Serum Acetyl Cholinesterase as a Biomarker of Arsenic Induced Neurotoxicity in Sprague-Dawley Rats


{kind=link}
Abstract
:Introduction
Materials and Methods
Chemicals
Animal Maintenance
Arsenic Treatment
Enzyme Analysis
Analysis
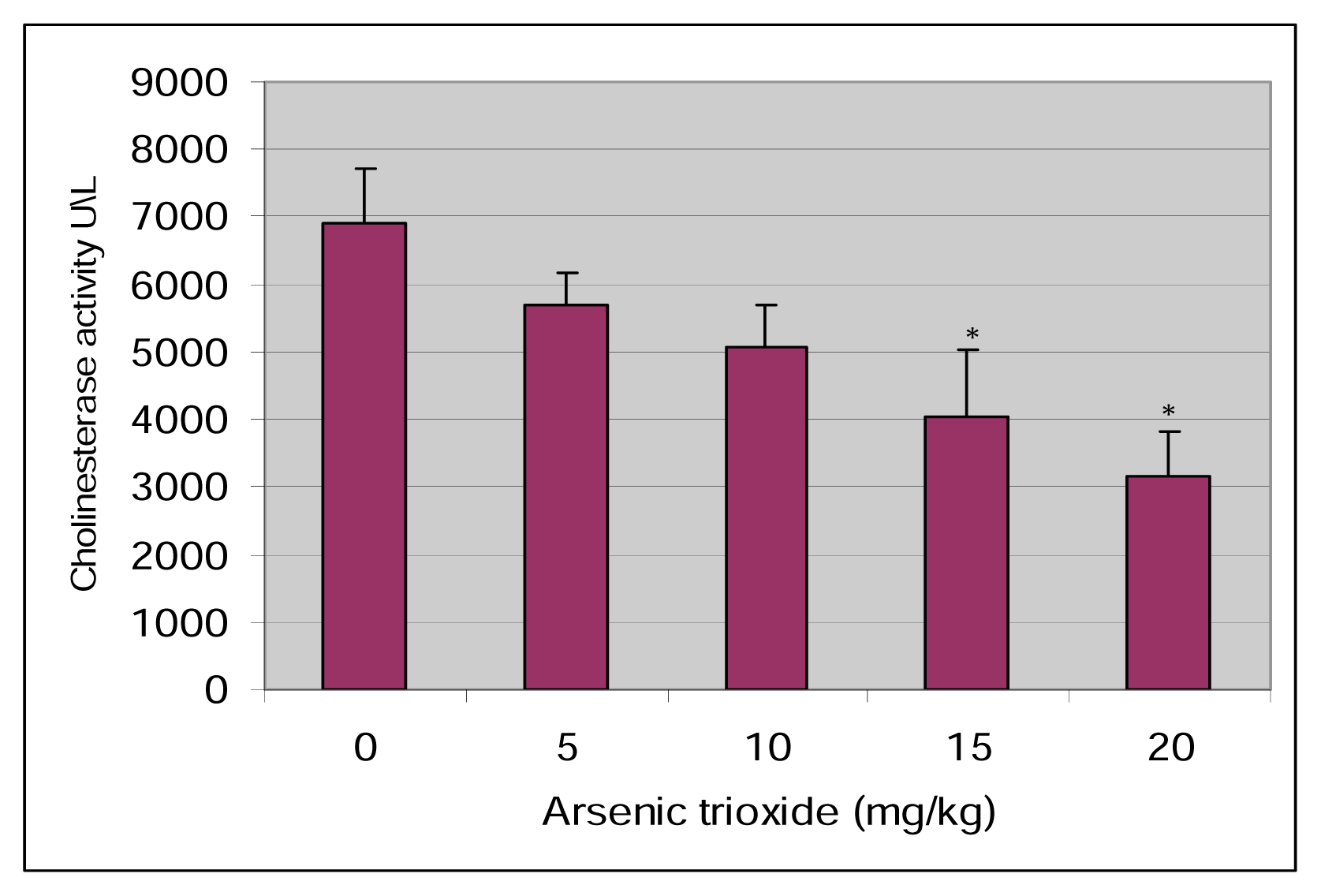
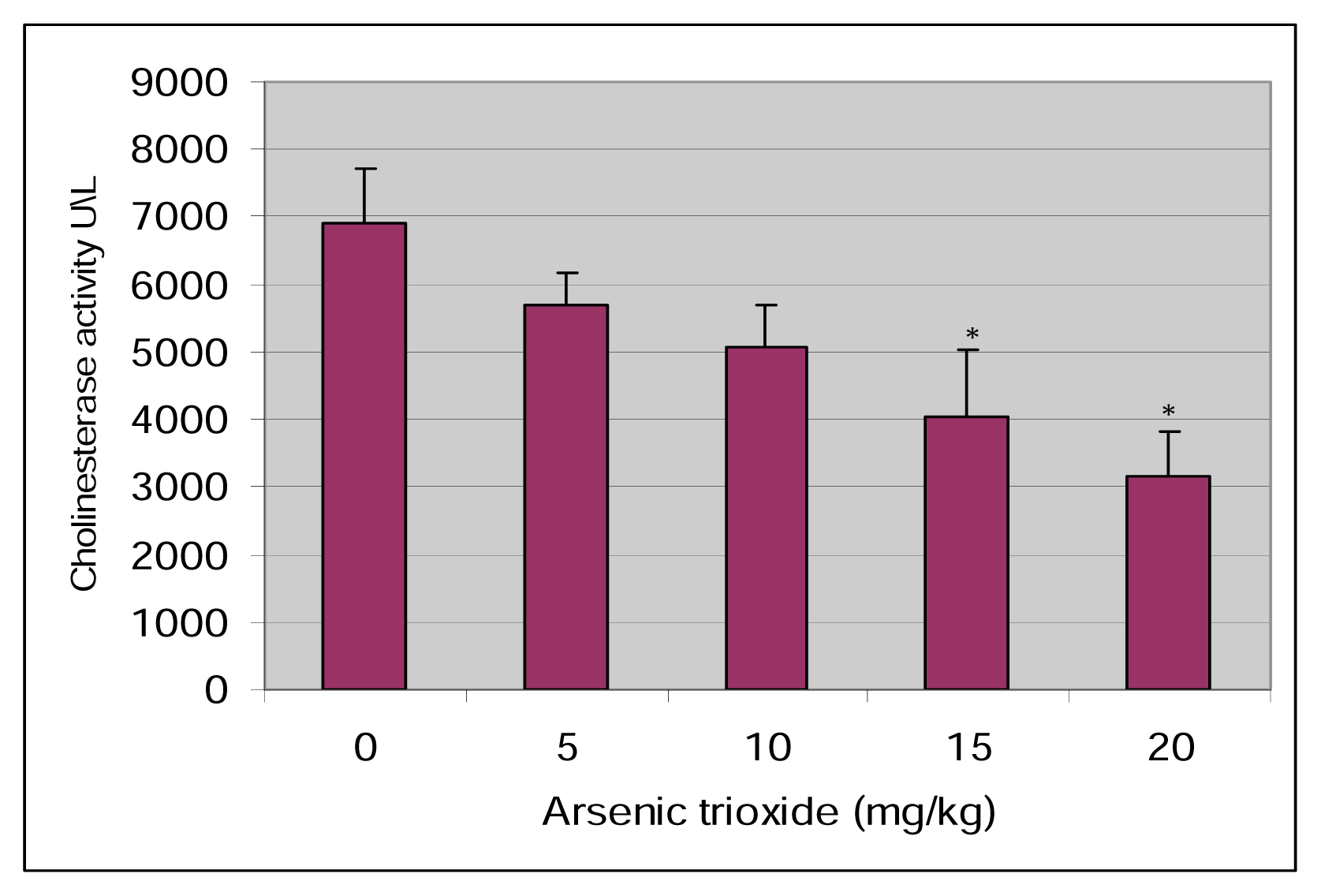
Results
Discussion
Acknowledgements
References
- Tchounwou, P. B.; Wilson, B. A.; Ishaque, A. Important considerations in the development of public health advisories for arsenic and arsenic containing compounds in drinking water. Rev. Environ. Hlth 1999, 14, 1–19. [Google Scholar]
- National Academy of Science, Arsenic; Washington, D. C, 1977.
- Rousselot, P.; Laboume, S.; Marolleau, J. P.; Larghero, T.; Noguera, M. L.; Brouet, J. C.; Fermand, J. P. Arsenic trioxide and melarsoprol induce apoptosis in plasma cell lines and in plasma cells from myeloma patients. Cancer Res 1999, 59, 1041–1048. [Google Scholar]
- Tchounwou, P. B.; Centeno, J. A.; Patlolla, A. K. Arsenic toxicity, mutagenesis, and carcinogenesis- A health risk assessment and management approach. Mol. Cell. Biochem 2004, 255, 47–55. [Google Scholar]
- Mckinney, J. D. Metabolism and disposition of inorganic arsenic in laboratory animals and humans. Environ. Oeochem. Health 1992, 14, 43–48. [Google Scholar]
- Tchounwou, P. B.; Patlolla, A.K.; Centeno, J. A. Carcinogenic and systemic health effects associated with arsenic exposure – A Critical Review. Toxicol. Pathol 2003, 31(6), 575–88. [Google Scholar]
- Tchounwou, P. B.; Wilson, B. A.; Ishaque, A.; Schneider, J. Atrazine potentiation of arsenic trioxide induced cytotoxicity and gene expression in HepG2 cells. Mol. Cell. Biochem 2001, 222, 49–59. [Google Scholar]
- Wang, Z.; Rossman, T. G. Cheng, L.W., Ed.; The Toxicology of Metals; Volume 1, CRC Press: Boca Raton, Fl, 1996; pp. 221–243. [Google Scholar]
- Aposhian, H.V.; Gurzau, E. S.; Le, X.C.; Gurzau, A.; Healy, S. M.; Lu, X.; Ma, M.; Yip, L.; Zakharyan, R. A.; Maiorino, R. M.; Dart, R. C.; Tircus, M. G.; Gonzalez-Ramirez, D.; Morgan, D. L.; Avram, D.; Aposhian, M. M. Occurrence of monomethylarsonous acid [MMA (III)], excreted in human urine. Toxicol Appl Pharmacol 2000a, 165, 74–83. [Google Scholar]
- Styblo, M.; Del Razo, L. M.; LeCluyse, E. L.; Hamilton, G. A.; Wang, C.; Cullen, W. R.; Thomas, D. J. Metabolism of arsenic in primary cultures of human and rat hepatocytes. Chem. Res. Toxicol 1999, 12, 560–565. [Google Scholar]
- Styblo, M.; Yamauchi, H.; Thomas, D.J. Comparative in vitro methylation of trivalent and pentavalent arsenicals. Toxicol. Appl. Pharmacol 1995, 135, 172–178. [Google Scholar]
- Bast, A. Anatomy and Toxicological Pathology of the nervous system. In Toxicology: Principles and Applications; Niesink, J. M., de Vries, J., Hollinger, M. A., Eds.; CRC Press: New York, 1996; pp. 974–1001. [Google Scholar]
- Pershagen, G. Sources of exposure and biological effects of arsenic. In Environmental Carcinogens Selected Methods of Analysis, Vol 8, some metals: As, Ni, Oi, Cr, Pb, Se, Zn; O’Neill, J. K., Schuller, P., Fishbein, L., Eds.; Lyon; IARC, 1986; pp. 45–61. [Google Scholar]
- Mazumder, D. N. G.; Chakraborty, A. K.; Ghose, A.; et al. Chronic arsenic toxicity from drinking tubewell water in rural West Bengal. Bulletin of the World Health Organization 1988, 66, 499–506. [Google Scholar]
- Beckett, W. S.; Moore, J. L.; Keogh, J. P.; Blecker, M. L. Acute encephalopathy due to occupational exposure to arsenic. British Journal of Industrial Medicine 1986, 43, 66–7. [Google Scholar]
- Fowler, B. A.; Woods, J. S. The effects of prolonged oral arsenate exposure on liver mitochondria of mice:morphometric and biochemical studies. Toxicol. Appl. Pharmocol 1979, 50, 177–87. [Google Scholar]
- Nagaraju, T. N.; Desiraju, T. Regional alterations in the levels of brain biogenic amines, glutamate, GABA and GAD activity due to chronic consumption of inorganic arsenide in developing and adult rats. Bull. of Environ. Contam. Toxicol 1993, 50, 100–7. [Google Scholar]
- Valkonen, S.; Savolainen, H.; Jarvisalo, J. Arsenic distribution and neurochemical effects in peroral sodium arsenite exposure of rats. Bull. of Environ. Contam. Toxicol 1983, 30, 303–8. [Google Scholar]
- Maxwell, D. M.; Vlahacos, C. P.; Lenz, D. E. A pharmacodynamic model for soman in the rat. Toxicol. lett 1988, 43, 175–188. [Google Scholar]
- Gearhart, J. M.; Jepson, G. W.; Clewell, H. J., III; Anderson, M. E.; Conolly, R. B. Physiologically based pharmacokinetic and pharmacodynamic model for the inhibition of acetylcholinesterase by diisopropylfluorophosphate. Toxicol. Appl. Pharmocol 1990, 106, 295–310. [Google Scholar]
- McCarty, L. S.; Mackay, D. Enhancing Eco-toxicological Modeling and assessment. Environ. Sci. Tech 1993, 27, 1719–1728. [Google Scholar]
- Abbas, R.; Hayton, W. L. A physiologically based pharmacokinetic and pharmacodynamic model for paraoxon in rainbow trout. Toxicol. Appl. Pharmacol 1997, 145(1), 192–201. [Google Scholar]
- Nagaraju, T. N.; Desiraju, T. Effects on operant learning and brain acetylcholine esterase activity in rats following chronic inorganic arsenic intake. Human. Exper. Toxicol 1994, 13, 353–356. [Google Scholar]
- Repetto, G.; Sanz, P.; Repetto, M. Comparative in vitro effects of sodium arsenite and sodium arsenate on neuroblastoma cells. Toxicol 1994, 92, 143–153. [Google Scholar]
- Tripathi, N.; Kannan, G. M.; Pant, B. P.; Jaiswal, D. K.; Malhotra, P. R.; Flora, S. J. S. Arsenic-induced changes in certain neurotransmitter levels and their recoveries following chelation in rat whole brain. Toxicol. Lett 1997, 92, 201–208. [Google Scholar]
- Liao, C. N.; Lin, M. C. Acute toxicity modeling of rainbow trout and silver sea bream exposed to waterborn metals. Environ. Toxicol 2001, 16, 349–360. [Google Scholar]
- Boyer, P. D.; Lardy, H.; Myrback, K. (Eds.) The Enzymes, 2nd ed.; Academic Press, 1963; New York.
- Mounter, L. A.; Whittaker, V. P. The effect of thiol and other group- specific reagents on erythrocyte and plasma cholinesterases. Biochem. J 1953, 53, 167–173. [Google Scholar]
- Rosenberry, T. L. Acetylcholinesterase- A Review. Advan. Enzymol 1975, 43, 103–218. [Google Scholar]
- Rusyniak, D. E.; Nanagas, K. A. Organophosphate Poisoning. Semin. Neurol 2004, 24(2), 197–204. [Google Scholar]
- Yang, L.; Heng-Yi, H.; Zhang, X. J. Increased expression of intranuclear AChE involved in apoptosis of SK-N-SH cells. Neurosci Res 2002, 42, 261–68. [Google Scholar]
- Calderon, F. H.; Von, B. R.; De Ferrari, G.; et al. Toxic effects of acetylcholinesterase on neuronal and glial-like cells in vitro. Mol. Psychiatry 1998, 3, 247–55. [Google Scholar]
- Andres, C.; Seidman, S.; Beeri, R.; et al. Transgenic acetylcholinesterase induces enlargement of murine neuromuscular junctions but leaves spinal cord synapses intact. Neurochem. Int 1998, 32, 449–56. [Google Scholar]
© 2005 MDPI. All rights reserved.
Share and Cite
Patlolla, A.K.; Tchounwou, P.B. Serum Acetyl Cholinesterase as a Biomarker of Arsenic Induced Neurotoxicity in Sprague-Dawley Rats. Int. J. Environ. Res. Public Health 2005, 2, 80-83. https://doi.org/10.3390/ijerph2005010080
Patlolla AK, Tchounwou PB. Serum Acetyl Cholinesterase as a Biomarker of Arsenic Induced Neurotoxicity in Sprague-Dawley Rats. International Journal of Environmental Research and Public Health. 2005; 2(1):80-83. https://doi.org/10.3390/ijerph2005010080
Chicago/Turabian StylePatlolla, Anita K., and Paul B. Tchounwou. 2005. "Serum Acetyl Cholinesterase as a Biomarker of Arsenic Induced Neurotoxicity in Sprague-Dawley Rats" International Journal of Environmental Research and Public Health 2, no. 1: 80-83. https://doi.org/10.3390/ijerph2005010080
APA StylePatlolla, A. K., & Tchounwou, P. B. (2005). Serum Acetyl Cholinesterase as a Biomarker of Arsenic Induced Neurotoxicity in Sprague-Dawley Rats. International Journal of Environmental Research and Public Health, 2(1), 80-83. https://doi.org/10.3390/ijerph2005010080