J.; Feng, R.; Zhang, X.; Wang, L. HPW/MCM-41 Catalyzed Isomerization and Dimerization of Pure Pinene and Crude Turpentine. Catal. Today 2014, 234, 271–277. [Google Scholar] [CrossRef]
- Zhang, S.; Xu, C.; Zhai, G.; Zhao, M.; Xian, M.; Jia, Y.; Yu, Z.; Liu, F.; Jian, F.; Sun, W. Bifunctional Catalyst Pd–Al-MCM-41 for Efficient Dimerization–Hydrogenation of β-Pinene in One Pot. RSC Adv. 2017, 7, 47539–47546. [Google Scholar] [CrossRef]
- Andrea Merino, N.; Cecilia Avila, M.; Alejandra Comelli, N.; Natalia Ponzi, E.; Isabel Ponzi, M. Dimerization of α-Pinene, Using Phosphotungstic Acid Supported on SiO2 as Catalyst. Curr. Catal. 2014, 3, 240–243. [Google Scholar] [CrossRef]
- Dabbawala, A.A.; Mishra, D.K.; Hwang, J.-S. Sulfated Tin Oxide as an Efficient Solid Acid Catalyst for Liquid Phase Selective Dehydration of Sorbitol to Isosorbide. Catal. Commun. 2013, 42, 1–5. [Google Scholar] [CrossRef]
- Suzuki, T.; Yokoi, T.; Otomo, R.; Kondo, J.N.; Tatsumi, T. Dehydration of Xylose Over Sulfated Tin Oxide Catalyst: Influences of the Preparation Conditions on the Structural Properties and Catalytic Performance. Appl. Catal. A Gen. 2011, 408, 117–124. [Google Scholar] [CrossRef]
- Moreno, J.; Jaimes, R.; Gómez, R.; Niño-Gómez, M. Evaluation of Sulfated Tin Oxides in the Esterification Reaction of Free Fatty Acids. Catal. Today 2011, 172, 34–40. [Google Scholar] [CrossRef]
- Ahmed, A.I.; El-Hakam, S.; Khder, A.; El-Yazeed, W.A. Nanostructure Sulfated Tin Oxide as an Efficient Catalyst for the Preparation of 7-Hydroxy-4-Methyl Coumarin by Pechmann Condensation Reaction. J. Mol. Catal. A Chem. 2013, 366, 99–108. [Google Scholar] [CrossRef]
- Bhure, M.H.; Kumar, I.; Natu, A.D.; Rode, C.V. Facile and Highly Selective Deprotection of Tert-Butyldimethyl Silyl Ethers Using Sulfated SnO2 as a Solid Catalyst. Synth. Commun. 2008, 38, 346–353. [Google Scholar] [CrossRef]
- Gutierrez-Baez, R.; Toledo-Antonio, J.; Cortes-Jacome, M.; Sebastian, P.; Vázquez, A. Effects of the SO4 Groups on the Textural Properties and Local Order Deformation of SnO2 Rutile Structure. Langmuir 2004, 20, 4265–4271. [Google Scholar] [CrossRef]
- Khder, A.; El-Sharkawy, E.; El-Hakam, S.; Ahmed, A. Surface Characterization and Catalytic Activity of Sulfated Tin Oxide Catalyst. Catal. Commun. 2008, 9, 769–777. [Google Scholar] [CrossRef]
- Qi, X.; Watanabe, M.; Aida, T.M.; Smith, R.L., Jr. Sulfated Zirconia as a Solid Acid Catalyst for the Dehydration of Fructose to 5-Hydroxymethylfurfural. Catal. Commun. 2009, 10, 1771–1775. [Google Scholar] [CrossRef]
- Trickett, C.A.; Popp, T.M.O.; Su, J.; Yan, C.; Weisberg, J.; Huq, A.; Urban, P.; Jiang, J.; Kalmutzki, M.J.; Liu, Q. Identification of the Strong Brønsted Acid Site in a Metal–Organic Framework Solid Acid Catalyst. Nat. Chem. 2019, 11, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Kassaye, S.; Pagar, C.; Pant, K.K.; Jain, S.; Gupta, R. Depolymerization of Microcrystalline Cellulose to Value Added Chemicals Using Sulfate Ion Promoted Zirconia Catalyst. Bioresour. Technol. 2016, 220, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Wang, Y.; Pang, D.; Ouyang, F.; Zhang, C. SO42−–Mn–Co–Ce Supported on TiO2/SiO2 with High Sulfur Durability for Low-Temperature SCR of NO with NH3. Catal. Commun. 2016, 78, 22–25. [Google Scholar] [CrossRef]
- Saravanan, K.; Park, K.S.; Jeon, S.; Bae, J.W. Aqueous Phase Synthesis of 5-Hydroxymethylfurfural from Glucose over Large Pore Mesoporous Zirconium Phosphates: Effect of Calcination Temperature. ACS Omega 2018, 3, 808–820. [Google Scholar] [CrossRef]
- Zhang, L.; Zou, W.; Ma, K.; Cao, Y.; Xiong, Y.; Wu, S.; Tang, C.; Gao, F.; Dong, L. Sulfated Temperature Effects on the Catalytic Activity of CeO2 in NH3-Selective Catalytic Reduction Conditions. J. Phys. Chem. C 2015, 119, 1155–1163. [Google Scholar] [CrossRef]
- Yadav, M.K.; Chudasama, C.D.; Jasra, R.V. Isomerisation of α-Pinene Using Modified Montmorillonite Clays. J. Mol. Catal. A Chem. 2004, 216, 51–59. [Google Scholar] [CrossRef]
- Kitano, T.; Okazaki, S.; Shishido, T.; Teramura, K.; Tanaka, T. Brønsted Acid Generation of Alumina-Supported Molybdenum Oxide Calcined at High Temperatures: Characterization by Acid-Catalyzed Reactions and Spectroscopic Methods. J. Mol. Catal. A Chem. 2013, 371, 21–28. [Google Scholar] [CrossRef]
- Da Silva Rocha, K.A.; Robles-Dutenhefner, P.A.; Kozhevnikov, I.V.; Gusevskaya, E.V. Phosphotungstic Heteropoly Acid as Efficient Heterogeneous Catalyst for Solvent-Free Isomerization of α-Pinene and Longifolene. Appl. Catal. A Gen. 2009, 352, 188–192. [Google Scholar] [CrossRef]
- Wang, J.; Hua, W.; Yue, Y.; Gao, Z. MSU-S Mesoporous Materials: An Efficient Catalyst for Isomerization of α-Pinene. Bioresour. Technol. 2010, 101, 7224–7230. [Google Scholar] [CrossRef]
- Akgül, M.; Özyağcı, B.; Karabakan, A. Evaluation of Fe-And Cr-Containing Clinoptilolite Catalysts for the Production of Camphene from α-Pinene. J. Ind. Eng. Chem. 2013, 19, 240–249. [Google Scholar] [CrossRef]
- Comelli, N.A.; Ponzi, E.N.; Ponzi, M.I. α-Pinene Isomerization to Camphene: Effect of Thermal Treatment on Sulfated Zirconia. Chem. Eng. J. 2006, 117, 93–99. [Google Scholar] [CrossRef]
- Comelli, N.A.; Ponzi, E.N.; Ponzi, M.I. Isomerization of α-Pinene, Limonene, α-Terpinene, and Terpinolene on Sulfated Zirconia. J. Am. Oil Chem. Soc. 2005, 82, 531–535. [Google Scholar] [CrossRef]
- Wróblewska, A.; Miądlicki, P.; Makuch, E. The Isomerization of α-Pinene over the Ti-SBA-15 Catalyst—The Influence of Catalyst Content and Temperature. React. Kinet. Mech. Catal. 2016, 119, 641–654. [Google Scholar] [CrossRef]
- Yamamoto, T.; Matsuyama, T.; Tanaka, T.; Funabiki, T.; Yoshida, S. Generation of Acid Sites on Silica-Supported Rare Earth Oxide Catalysts: Structural Characterization and Catalysis for α-Pinene Isomerization. Phys. Chem. Chem. Phys. 1999, 1, 2841–2849. [Google Scholar] [CrossRef]
- Lopes, C.; Lourenco, J.; Pereira, C.; Marcelo-Curto, M. Aromatization of Limonene with Zeolites Y. In Natural Products in the New Millennium: Prospects and Industrial Application; Springer: Berlin, Germany, 2002; pp. 429–436. [Google Scholar]
- Salacinski, E.J. Acid-Catalyzed Isomerization of the p-Menthadienes; The University of Arizona: Tucson, AZ, USA, 1966. [Google Scholar]
- McCormick, J.; Barton, D.L. Studies in 85% H3PO4—II: On the Role of the α-Terpinyl Cation in Cyclic Monoterpene Genesis. Tetrahedron 1978, 34, 325–330. [Google Scholar] [CrossRef]
- Behr, A.; Wintzer, A. Hydroaminomethylation of the Renewable Limonene with Ammonia in an Aqueous Biphasic Solvent System. Chem. Eng. Technol. 2015, 38, 2299–2304. [Google Scholar] [CrossRef]
- Al-Wadaani, F.; Kozhevnikova, E.F.; Kozhevnikov, I.V. Zn (II)–Cr (III) Mixed Oxide as Efficient Bifunctional Catalyst for Dehydroisomerisation of α-Pinene to P-Cymene. Appl. Catal. A Gen. 2009, 363, 153–156. [Google Scholar] [CrossRef]
- Zhao, C.; Gan, W.; Fan, X.; Cai, Z.; Dyson, P.J.; Kou, Y. Aqueous-Phase Biphasic Dehydroaromatization of Bio-Derived Limonene into P-Cymene by Soluble Pd Nanocluster Catalysts. J. Catal. 2008, 254, 244–250. [Google Scholar] [CrossRef]
- Golets, M.; Ajaikumar, S.; Mohln, M.; Wärnå, J.; Rakesh, S.; Mikkola, J.-P. Continuous Production of the Renewable ρ-Cymene from α-Pinene. J. Catal. 2013, 307, 305–315. [Google Scholar] [CrossRef]
- Linnekoski, J.A.; Asikainen, M.; Heikkinen, H.; Kaila, R.K.; Räsänen, J.; Laitinen, A.; Harlin, A. Production of P-Cymene from Crude Sulphate Turpentine with Commercial Zeolite Catalyst Using a Continuous Fixed Bed Reactor. Org. Process Res. Dev. 2014, 18, 1468–1475. [Google Scholar] [CrossRef]
- Martin-Luengo, M.; Yates, M.; Rojo, E.S.; Arribas, D.H.; Aguilar, D.; Hitzky, E.R. Sustainable P-Cymene and Hydrogen from Limonene. Appl. Catal. A Gen. 2010, 387, 141–146. [Google Scholar] [CrossRef]
- Arias-Ugarte, R.; Wekesa, F.S.; Schunemann, S.; Findlater, M. Iron (III)-Catalyzed Dimerization of Cycloolefins: Synthesis of High-Density Fuel Candidates. Energy Fuels 2015, 29, 8162–8167. [Google Scholar] [CrossRef]
- Fernandes, C.; Catrinescu, C.; Castilho, P.; Russo, P.; Carrott, M.; Breen, C. Catalytic Conversion of Limonene over ACID ACTIVATED SERRA de Dentro (SD) Bentonite. Appl. Catal. A Gen. 2007, 318, 108–120. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Intact SnO2 1 | SO42−/SnO2 | |||
|---|---|---|---|---|---|
| 450 °C 2 | 550 °C | 600 °C | 650 °C | ||
| NH3 uptake (μmol g−1) | 171.1 3 | 1161.8 | 760.2 | 400.1 | 425.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, S.-M.; Hong, C.-Y.; Park, S.-Y.; Lee, D.-S.; Choi, J.-H.; Koo, B.; Choi, I.-G. Application of Sulfated Tin (IV) Oxide Solid Superacid Catalyst to Partial Coupling Reaction of α-Pinene to Produce Less Viscous High-Density Fuel. Energies 2019, 12, 1905. https://doi.org/10.3390/en12101905
Cho S-M, Hong C-Y, Park S-Y, Lee D-S, Choi J-H, Koo B, Choi I-G. Application of Sulfated Tin (IV) Oxide Solid Superacid Catalyst to Partial Coupling Reaction of α-Pinene to Produce Less Viscous High-Density Fuel. Energies. 2019; 12(10):1905. https://doi.org/10.3390/en12101905
Chicago/Turabian StyleCho, Seong-Min, Chang-Young Hong, Se-Yeong Park, Da-Song Lee, June-Ho Choi, Bonwook Koo, and In-Gyu Choi. 2019. "Application of Sulfated Tin (IV) Oxide Solid Superacid Catalyst to Partial Coupling Reaction of α-Pinene to Produce Less Viscous High-Density Fuel" Energies 12, no. 10: 1905. https://doi.org/10.3390/en12101905