
Influence of Catalytic Formulation and Operative Conditions on Coke Deposition over CeO2-SiO2 Based Catalysts for Ethanol Reforming




Department of Industrial Engineering, University of Salerno, via Giovanni Paolo II 132, 83040 Fisciano (SA), Italy
*
Author to whom correspondence should be addressed.
Energies 2017, 10(7), 1030; https://doi.org/10.3390/en10071030
Submission received: 8 June 2017
/
Revised: 2 July 2017
/
Accepted: 14 July 2017
/
Published: 19 July 2017
(This article belongs to the Collection Bioenergy and Biofuel)
Abstract
:In this work, a series of CeO2-SiO2 (30 wt % of ceria)-based catalysts was prepared by the wetness impregnation method and tested for ESR (ethanol steam reforming) at 450–500 °C, atmospheric pressure and a water/ethanol ratio increasing from 4 to 6 (the ethanol concentration being fixed to 10 vol %); after every test, coke gasification measurements were performed at the same water partial pressure, and the temperature of the test and the gasified carbon was measured from the areas under the CO and CO2 profiles. Finally, oxidation measurements under a 5% O2/N2 stream made it possible to calculate the total carbon deposited. In an attempt to improve the coke resistance of a Pt-Ni/CeO2-SiO2 catalyst, the effect of support basification by alkali addition (K and Cs), as well as Pt substitution by Rh was investigated. The novel catalysts, especially those containing Rh, displayed a lowering in the carbon formation rate; however, a faster reduction of ethanol conversion with time-on-stream and lessened hydrogen selectivities were recorded. In addition, no significant gain in terms of coke gasification rates was observed. The most active catalyst (Pt-Ni/CeO2-SiO2) was also tested under different operative conditions, in order to study the effect of temperature and water/ethanol ratio on carbon formation and gasification. The increase in the water content resulted in an enhanced reactor-plugging time due to reduced carbonaceous deposits formation; however, no effect of steam concentration on the carbon gasification rate were recorded. On the other hand, the increase in temperature from 450–500 °C lowered the coke selectivity by almost one order of magnitude improving, at the same time, the contribution of the gasification reactions.
1. Introduction
In order to encourage an economy based on clean energy, the demand for hydrogen fuel cells is expected to grow rapidly in the near future [1]. Hydrogen is a valid alternative to conventional fuels due to its ability to burn without emitting environmental pollutants, as well as its high energy content per unit of weight (142 KJ/g [2], 2.75 times greater than that of hydrocarbon fuels). Nowadays, methane steam reforming is the most conventional and economical process for hydrogen production [3,4] but other technologies, including biofuels conversion, have been intensively investigated, also due to the bio-feedstock’s ability to act as an emission sink within a carbon-balanced life-cycle [5,6]. Moreover, among the various renewable sources, biomass is highly promising due to its abundance and low cost [7,8]. Biofuels can be produced both from food crops (first generation), as well as the residuals of agricultural and forestry wastes (second generation). Bioethanol presents several advantages, such as a high octane number, high heat of vaporization and, most importantly, reduction of greenhouse gas emissions. In fact, the CO2 generated during bio-fuels conversion can be recycled for growing plants through photosynthesis, resulting in a carbon neutral process. In addition, bioethanol provides an important route for gasoline replacing, due to its easy handling and hydrogen storage ability [9]. Currently, starch and sugar-rich materials are mainly employed for large-scale bioethanol production which, however, are not desirable due to their value as food [10]. On the other hand, lignocellulosic biomass is cheap and widely available and can be converted to bioethanol by a pre-treatment-hydrolysis-fermentation cycle [11]. Ethanol can be efficiently converted into H2 by means of its catalytic reaction with steam, according to Equation 1:
Despite ethanol steam reforming (ESR) theoretically producing only H2 and CO2, other reactions, such as methanation, water gas shift, dehydration, dehydrogenation, and cracking, can led to the formation of several side products, including methane, carbon monoxide, acetaldehyde, ethylene, acetone, and coke [12,13]. For example, unsaturated hydrocarbon (C2H4) can be polymerized to coke while the decomposition of saturated hydrocarbons, such as CH4, promote carbidic species formation that act as intermediates in the filamentous carbon growth [14]. Hence, major challenges of catalyst design for ethanol steam reforming is to maximize hydrogen productivity, inhibiting, at the same time, coke formation. In addition, operating at low-temperatures are highly desired from the point of view of the hydrogen economy and energy consumption [15].
Several metals have been effectively used for the steam reforming of ethanol, due to their capability to break C2H5OH molecule. In particular, less-oxophilic metals (Pd and Pt) are known to activate α-C–H bonds, while more-oxophilic metals (Co, Ni, Rh, Ru) promote molecule activation via O–H [16]. Despite noble metals catalysts being more active and stable than base metal catalysts in ESR reaction, the high costs hinder their practical applications. Conversely, nickel and cobalt-based catalysts are widely used and recognized as appropriate catalysts for ethanol reforming due to their low price and high activity towards C–C bond cleavage [17,18]. However, to overcome the considerable coke selectivity observed over these catalysts and to increase the hydrogen yield, the synergistic combination of transition metals with very low amounts of noble metals has been reported as a viable route [19,20,21]. It was also shown that adding promoters to Ni-based catalysts is an effective method to prevent carbon deposition. In fact, alkali metals (K, Cs, Na, Li), being electron donators, display excess mobile electrons, which enhances hydrogen spillover on the catalyst surface, thus reducing the amount of coke deposits [22,23]. Moreover, due to the neutralization of catalyst acid sites, alkali metals are able to suppress hydrocarbon (CH4 and ethylene) decomposition and Boudouard reaction. Rass-Hansen et al. [24] found an optimum potassium doping (0.5%) over Ni/MgAl2O4 for maximizing steam adsorption onto the catalyst surface and reduce the carbon formation while Akiyama et al. reported that the cesium-doped Ni/ZrO2 catalysts improve H2 yield with respect to Li- and K-based samples and were more effective in decreasing carbon deposition [25].
The nature of the support is also of great importance for the final catalyst performances. It was shown that acidic supports, such as Al2O3, preferentially promote dehydration while basic support (MgO) favor dehydrogenation reactions. Conversely, reducible supports, including rare earth oxides, display low by-products selectivity and improved hydrogen yield. The superior activity of CeO2-based support can be linked to the good metals particles dispersion and the prevention of sintering phenomena; moreover, their capability to add and remove oxygen in a reversible manner facilitates carbon gasification [26]. Mobile oxygen of ceria can also activate water, with regard to the formation of hydroxyl groups, resulting in higher ESR efficiency [27]. In particular, ceria was shown to accelerate the reaction of steam and absorbed species at the metal-support interface, giving place to –O and –HO species, which are transferred to the surface carbon, thus promoting coke conversion to gaseous products (CO, H2, and CO2) [28,29]. However, the low intrinsic surface area of reducible supports has encouraged the deposition of thin films of these oxides on porous supports. To that end, silica is a very interesting material due to its high surface area and chemically-inert nature [30]. Moreover, the high number of oxygen vacancies in CeO2-SiO2 mixed oxides may increase the lattice oxygen mobility and improve reducibility, activity, and stability of the catalyst [31].
In our previous works [32,33], we demonstrated the efficiency of a bimetallic Pt-Ni catalyst supported on CeO2-SiO2 for hydrogen production via ethanol reforming: the combination of two metals, as well as the choice of a mixed oxide as a support having enhanced redox properties with respect to ceria alone resulted in interesting activity and stability.
In this study, a series of CeO2-SiO2 based catalysts were prepared and characterized. The effect of alkali metals (K and Cs) modification, as well as the addition of Rh (or Pt substitution by Rh) on catalyst activity and stability for ethanol steam reforming was investigated. Moreover, once selected the most interesting sample, coke formation and gasification rates were evaluated and compared at different operative conditions (temperature and water/ethanol ratio).
2. Experimental
2.1. Catalyst Preparation and Characterization
Five CeO2-SiO2 based catalysts (Pt-Ni, Rh-Pt-Ni, Rh-Ni, Pt-Ni-K, Pt-Ni-Cs) were prepared by sequential wetness impregnation. More details about the preparation method are reported in [32]. CeO2 content in the catalyst was previously optimized and fixed to 30 wt %; Ni and noble metals loading (in the bimetallic catalysts) were equal to 10 wt % and 3 wt %, respectively, calculated on the basis of ceria mass. K, Cs, and Rh content in trimetallic catalyst was 0.5 wt % on ceria and potassium hydroxide, caesium chloride (both from Sigma-Aldrich, Saint Louis, MO, USA), and rhodium chloride (Strem Chemicals, Newburyport, MA, USA) were selected as metals salt precursors. K and Cs were directly impregnated on the support, followed by Ni and then Pt while the trimetallic catalysts were prepared by depositing Rh on the Pt-Ni/CeO2/SiO2 sample.
The CeO2, as well as the metal loading (wt %), were determined by X-ray fluorescence on an ARL (Air Resources Laboratory) QUANT'X ED-XRF (energy-dispersive X-ray diffraction) spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). N2 adsorption-desorption isotherms at −196 °C were obtained for the determination of the specific surface area (SSA) of the catalytic samples, according to Branauer-Emmet-Teller (BET) method by using a ThermoScientific Surfer (Thermo Fisher Scientific, Waltham, MA, USA). The calcined catalysts, as well as the bare support, were characterized by powder XRD (X-ray diffraction, model D8 Advance Bruker (Bruker, Billerica, MA, USA) using nickel-filtered CuKα radiation over the 2ϴ range 20–80°. The Scherrer equation, (d = 0.9·λ·β−1·cosθ−1), where λ is the wavelength of incident radiation (nm), β is the half-height width of the most intense peak for the species (radians) and ϴ is the Bragg angle (°) of that peak, was employed to determine the particle size of the various phases based on their most intense diffraction peaks. The hydrogen temperature programmed reduction (H2-TPR) experiments were performed in the laboratory apparatus described in the Section 2.2. The reduction was investigated in the temperature range of 25–500 °C using a constant flow (500 N·cm3·min−1) of 5% H2-N2 gas mixture. The maximum temperature was held for 1 h in order to assure complete reduction of metals oxides. The H2 consumption was obtained by integrating the area under the TPR profile.
2.2. Catalyst Testing
An amount of the calcined sample (4.5 g) was pressed, sieved into a 45–80 mesh, and loaded in a stainless steel tubular reactor (Officina Elettromeccanica Mormile, Casavatore, Italy) having an annular configuration (internal diameter = 15 mm, external diameter = 17 mm). The sample was treated in an H2-N2 mixture as described above and the catalytic activity was evaluated at atmospheric pressure, 450 °C and water/ethanol molar ratio f.r. (feeding ratio) of 3 (10 vol % of ethanol). Further tests on the Pt-Ni/CeO2/SiO2 sample were carried out at 500 °C and f.r. of 4 and 6. The reaction temperature was measured by means of a thermocouple that was inserted into the tube reactor, in contact with the catalyst bed. A differential pressure sensor (Nagano Sensortechnik, Ottendorf-Okrilla, Germany) allowed monitoring the pressure drop through the catalytic bed. The test was performed until measuring a pressure drop of 500 mbar; then, a N2-steam mixture having the same water concentration of the feeding substituted the reacting stream and gasification measurements were carried out. Water/ethanol mixture, stored in a tank under N2 pressure, was fed into a boiler (T = 200 °C, Officina Elettromeccanica Mormile, Casavatore, Italy) and then into the reactor by means of a mass flow controller for liquids (Bronkhorst High-Tech, Bronkhorst, Ruurlo, The Netherlands) with a flow-rate of 13.4 g·h−1. N2 (300 N·cm3∙min−1) was supplied into the boiler as a carrier gas; the resulting weight hourly space velocity (WHSV), defined as the ratio of the ethanol mass flow-rate and catalytic mass, was equal to 4.1 h−1. The inlet and the outlet lines of the reactor were heated to 160 °C for complete vaporization of the reactant species. The composition of the outlet gas (i.e., CO, CO2, CH4, H2O, and C2H5OH concentration) was analysed with an on-line FT-IR (Fourier transform-infrared spectroscopy) Spectrophotometer (Antaris IGS Analyser, Thermo Fisher Scientific, Waltham, MA, USA). Unreacted water and ethanol as well as condensable reaction products eventually present were trapped in a condensate collector while the hydrogen concentration in the dry outlet gas was measured with an advanced optima gas analyser from ABB (Caldos 27, Alamo, TX, USA). Ethanol conversion (X), hydrogen yield (Y), carbon formation (CFR), and carbon gasification (CGR) rates were calculated following Equations (2)–(5), respectively, where masscoke,oxidised (in grams) is determined through thermo-gravimetric analysis carried out on the spent catalyst after the reaction + gasification step; masscatalyst (in grams) stands for the catalytic mass; masscarbon,fed (mc,fed in grams) refers to the total mass of carbon fed as ethanol during the test; and timereaction is the time-on-stream in hours. Concerning CGR, the gasification time was set to 60 min (timegasification) and the mass of carbon oxidized (masscoke,oxidized) was evaluated by integrating the area under CO and CO2 signals.
3. Results and Discussion
3.1. Textural/Structural Properties and H2-TPR Measurements
The textural and structural properties of the bare CeO2-SiO2 (CeSi) and of all the catalysts are summarized in Table 1. The ceria content in all the samples was close to the nominal value. A fairly good agreement was also observed for Ni and noble metals loading (the expected values of 10 wt % and 3 wt %, respectively, refers to ceria mass). Due to the very low content of Rh in the trimetallic catalyst and of the alkali metals, their value was not verified by means of XRF (X-ray fluorescence). BET surface area decreases because of active species composition, except for the Pt-Ni/CeO2/SiO2 catalyst, which displayed a negligible area variation with respect to the bare support. Upon Rh deposition on Ni/CeO2-SiO2 sample, a decrease in BET area was observed, probably due to partial pore blockage and to a worst dispersion of rhodium with respect to platinum. The trimetallic catalyst containing Rh, as well as the alkali-doped samples, displayed lower surface area than the bimetallic sample and this difference is ascribable to the further calcination step. However, the lowest area was recorded over the K-containing sample while a similar area was determined for the Pt-Ni-Cs and Rh-Pt-Ni catalysts.
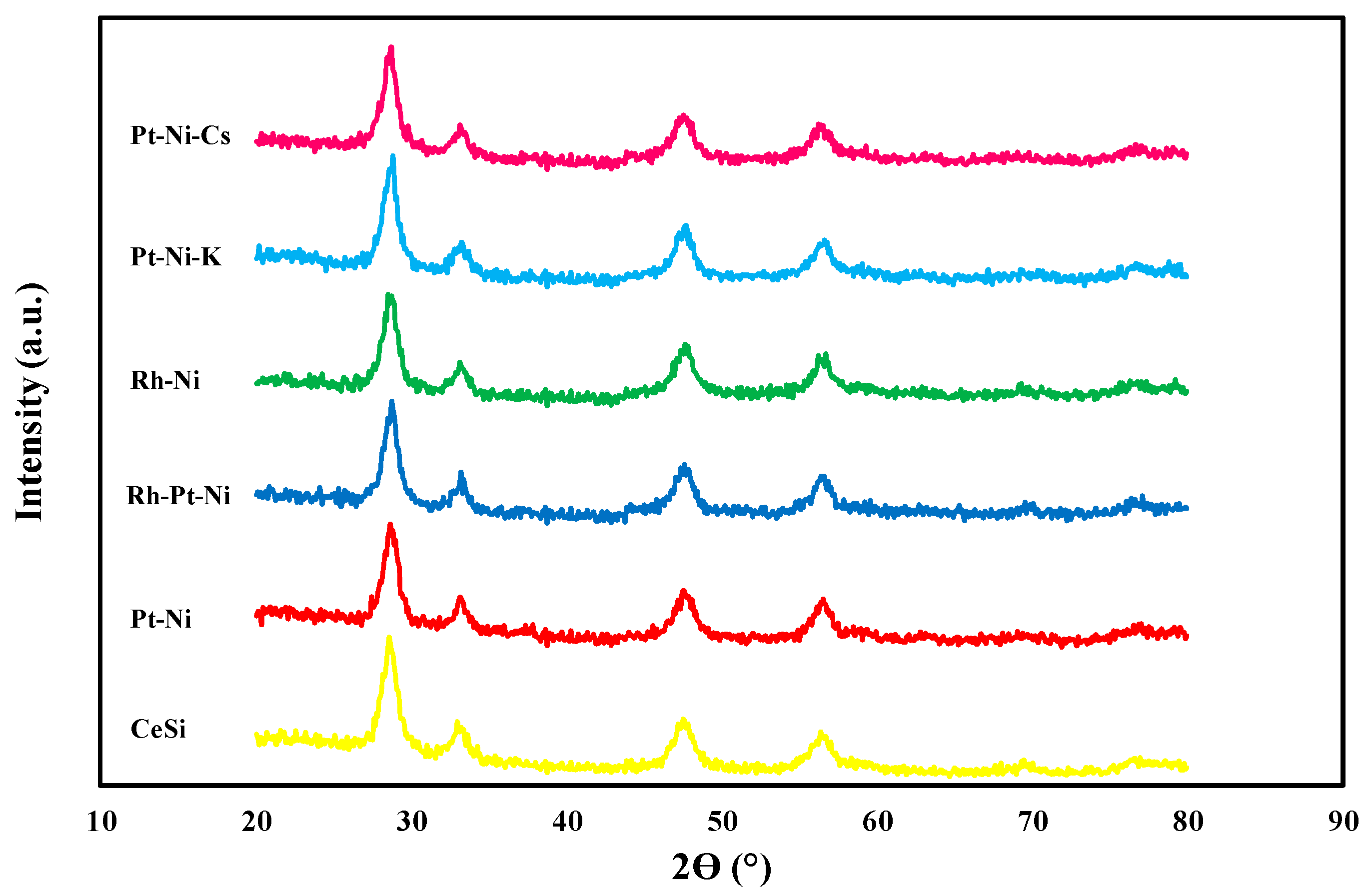
The crystalline structure of the calcined support and of all the catalysts is reported in Figure 1. The broad peaks originated from about 2ϴ = 15° to 35° are the reflections ascribable to amorphous silica framework [34]. In addition, the XRD data of all the samples display the presence of characteristic lines of ceria cubic phase [35] (at 2ϴ = 28.5°, 33.0°, 47.4°, and 56.3°). No other peaks were detected over the samples after active species deposition: NiO and PtOx phase are not visible and this may be due to the low metals amount (the experimental composition of the samples is reported in Table 1) or well-dispersed oxides [36]. The crystallite sizes of ceria particles (d) were not affected by active species deposition and by the number of calcination steps occurred during catalyst preparation; as a result, all the samples displayed ceria particles between 7 and 8 nm.
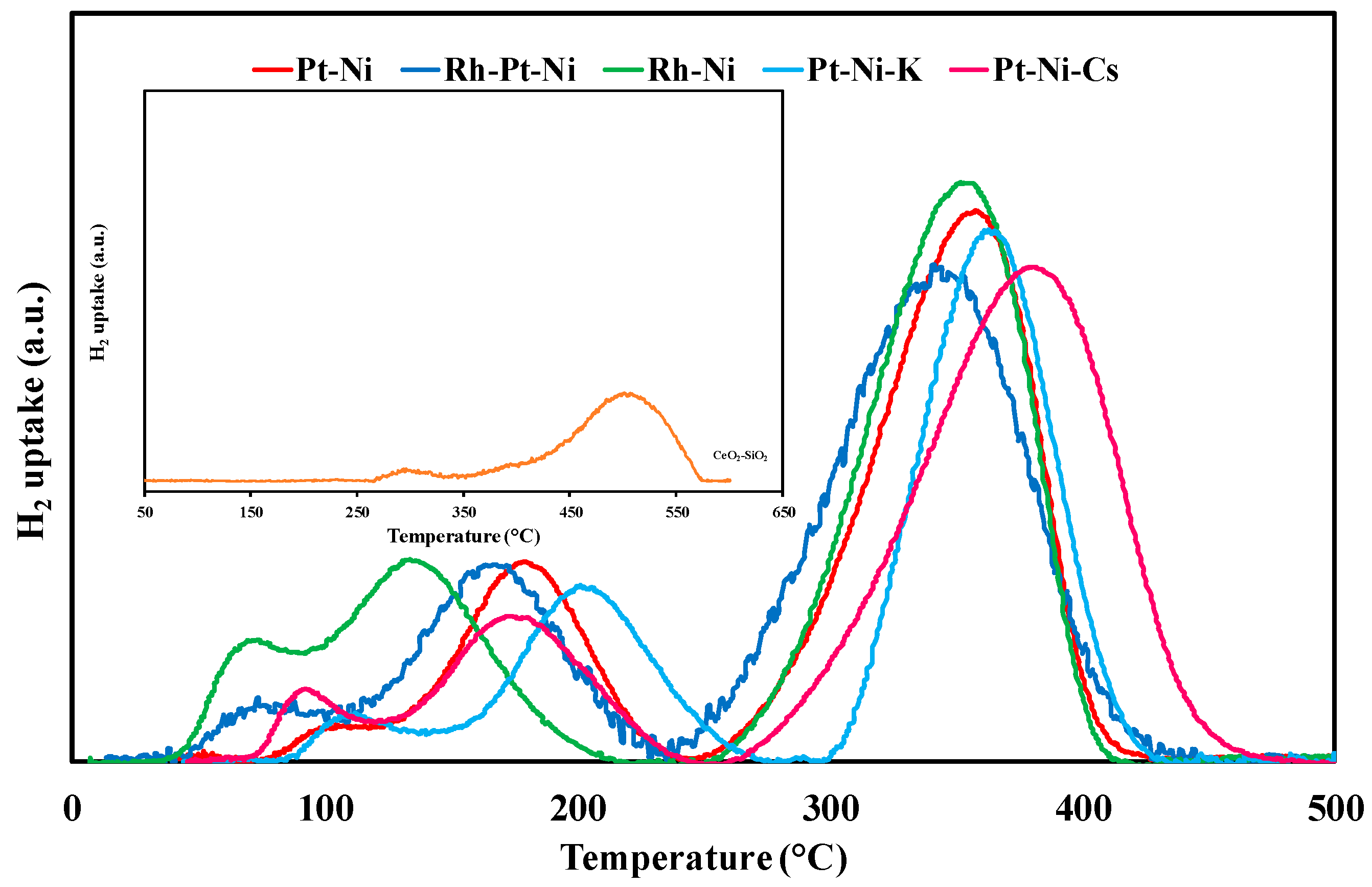
TPR measurements were carried out in order to investigate the reducibility of the calcined support and to examine the interaction strength of metal species with the surface of the support. H2-uptake profiles of CeO2-SiO2 based catalysts in comparison with the TPR curve of the bare support are shown in Figure 2. The Gaussian-type deconvolution of the profile was also carried out, thus allowing the evaluation of hydrogen consumption. All the catalytic samples had similar reduction behaviour with two broad reduction zones between 50–250 °C and above 250 °C, respectively ascribable to noble [37] metals (rhodium oxides are commonly reduced at lower temperatures compared to the Pt ones [38]) and non-noble metal [39] reduction. An additional shoulder always accompanies the major peak observed at lower temperature. In fact, the well-dispersed noble metals nanoparticles were easily reduced below 130 °C while the Pt and/or Rh nanoparticles strongly interacting with Ni and/or with CeO2-SiO2 support were reduced in the temperature range of 150–300 °C [36,40]. On the other hand, for all the prepared catalysts, the reduction band observed at high temperature can be deconvoluted into two hydrogen consumption peaks, which indicates that NiO particles differently interacted with the support: free NiO particles can be reduced earlier than NiO strongly bonded on CeO2-SiO2 support [41].
Rh-containing samples displayed the lowest temperature for consumption peaks, due to the improved catalyst reducibility upon Rh addition [42]. Conversely, the support doping by alkali addition (especially in the case of potassium) was accompanied by a leftward shift peaks, which attested that catalyst basification may increase the metals-support interactions and retard oxide reduction [43].
The results shown in Table 2 demonstrate that the quantity of consumed H2 during the TPR analysis surpassed the value needed for complete reduction of deposited NiO, PtOx, and/or RhOx to metals, according to the theoretical chemical composition of the tested materials. Consequently, partial reduction of the support must have occurred simultaneously to metal oxide reduction because of hydrogen spillover. The latter effect [44], in fact, promotes the migration of hydrogen atoms from the reduced metal particles (especially Rh and Pt) to the nearby ceria surface, thus assuring the reduction of lattice oxygen in the ceria surface, as well as bulk, along with the metal oxides [45]. In fact, the reduction of the Ce4+-O ions is shifted towards lower temperature range. Moreover, in the case of CeO2-SiO2 supported catalysts, the high oxygen storage capacity (OSC) of ceria, as well as oxygen mobility improved by silica, further increased catalyst reducibility [46]. The H2 consumption at low temperature recorded over Pt-Ni catalyst increased by almost 200 µmol/gcat upon Rh deposition; rhodium addition to the catalyst also improved the reducibility of NiO particles and high-temperature uptake grew from 1636 to 2089 µmol/gcat. The highest hydrogen consumption was measured over the bimetallic catalyst containing rhodium (3209 µmol/gcat), again demonstrating the better reducibility properties of Rh over Pt. Conversely, alkali addition partially inhibited Pt-Ni/CeO2/SiO2 reducibility, lowering the H2 uptake of almost 10% in the case of potassium-based sample. The TPR analysis of the bare support (Figure 2 and Table 2) also confirmed the marked effect of the spillover phenomena: for the CeO2-SiO2 sample, a very low H2 consumption was calculated (298 µmol/gcat), with it being interesting to highlight that reduction peaks are observed at higher temperatures compared with the metal-containing sample. This result highlights the key role of the active component in promoting support reduction.
3.2. Ethanol Steam Reforming Tests: Effect of Catalytic Formulation
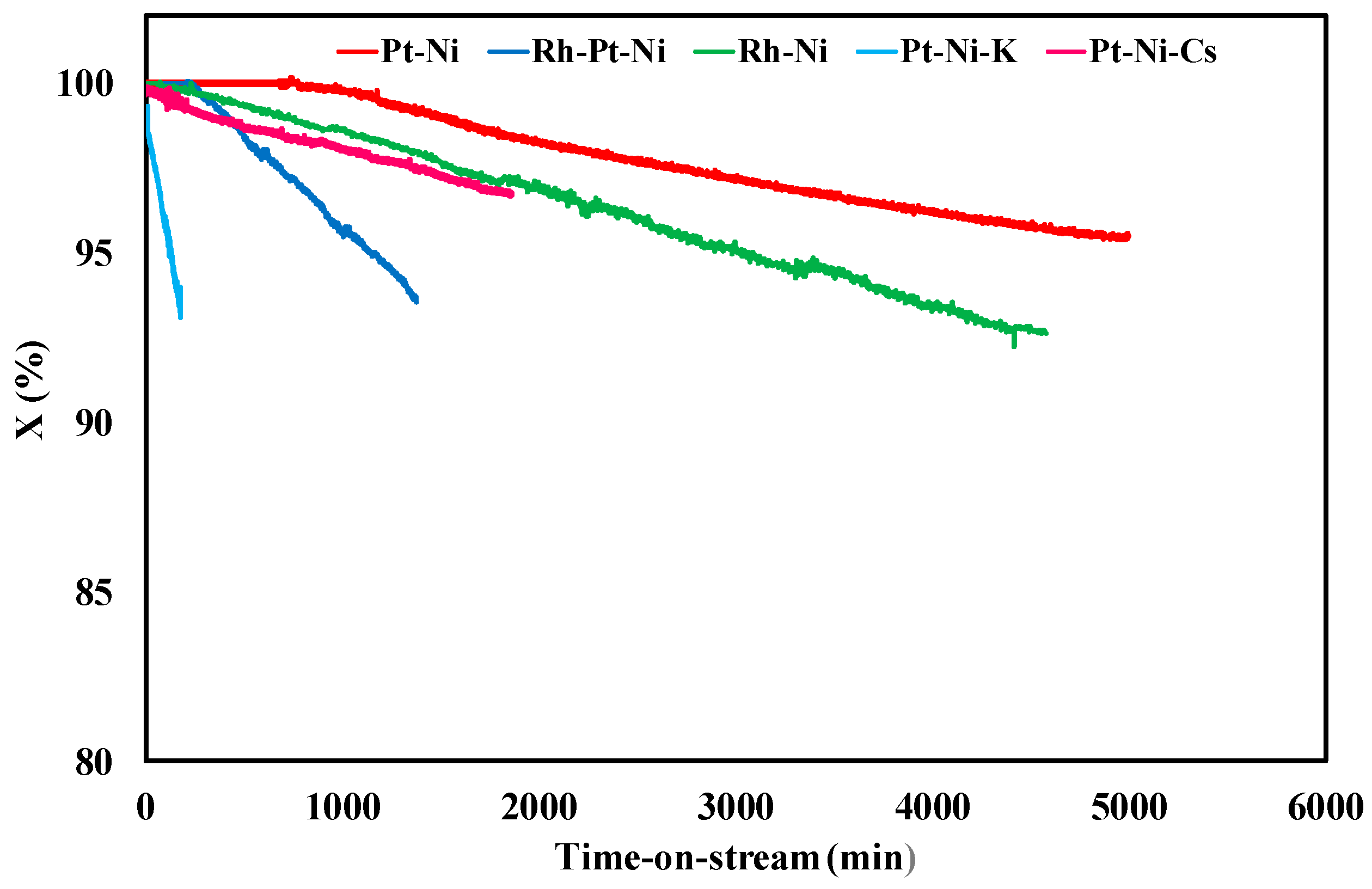
The stability tests carried out at 450 °C and f.r. = 3 were performed for different times, selected in order to reach 500 mbar of pressure drops through the bed and guarantee almost the same mass of carbon deposited. The results shown in Table 3 revealed a very fast reactor plugging for the K-based catalyst, which also displayed a CFR of 0.017 gcoke,oxidized·gcat−1·gc,fed−1·h−1. A quite high coke selectivity was also observed over the Pt-Ni sample: pressure drops reached the critical value of 500 mbar within 310 min. However, it is interesting to observe that Rh deposition on Pt-Ni/CeO2-SiO2 catalyst reduced carbon formation rate from 0.0030 to 0.00084 gcoke,oxidized·gcat−1·gc,fed−1·h−1. On the other hand, over the sample basified by Cs deposition, CFR was halved with respect to the Rh-Pt-Ni sample. The lowest activity towards carbon formation reactions was measured over the bimetallic catalyst containing Rh: also other authors found that Rh is less selective than Pt towards coke precursors formation during reforming [47]. However, looking at the coke gasification measurements, differently form the result previously reported about the clear role of alkali metals [48] as well as Rh in carbon gasification reactions [49], CGR was unaffected by catalyst formulation and very similar values were recorded over the five catalysts (Table 3). Figure 3 displays the trend of ethanol conversion as a function of time on stream: TOS (time-on-stream) for all the samples was equal to critical plugging time, except for the Pt-Ni catalyst, which was tested for 5000 min, in order to evaluate in more detail the dependence of X vs TOS. It was found that Pt-Ni and Rh-Pt-Ni catalysts maintained initial catalytic performances for at least 240 min without apparent deactivation. Conversely, similar C2H5OH profiles were recorded over the Rh-Pt-Ni and Pt-Ni-Cs samples, with an activity reduction from 100% to 97% in the time range of 0–1900 min. The K-containing catalyst displayed the worst performances among the prepared catalysts with a very fast conversion decrease to 93%. The Pt-Ni conversion profile was higher than that recorded over the other samples, with an X value of 95% after 5000 min. It is also interesting to note that the bimetallic catalyst containing Pt also assures an initial hydrogen yield better than the other samples (26.5%) and very close to the value predicted by thermodynamic equilibrium. The unsatisfactory behaviour of the Pt-Ni-K catalyst can be ascribed to its low surface area (Table 1) and a subsequent worse active species dispersion, which reduced ethanol conversion in reforming reactions [50].
On the other hand, Rh addition had no positive effect on the stability of Pt-Ni catalyst while the bimetallic catalyst containing Rh showed slightly improved activity with respect to the Cs-containing sample. The described results suggest that, despite Rh and alkali metals are able to reduce coke formation rate and, consequently, enhance reactor-plugging time, no improvements can be drawn under the point of view of gasification reactions, which spurs the investigation of other parameters suspected to affect coke removal by steam (water partial pressure and reaction temperature). Moreover, a negative effect of support basification, Rh addition as well as Pt substitution by Rh on the long-term stability and H2 yield of Pt-Ni/CeO2-SiO2 catalyst was observed. Based on the above discussion, the Pt-Ni catalyst was selected as the most interesting sample in terms of catalytic performances and employed for further tests.
3.3. Ethanol Steam Reforming Tests: Effect of Operative Conditions
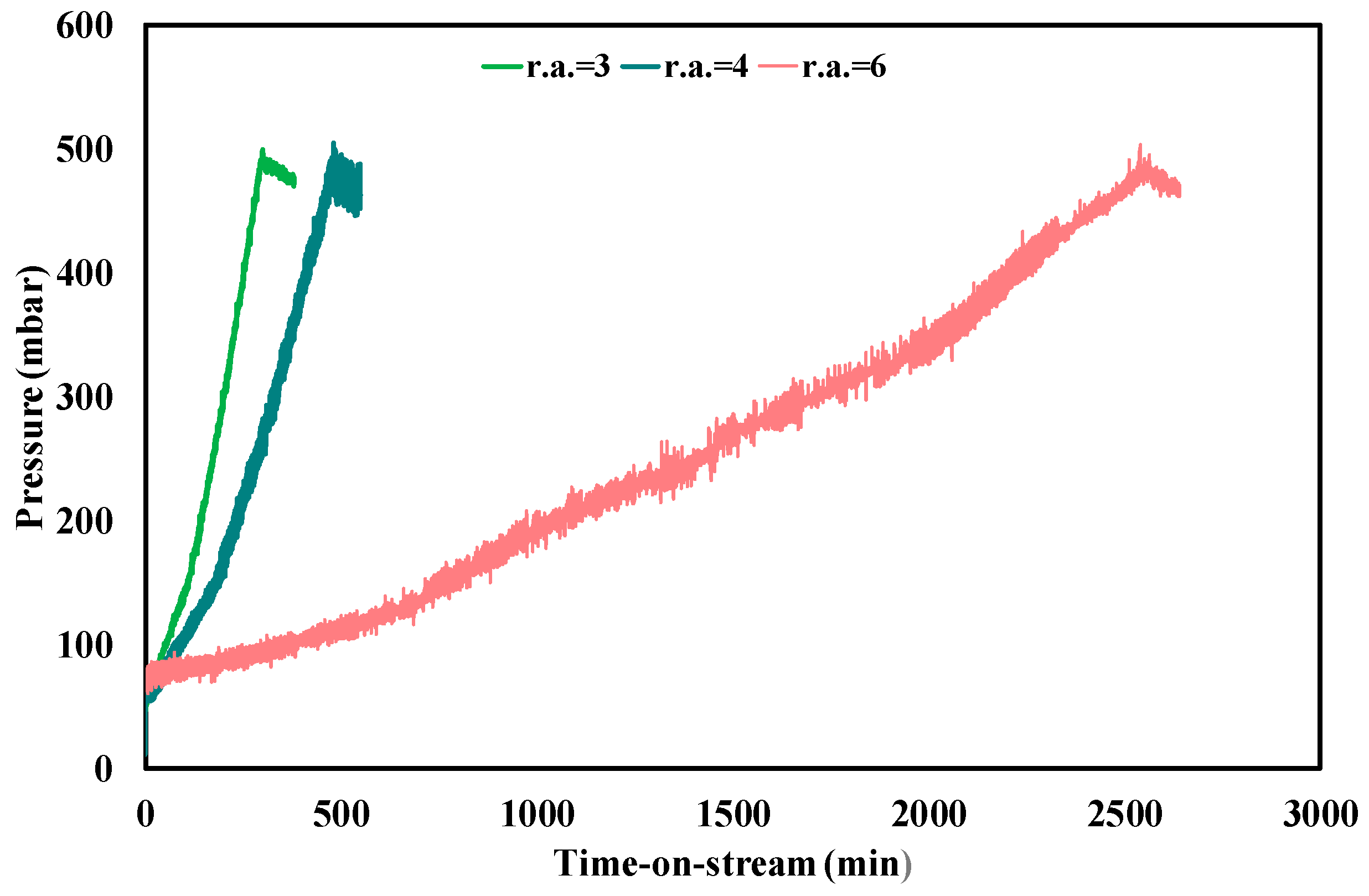
The impact of different amount of steam and the influence of reaction temperatures on the Pt-Ni/CeO2-SiO2 catalyst performances during ethanol steam reforming at WHSV = 4.1 h−1 was also investigated. C2H5OH conversion was not affected by water partial pressure change and 100% of conversion (as predicted by thermodynamic analysis) was measured during all the tests, carried out for a TOS equal to the critical plugging time. A stable product gas distribution was also recorded, with a mean hydrogen yield increasing from 26.5% at f.r. = 3 to 30% and 36%, at f.r. = 4 and 6, respectively; this trend is reasonable since the added steam could favour reforming reactions [51]. In addition, a strong variation in the pressure drop profile with the growth of the amount of steam was observed (Figure 4). The plugging time measured during the test at f.r. = 6 was almost eight times higher than the value recorded at stoichiometric feeding conditions (Table 4). Moreover, for a water concentration growth in the range 40–60 vol % in the reacting mixture, CFR was reduced of almost one order of magnitude (from 0.0013 to 0.00011 gcoke,oxidized·gcat−1·gc,fed−1·h−1). However, the carbon gasification rate, as it is possible to observe also from the slope of pressure profile during gasification measurements (Figure 4) and from the data shown in Table 4, was not affected by water partial pressure. This phenomenon can be explained considering that, even when the steam to ethanol molar ratio is adjusted to favour the gasification reaction, the kinetics of the reaction can be very slow, resulting in a negligible effect of water partial pressure [52]. Based on the above results, the effect of temperature was also studied and the results are shown in Figure 5 and Table 4.
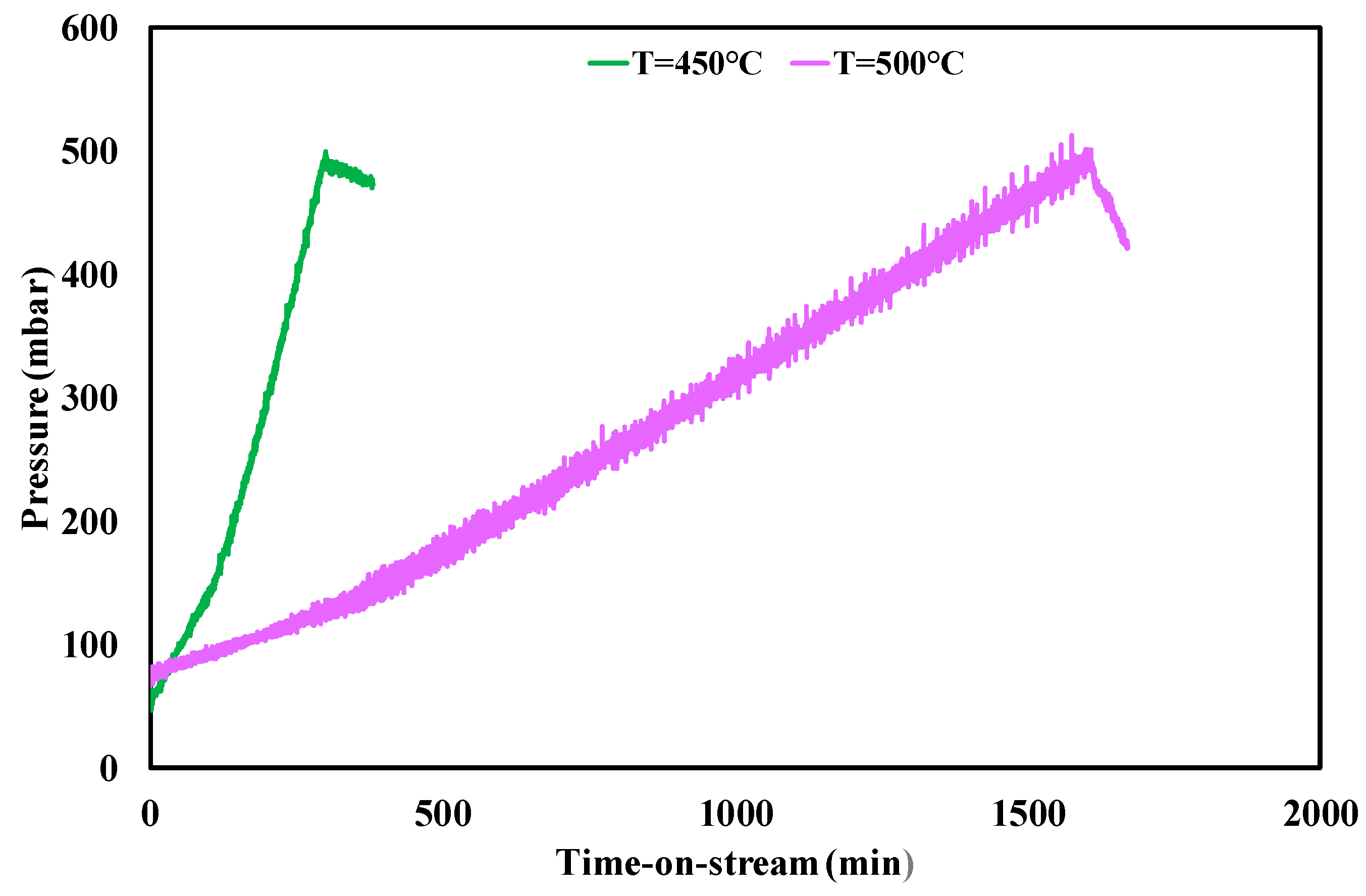
The trend of pressure drops during stability tests and the carbon formation rate variation with temperature in the range 450–500 °C suggest that the most favourable conditions for carbon deposition are always at the lowest catalyst temperature, where the reactions leading to coke formation are much higher than the rates of carbon gasification reactions [53]. The temperature growth from 450 to 500 °C reduced CFR, due to the lower activity towards coke deposits formation and/or the increased contribution of gasification reaction during the test. Moreover, the results of gasification measurements revealed a strong kinetics improvement, which allowed the increase of CGR from 0.035 to 0.25 gcoke,gasified·gcat−1·h−1.
4. Conclusions
CeO2-SiO2-based catalysts prepared by wetness impregnation have been tested in an ethanol steam reforming reaction. The physiochemical properties of the prepared catalysts (Pt-Ni, Rh-Pt-Ni, Rh-Ni, Pt-Ni-K, and Pt-Ni-Cs) and the effect of catalytic formulation, as well as test conditions on the performances for ESR, were investigated. Among the tested samples, the catalysts containing Rh and Cs displayed the lowest carbon formation rate. However, lessened ethanol conversions and hydrogen yields were measured over the above samples (the worse performances being observed over the K-based sample) while the Pt-Ni/CeO2-SiO2 catalyst displayed the best results in terms of activity and stability; the total conversion was recorded for more than 10 h and the initial hydrogen yield was very close to the thermodynamic predictions. The latter sample, which displayed relevant resistance towards deactivation, was further investigated under different operative conditions (H2O/C2H5OH ratio and T). The increase of water amount in the feed promoted hydrogen production and reduced the carbon formation rate over the Pt-Ni catalyst, without, however, bringing any relevant improvement in terms of carbon gasification. Conversely, the temperature rise enhanced both the plugging time and coke gasification rates.
Acknowledgments
The research leading to these results has received funding from the European Union's Seventh Framework Programme (FP7/2007-2013) for the Fuel Cells and Hydrogen Joint Technology Initiative under grant agreement No. 621196. The present publication reflects only the author’s views and the FCH JU (Fuel Cells and Hydrogen Joint Undertaking) and the Union are not liable for any use that may be made of the information contained therein.
Author Contributions
Vincenzo Palma and Concetta Ruocco conceived and designed the experiments; Concetta Ruocco performed the experiments; Vincenzo Palma, Concetta Ruocco and Antonio Ricca analyzed the data; Eugenio Meloni contributed reagents/materials/analysis tools; Concetta Ruocco wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Conte, M.; Di Mario, F.; Iacobazzi, A.; Mattucci, A.; Moreno, A.; Ronchetti, M. Hydrogen as future energy carrier: The ENEA point of view on technology and application prospects. Energies 2009, 2, 150. [Google Scholar] [CrossRef]
- Cifuentes, B.; Hernández, M.; Monsalve, S.; Cobo, M. Hydrogen production by steam reforming of ethanol on a RhPt/CeO2/SiO2 catalyst: Synergistic effect of the Si:Ce ratio on the catalyst performance. Appl. Catal. A Gen. 2016, 523, 283–293. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, Z.; Liu, L.; Wang, X. Heat recovery from high temperature slags: A review of chemical methods. Energies 2015, 8, 1917. [Google Scholar] [CrossRef]
- Zhang, N.; Chen, X.; Chu, B.; Cao, C.; Jin, Y.; Cheng, Y. Catalytic performance of Ni catalyst for steam methane reforming in a micro-channel reactor at high pressure. Chem. Eng. Proc. Proc. Intensif. 2017, 118, 19–25. [Google Scholar] [CrossRef]
- Hu, L.; Lin, L.; Liu, S. Chemoselective hydrogenation of biomass-derived 5-hydroxymethylfurfural into the liquid biofuel 2, 5-dimethylfuran. Ind. Eng. Chem. Res. 2014, 53, 9969–9978. [Google Scholar] [CrossRef]
- Magdeldin, M.; Kohl, T.; De Blasio, C.; Järvinen, M.; Won Park, S.; Giudici, R. The BioSCWG project: Understanding the trade-offs in the process and thermal design of hydrogen and synthetic natural gas production. Energies 2016, 9, 838. [Google Scholar] [CrossRef]
- Sisinni, M.; Di Carlo, A.; Bocci, E.; Micangeli, A.; Naso, V. Hydrogen-rich gas production by sorption enhanced steam reforming of woodgas containing TAR over a commercial Ni Catalyst and calcined dolomite as CO2 sorbent. Energies 2013, 6, 3167. [Google Scholar] [CrossRef] [Green Version]
- Inayat, A.; Ahmad, M.M.; Yusup, S.; Mutalib, M.I.A. Biomass steam gasification with in-situ CO2 capture for enriched hydrogen gas production: A reaction kinetics modelling approach. Energies 2010, 3, 1472. [Google Scholar] [CrossRef]
- Tonezzer, M.; Dang, T.T.L.; Tran, Q.H.; Iannotta, S. Dual-selective hydrogen and ethanol sensor for steam reforming systems. Sens. Actuators B Chem. 2016, 236, 1011–1019. [Google Scholar] [CrossRef]
- Yasuda, M.; Kurogi, R.; Tsumagari, H.; Shiragami, T.; Matsumoto, T. New approach to fuelization of herbaceous lignocelluloses through simultaneous saccharification and fermentation followed by photocatalytic reforming. Energies 2014, 7, 4087. [Google Scholar] [CrossRef]
- Hari Krishna, S.; Chowdary, G.V. Optimization of simultaneous saccharification and fermentation for the production of ethanol from lignocellulosic biomass. J. Agric. Food Chem. 2000, 48, 1971–1976. [Google Scholar] [CrossRef]
- Dantas, S.C.; Resende, K.A.; Ávila-Neto, C.N.; Noronha, F.B.; Bueno, J.M.C.; Hori, C.E. Nickel supported catalysts for hydrogen production by reforming of ethanol as addressed by in situ temperature and spatial resolved XANES analysis. Int. J. Hydrogen Energy 2016, 41, 3399–3413. [Google Scholar] [CrossRef]
- Carvalho, F.L.S.; Asencios, Y.J.O.; Bellido, J.D.A.; Assaf, E.M. Bio-ethanol steam reforming for hydrogen production over Co3O4/CeO2 catalysts synthesized by one-step polymerization method. Fuel Proc. Technol. 2016, 142, 182–191. [Google Scholar] [CrossRef]
- Sharma, Y.C.; Kumar, A.; Prasad, R.; Upadhyay, S.N. Ethanol steam reforming for hydrogen production: Latest and effective catalyst modification strategies to minimize carbonaceous deactivation. Renew. Sustain. Energy Rev. 2017, 74, 89–103. [Google Scholar] [CrossRef]
- Arslan, A.; Doğu, T. Effect of calcination/reduction temperature of Ni impregnated CeO2-ZrO2 catalysts on hydrogen yield and coke minimization in low temperature reforming of ethanol. Int. J. Hydrogen Energy 2016, 41, 16752–16761. [Google Scholar] [CrossRef]
- Cifuentes, B.; Valero, M.; Conesa, J.; Cobo, M. Hydrogen production by steam reforming of ethanol on Rh-Pt catalysts: Influence of CeO2, ZrO2, and La2O3 as Supports. Catalysts 2015, 5, 1872. [Google Scholar] [CrossRef] [Green Version]
- Yaakob, Z.; Bshish, A.; Ebshish, A.; Tasirin, S.; Alhasan, F. Hydrogen production by steam reforming of ethanol over nickel catalysts supported on sol gel made alumina: Influence of calcination temperature on supports. Materials 2013, 6, 2229. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Inaba, M. Steam reforming of bio-ethanol to produce hydrogen over Co/CeO2 catalysts derived from Ce1-xCoxO2-y precursors. Catalysts 2016, 6, 26. [Google Scholar] [CrossRef]
- Kugai, J.; Velu, S.; Song, C. Low-temperature reforming of ethanol over CeO2-supported Ni-Rh bimetallic catalysts for hydrogen production. Catal. Lett. 2005, 101, 255–264. [Google Scholar] [CrossRef]
- Palma, V.; Ruocco, C.; Castaldo, F.; Ricca, A.; Boettge, D. Ethanol steam reforming over bimetallic coated ceramic foams: Effect of reactor configuration and catalytic support. Int. J. Hydrogen Energy 2015, 40, 12650–12662. [Google Scholar] [CrossRef]
- Zhou, L.; Guo, Y.; Kameyama, H.; Basset, J.-M. An anodic alumina supported Ni-Pt bimetallic plate-type catalysts for multi-reforming of methane, kerosene and ethanol. Int. J. Hydrogen Energy 2014, 39, 7291–7305. [Google Scholar] [CrossRef]
- Carrero, A.; Calles, J.A.; Vizcaíno, A.J. Effect of Mg and Ca addition on coke deposition over Cu–Ni/SiO2 catalysts for ethanol steam reforming. Chem. Eng. J. 2010, 163, 395–402. [Google Scholar] [CrossRef]
- Wang, S.; Lu, G.Q. Effects of promoters on catalytic activity and carbon deposition of Ni/γ-Al2O3 catalysts in CO2 reforming of CH4. J. Chem. Technol. Biotechnol. 2000, 75, 589–595. [Google Scholar] [CrossRef]
- Rass-Hansen, J.; Christensen, C.H.; Sehested, J.; Helveg, S.; Rostrup-Nielsen, J.R.; Dahl, S. Renewable hydrogen: Carbon formation on Ni and Ru catalysts during ethanol steam-reforming. Green Chem. 2007, 9, 1016–1021. [Google Scholar] [CrossRef]
- Akiyama, M.; Oki, Y.; Nagai, M. Steam reforming of ethanol over carburized alkali-doped nickel on zirconia and various supports for hydrogen production. Catal. Today 2012, 181, 4–13. [Google Scholar] [CrossRef]
- Osorio-Vargas, P.; Campos, C.H.; Navarro, R.M.; Fierro, J.L.G.; Reyes, P. Improved ethanol steam reforming on Rh/Al2O3 catalysts doped with CeO2 or/and La2O3: Influence in reaction pathways including coke formation. Appl. Catal. A Gen. 2015, 505, 159–172. [Google Scholar] [CrossRef]
- Konsolakis, M.; Ioakimidis, Z.; Kraia, T.; Marnellos, G. Hydrogen production by ethanol steam reforming (ESR) over CeO2 supported transition metal (Fe, Co, Ni, Cu) catalysts: Insight into the structure-activity relationship. Catalysts 2016, 6, 9. [Google Scholar] [CrossRef]
- Tao, J.; Zhao, L.; Dong, C.; Lu, Q.; Du, X.; Dahlquist, E. Catalytic steam reforming of toluene as a model compound of biomass gasification tar using Ni-CeO2/SBA-15 catalysts. Energies 2013, 6, 3284. [Google Scholar] [CrossRef]
- Santander, J.A.; Tonetto, G.M.; Pedernera, M.N.; López, E. Ni/CeO2-MgO catalysts supported on stainless steel plates for ethanol steam reforming. Int. J. Hydrogen Energy 2017, 42, 9482–9492. [Google Scholar] [CrossRef]
- Parlett, C.M.A.; Aydin, A.; Durndell, L.J.; Frattini, L.; Isaacs, M.A.; Lee, A.F.; Liu, X.; Olivi, L.; Trofimovaite, R.; Wilson, K.; et al. Tailored mesoporous silica supports for Ni catalysed hydrogen production from ethanol steam reforming. Catal. Commun. 2017, 91, 76–79. [Google Scholar] [CrossRef]
- Hu, J.; Yu, C.; Bi, Y.; Wei, L.; Chen, J.; Chen, X. Preparation and characterization of Ni/CeO2-SiO2 catalysts and their performance in catalytic partial oxidation of methane to syngas. Chin. J. Catal. 2014, 35, 8–20. [Google Scholar] [CrossRef]
- Palma, V.; Ruocco, C.; Meloni, E.; Ricca, A. Oxidative steam reforming of ethanol on mesoporous silica supported PtNi/CeO2 catalysts. Int. J. Hydrogen Energy 2017, 42, 1598–1608. [Google Scholar] [CrossRef]
- Palma, V.; Ruocco, C.; Meloni, E.; Gallucci, F.; Ricca, A. Enhancing Pt-Ni/CeO2 performances for ethanol reforming by catalyst supporting on high surface silica. Catal. Today 2017, in press. [Google Scholar] [CrossRef]
- Qin, H.; Qian, X.; Meng, T.; Lin, Y.; Ma, Z. Pt/MOx/SiO2, Pt/MOx/TiO2, and Pt/MOx/Al2O3 Catalysts for CO Oxidation. Catalysts 2015, 5, 606. [Google Scholar] [CrossRef]
- Solsona, B.; Sanchis, R.; Dejoz, A.; García, T.; Ruiz-Rodríguez, L.; López Nieto, J.; Cecilia, J.; Rodríguez-Castellón, E. Total oxidation of propane using CeO2 and CuO-CeO2 catalysts prepared using templates of different nature. Catalysts 2017, 7, 96. [Google Scholar] [CrossRef]
- Chen, A.; Guo, H.; Song, Y.; Chen, P.; Lou, H. Recyclable CeO2-ZrO2 and CeO2-TiO2 mixed oxides based Pt catalyst for aqueous-phase reforming of the low-boiling fraction of bio-oil. Int. J. Hydrogen Energy 2017, 42, 9577–9588. [Google Scholar] [CrossRef]
- Hong, X.; Sun, Y.; Zhu, T.; Liu, Z. Pt-Au/MOx-CeO2 (M = Mn, Fe, Ti) catalysts for the co-oxidation of CO and H2 at room temperature. Molecules 2017, 22, 351. [Google Scholar] [CrossRef] [PubMed]
- Hérault, N.; Olivet, L.; Pirault-Roy, L.; Especel, C.; Vicerich, M.A.; Pieck, C.L.; Epron, F. Controlled preparation and characterization of Pt-Rh/Al2O3 bimetallic catalysts for reactions in reducing conditions. Appl. Catal. A Gen. 2016, 517, 81–90. [Google Scholar] [CrossRef]
- Atzori, L.; Cutrufello, M.G.; Meloni, D.; Cannas, C.; Gazzoli, D.; Monaci, R.; Sini, M.F. Highly active NiO-CeO2 catalysts for synthetic natural gas production by CO2 methanation. Catal. Today 2017, in press. [Google Scholar] [CrossRef]
- Cobo, M.; Pieruccini, D.; Abello, R.; Ariza, L.; Córdoba, L.F.; Conesa, J.A. Steam reforming of ethanol over bimetallic RhPt/La2O3: Long-term stability under favorable reaction conditions. Int. J. Hydrogen Energy 2013, 38, 5580–5593. [Google Scholar] [CrossRef]
- Carrero, A.; Calles, J.; García-Moreno, L.; Vizcaíno, A. Production of renewable hydrogen from glycerol steam reforming over bimetallic Ni-(Cu,Co,Cr) catalysts supported on SBA-15 Silica. Catalysts 2017, 7, 55. [Google Scholar] [CrossRef]
- Ocsachoque, M.; Pompeo, F.; Gonzalez, G. Rh–Ni/CeO2-Al2O3 catalysts for methane dry reforming. Catal. Today 2011, 172, 226–231. [Google Scholar] [CrossRef]
- Xiong, H.; Motchelaho, M.A.; Moyo, M.; Jewell, L.L.; Coville, N.J. Effect of group I alkali metal promoters on Fe/CNT catalysts in Fischer-Tropsch synthesis. Fuel 2015, 150, 687–696. [Google Scholar] [CrossRef]
- Aboud, M.; ALOthman, Z.; Habila, M.; Zlotea, C.; Latroche, M.; Cuevas, F. Hydrogen storage in pristine and d10-block metal-anchored activated carbon made from local wastes. Energies 2015, 8, 3578. [Google Scholar] [CrossRef]
- Mortola, V.B.; Damyanova, S.; Zanchet, D.; Bueno, J.M.C. Surface and structural features of Pt/CeO2-La2O3-Al2O3 catalysts for partial oxidation and steam reforming of methane. Appl. Catal. B Environ. 2011, 107, 221–236. [Google Scholar] [CrossRef]
- Mamontov, G.V.; Grabchenko, M.V.; Sobolev, V.I.; Zaikovskii, V.I.; Vodyankina, O.V. Ethanol dehydrogenation over Ag-CeO2/SiO2 catalyst: Role of Ag-CeO2 interface. Appl. Catal. A Gen. 2016, 528, 161–167. [Google Scholar] [CrossRef]
- de Caprariis, B.; de Filippis, P.; Palma, V.; Petrullo, A.; Ricca, A.; Ruocco, C.; Scarsella, M. Rh, Ru and Pt ternary perovskites type oxides BaZr(1 − x)MexO3 for methane dry reforming. Appl. Catal. A Gen. 2016, 517, 47–55. [Google Scholar] [CrossRef]
- Corella, J.; Toledo, J.M.; Molina, G. Steam gasification of coal at low-medium (600−800 °C) temperature with simultaneous CO2 capture in fluidized bed at atmospheric pressure: The effect of inorganic species. 1. Literature review and comments. Ind. Eng. Chem. Res. 2006, 45, 6137–6146. [Google Scholar] [CrossRef]
- Hou, T.; Yu, B.; Zhang, S.; Xu, T.; Wang, D.; Cai, W. Hydrogen production from ethanol steam reforming over Rh/CeO2 catalyst. Catal. Commun. 2015, 58, 137–140. [Google Scholar] [CrossRef]
- Zhao, X.; Lu, G. Improving catalytic activity and stability by in-situ regeneration of Ni-based catalyst for hydrogen production from ethanol steam reforming via controlling of active species dispersion. Int. J. Hydrogen Energy 2016, 41, 13993–14002. [Google Scholar] [CrossRef]
- de Lima, S.M.; da Cruz, I.O.; Jacobs, G.; Davis, B.H.; Mattos, L.V.; Noronha, F.B. Steam reforming, partial oxidation, and oxidative steam reforming of ethanol over Pt/CeZrO2 catalyst. J. Catal. 2008, 257, 356–368. [Google Scholar] [CrossRef]
- Trimm, D.L. Coke formation and minimisation during steam reforming reactions. Catal. Today 1997, 37, 233–238. [Google Scholar] [CrossRef]
- Coll, R.; Salvadó, J.; Farriol, X.; Montané, D. Steam reforming model compounds of biomass gasification tars: Conversion at different operating conditions and tendency towards coke formation. Fuel Proc. Technol. 2001, 74, 19–31. [Google Scholar] [CrossRef]
Figure 1.
X-ray diffraction patterns of the CeO2-SiO2 support and the catalysts.

Figure 2.
H2 consumption profiles of the CeO2-SiO2 based catalysts; (insertion: H2 consumption over the bare support).
Figure 2.
H2 consumption profiles of the CeO2-SiO2 based catalysts; (insertion: H2 consumption over the bare support).
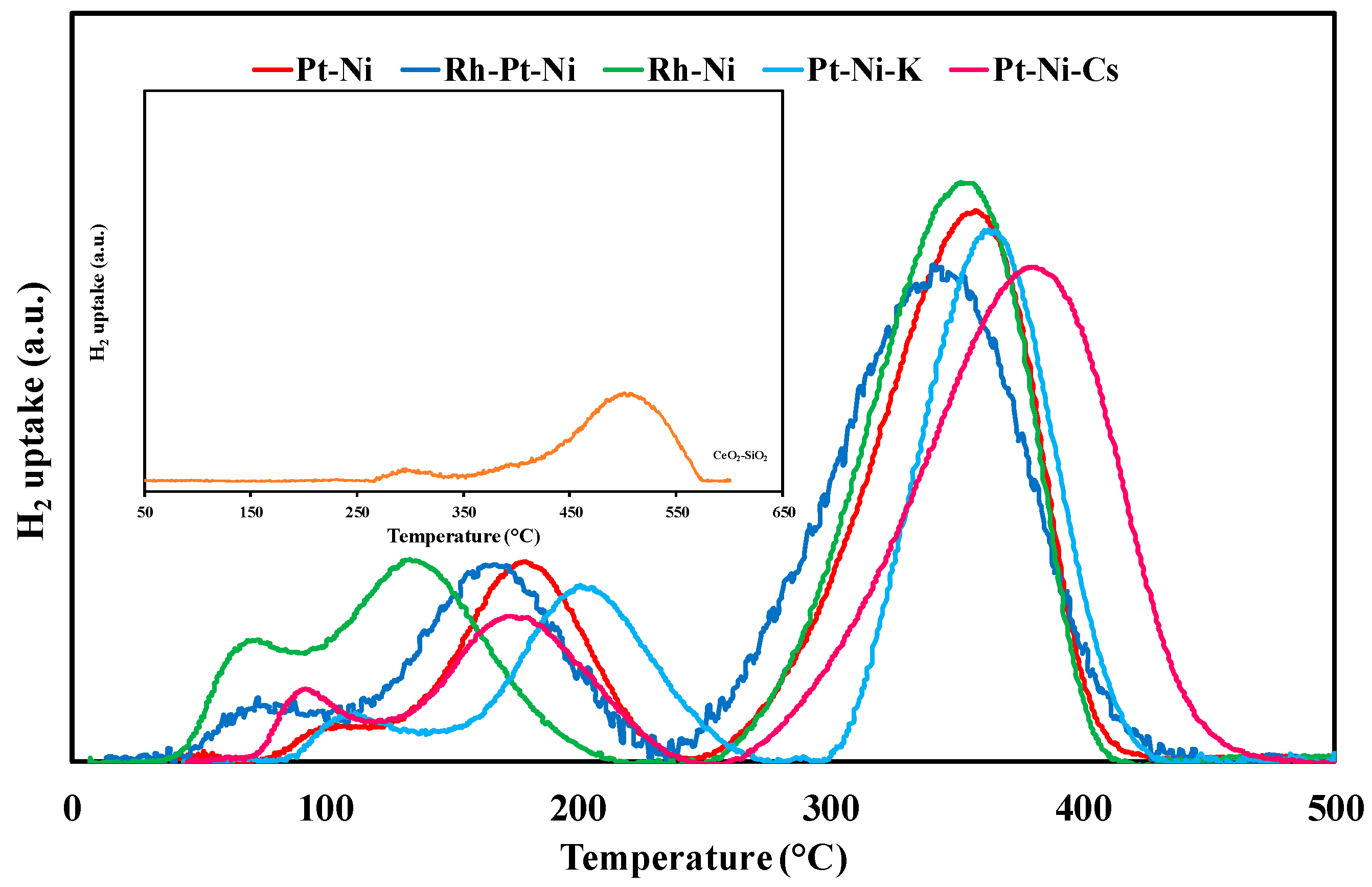
Figure 3.
Results of stability tests on CeO2-SiO2 based catalysts at 450 °C, f.r. = 3 and WHSV = 4.1 h−1.
Figure 3.
Results of stability tests on CeO2-SiO2 based catalysts at 450 °C, f.r. = 3 and WHSV = 4.1 h−1.
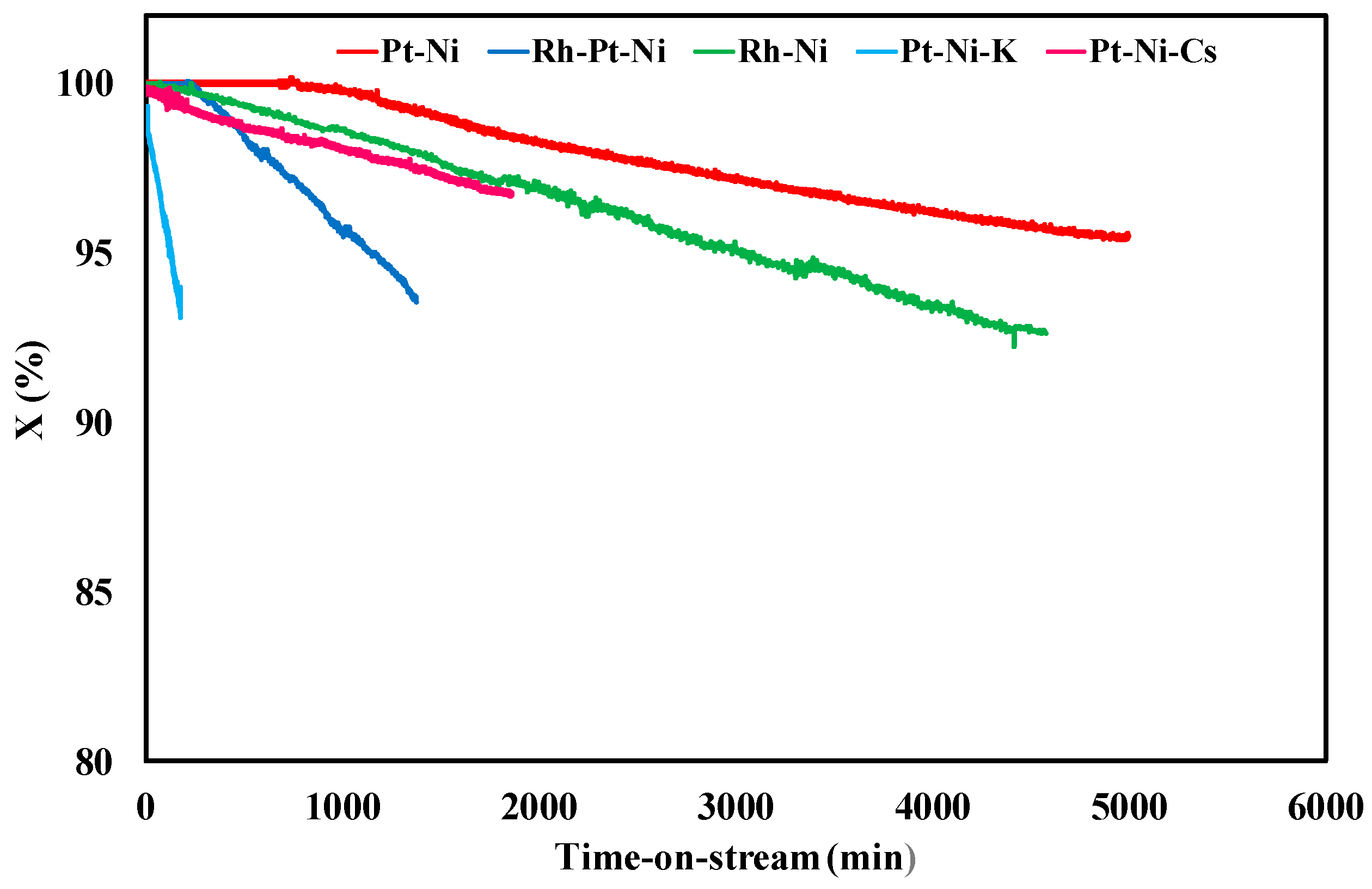
Figure 4.
Pressure drops profile through the catalytic bed during stability tests and gasification measurements at 450 °C, f.r. between 3 and 6, WHSV = 4.1 h−1.
Figure 4.
Pressure drops profile through the catalytic bed during stability tests and gasification measurements at 450 °C, f.r. between 3 and 6, WHSV = 4.1 h−1.
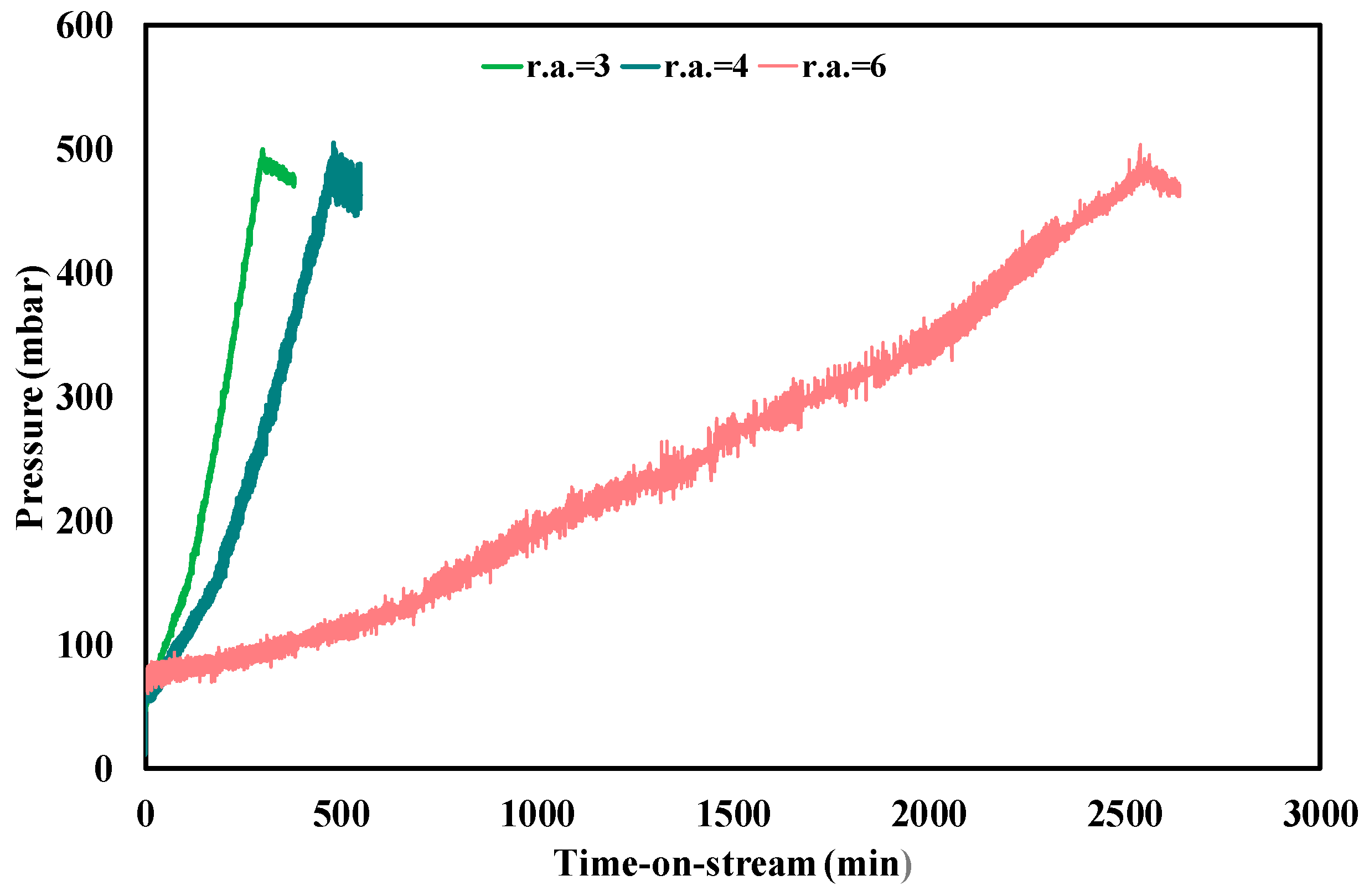
Figure 5.
Pressure drops profile through the catalytic bed during stability tests and gasification measurements at f.r. = 3 and different temperatures; WHSV = 4.1 h−1.
Figure 5.
Pressure drops profile through the catalytic bed during stability tests and gasification measurements at f.r. = 3 and different temperatures; WHSV = 4.1 h−1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Chemical composition, CeO2 crystallite sizes (d), and specific surface area of the calcined samples.
Table 1.
Chemical composition, CeO2 crystallite sizes (d), and specific surface area of the calcined samples.
| Sample | SiO2 (wt %) | CeO2 (wt %) | Ni (wt %) | Pt (or Rh) (wt %) | SSA (m2∙g−1) | d (Å) |
|---|---|---|---|---|---|---|
| CeSi | 68.9 | 31.1 | - | - | 254 | 78 |
| Pt-Ni | 65.3 | 30.3 | 3.4 | 1 | 255 | 73 |
| Rh-Pt-Ni | 65.3 | 30.3 | 3.4 | 1 | 197 | 68 |
| Rh-Ni | 65 | 30.3 | 3.6 | 1.1 | 223 | 73 |
| Pt-Ni-K | 65.3 | 30.2 | 3.5 | 1 | 155 | 68 |
| Pt-Ni-Cs | 65.3 | 30.3 | 3.3 | 1.1 | 203 | 69 |
Table 2.
Comparison between experimental and theoretical amounts of hydrogen consumption during TPR.
Table 2.
Comparison between experimental and theoretical amounts of hydrogen consumption during TPR.
| Sample | T (°C) | H2 Uptake (µmol/gcat) | ||
|---|---|---|---|---|
| Experimental | Total Experimental | Total Expected | ||
| CeO2-SiO2 | - | 300 | - | - |
| - | 448 | - | - | |
| - | 509 | 298 | 6210 | |
| Pt-Ni | 109 | 119 (PtOx) | - | - |
| 179 | 943 (PtOx) | - | - | |
| 334 | 1032 (NiO) | - | - | |
| 366 | 641 (NiO) | 2735 | 2012 | |
| Rh-Pt-Ni | 70 | 95 (RhOx) | - | - |
| 96 | 154 (PtOx) | - | - | |
| 167 | 1029 (PtOx) | - | - | |
| 315 | 836 (NiO) | - | - | |
| 357 | 967 (NiO) | 3081 | 2109 | |
| Rh-Ni | 69 | 286 (RhOx) | - | - |
| 134 | 1184 (RhOx) | - | - | |
| 339 | 1189 (NiO) | - | - | |
| 360 | 550 (NiO) | 3209 | 2287 | |
| Pt-Ni-K | 116 | 179 (PtOx) | - | - |
| 204 | 925 (NiO) | - | - | |
| 363 | 1367 (NiO) | 2471 | 2012 | |
| Pt-Ni-Cs | 95 | 151 (PtOx) | - | - |
| 174 | 742 (PtOx) | - | - | |
| 353 | 1024 (NiO) | - | - | |
| 393 | 771 (NiO) | 2688 | 2012 | |
Table 3.
Initial hydrogen yield (Y), plugging time corresponding to 500 mbar (tplugging), carbon formation rate (CFR), and carbon gasification rate (CGR) during stability tests carried out at 450 °C, f.r. = 3, and WHSV = 4.1 h−1.
Table 3.
Initial hydrogen yield (Y), plugging time corresponding to 500 mbar (tplugging), carbon formation rate (CFR), and carbon gasification rate (CGR) during stability tests carried out at 450 °C, f.r. = 3, and WHSV = 4.1 h−1.
| Sample | Y (%) | tplugging (min) | CFR (gcoke,oxidized·gcat−1·gc,fed−1·h−1) | CGR (gcoke,gasified·gcat−1·h−1) |
|---|---|---|---|---|
| Pt-Ni | 26.5 | 310 | 0.0030 | 0.035 |
| Rh-Pt-Ni | 23.9 | 1300 | 0.00084 | 0.033 |
| Rh-Ni | 23.2 | 4900 | 0.000065 | 0.035 |
| Pt-Ni-K | 20.1 | 200 | 0.017 | 0.037 |
| Pt-Ni-Cs | 24.7 | 1600 | 0.00039 | 0.036 |
Table 4.
Plugging time corresponding to 500 mbar (tplugging), carbon formation rate (CFR) and carbon gasification rate (CGR) during stability tests carried out different operative conditions; WHSV = 4.1 h−1.
Table 4.
Plugging time corresponding to 500 mbar (tplugging), carbon formation rate (CFR) and carbon gasification rate (CGR) during stability tests carried out different operative conditions; WHSV = 4.1 h−1.
| Test Condition | tplugging (min) | CFR (gcoke,oxidized·gcat−1·gc,fed−1·h−1) | CGR (gcoke,gasified·gcat−1·h−1) |
|---|---|---|---|
| f.r. = 3, T = 450 °C | 310 | 0.0030 | 0.035 |
| f.r. = 4, T = 450 °C | 480 | 0.0013 | 0.035 |
| f.r. = 6, T = 450 °C | 2550 | 0.00011 | 0.033 |
| f.r. = 3, T = 500 °C | 1600 | 0.00025 | 0.25 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Palma, V.; Ruocco, C.; Meloni, E.; Ricca, A. Influence of Catalytic Formulation and Operative Conditions on Coke Deposition over CeO2-SiO2 Based Catalysts for Ethanol Reforming. Energies 2017, 10, 1030. https://doi.org/10.3390/en10071030
AMA Style
Palma V, Ruocco C, Meloni E, Ricca A. Influence of Catalytic Formulation and Operative Conditions on Coke Deposition over CeO2-SiO2 Based Catalysts for Ethanol Reforming. Energies. 2017; 10(7):1030. https://doi.org/10.3390/en10071030
Chicago/Turabian StylePalma, Vincenzo, Concetta Ruocco, Eugenio Meloni, and Antonio Ricca. 2017. "Influence of Catalytic Formulation and Operative Conditions on Coke Deposition over CeO2-SiO2 Based Catalysts for Ethanol Reforming" Energies 10, no. 7: 1030. https://doi.org/10.3390/en10071030
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.