Effect of Light Irradiation and Sex Hormones on Jurkat T Cells: 17β-Estradiol but Not Testosterone Enhances UVA-Induced Cytotoxicity in Jurkat Lymphocytes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction
Materials and Methods
Reagents and Cell Lines
Ultraviolet A Treatment
Cell Treatment for Acute Cytotoxicity
Cell Treatment for Genotoxicity Assay
DNA Laddering
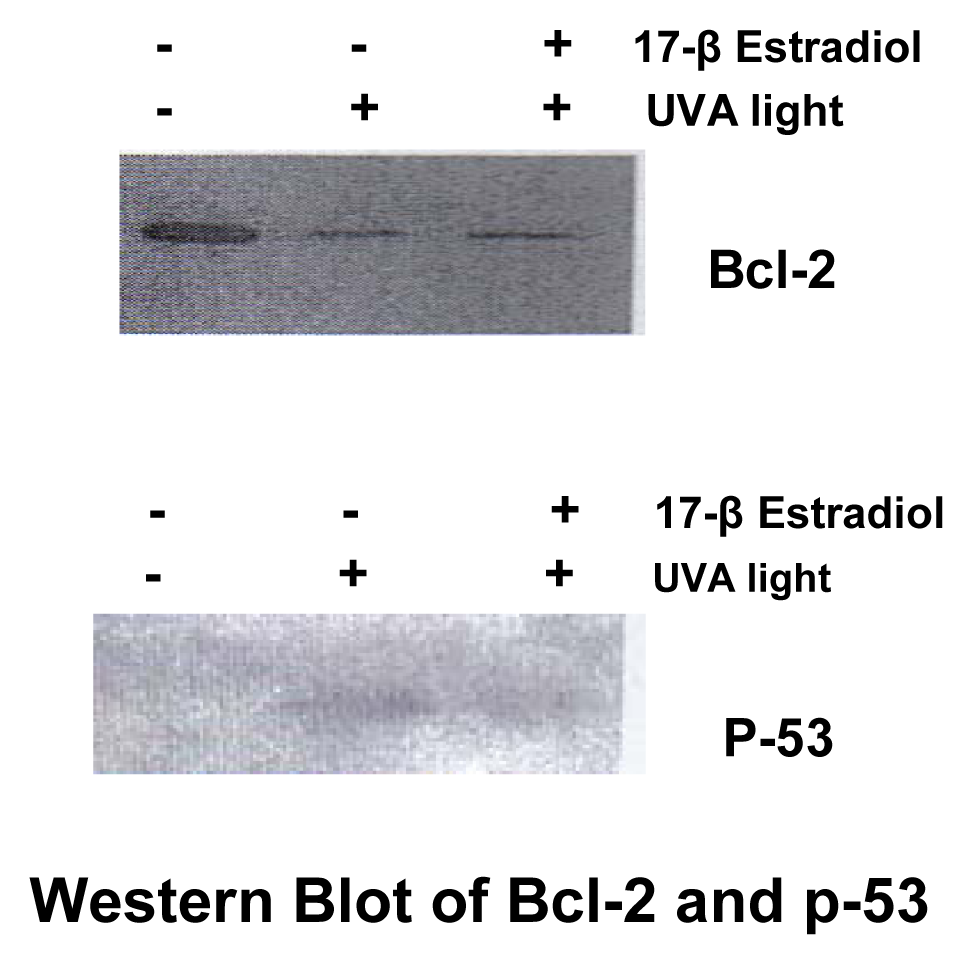
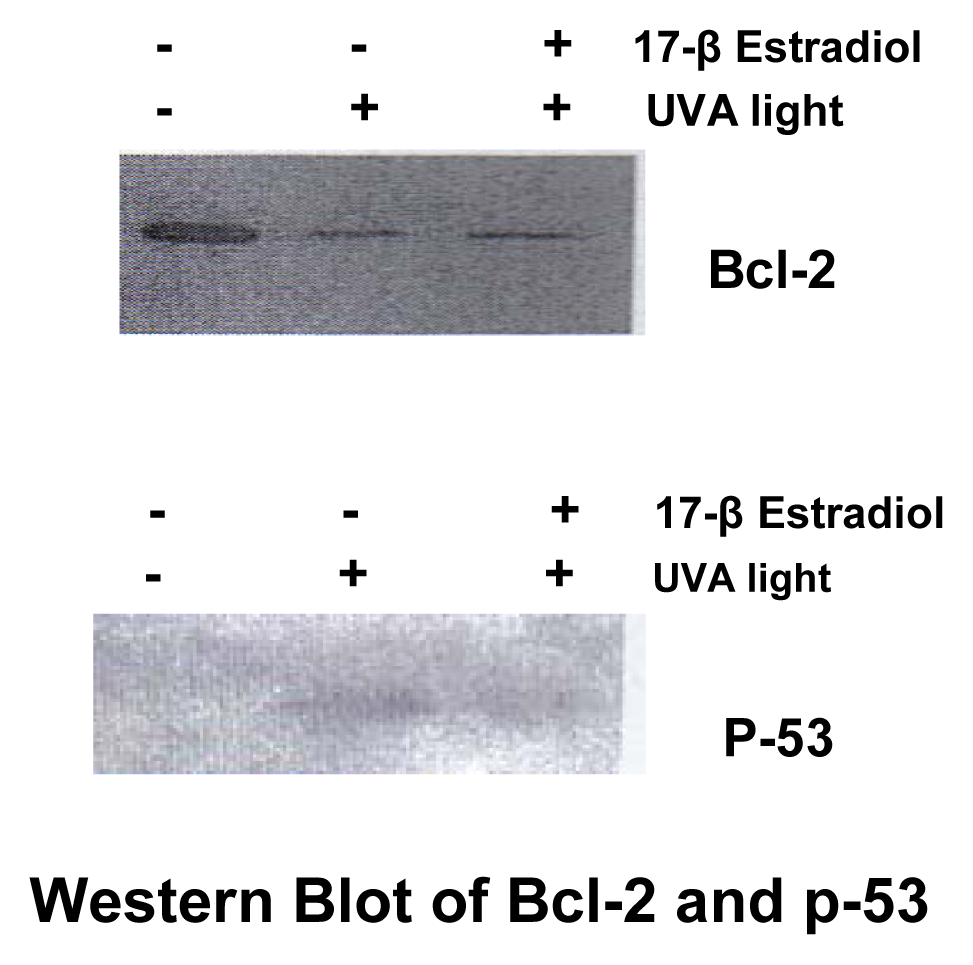
Western Blotting
Statistical Analysis
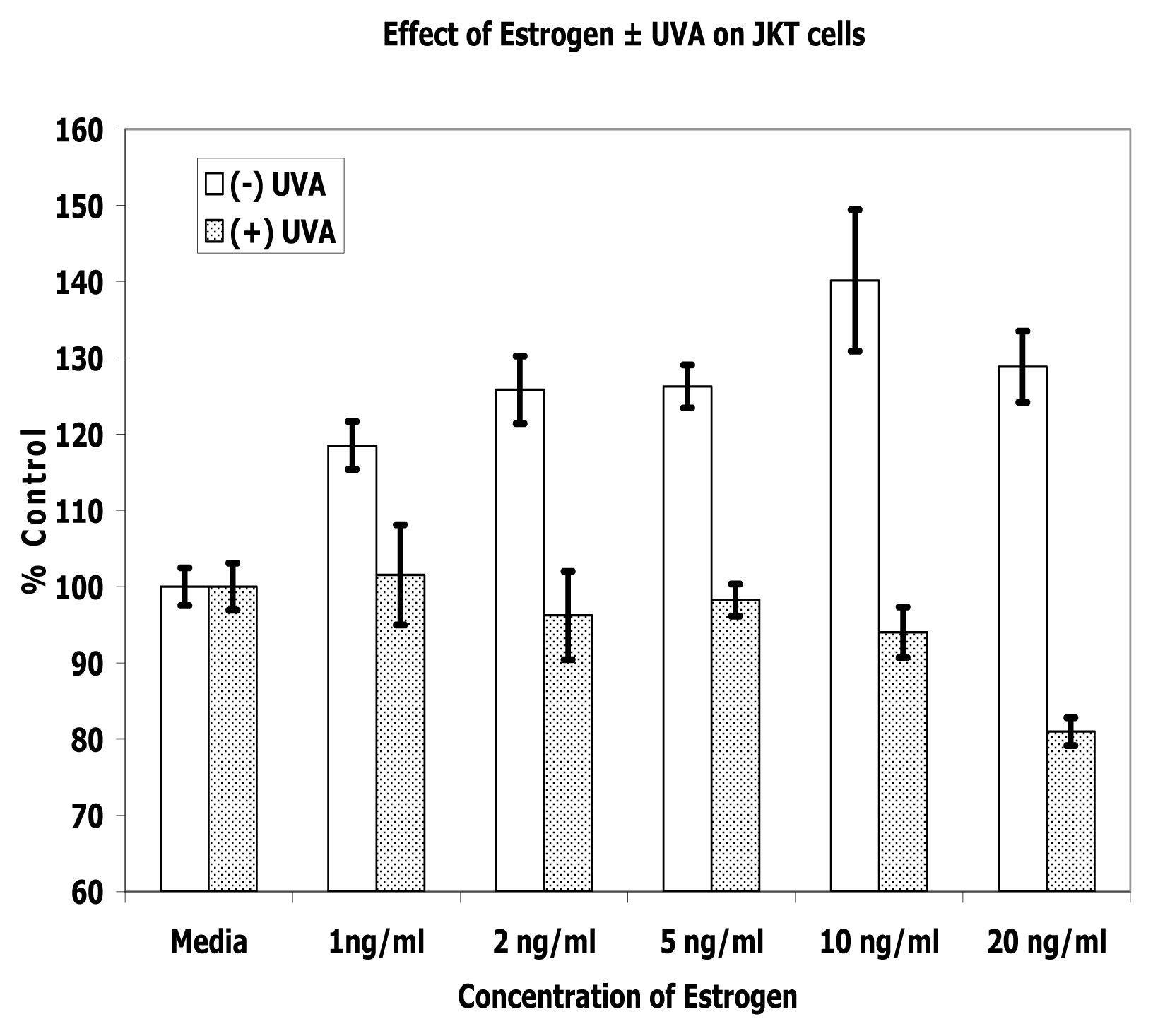
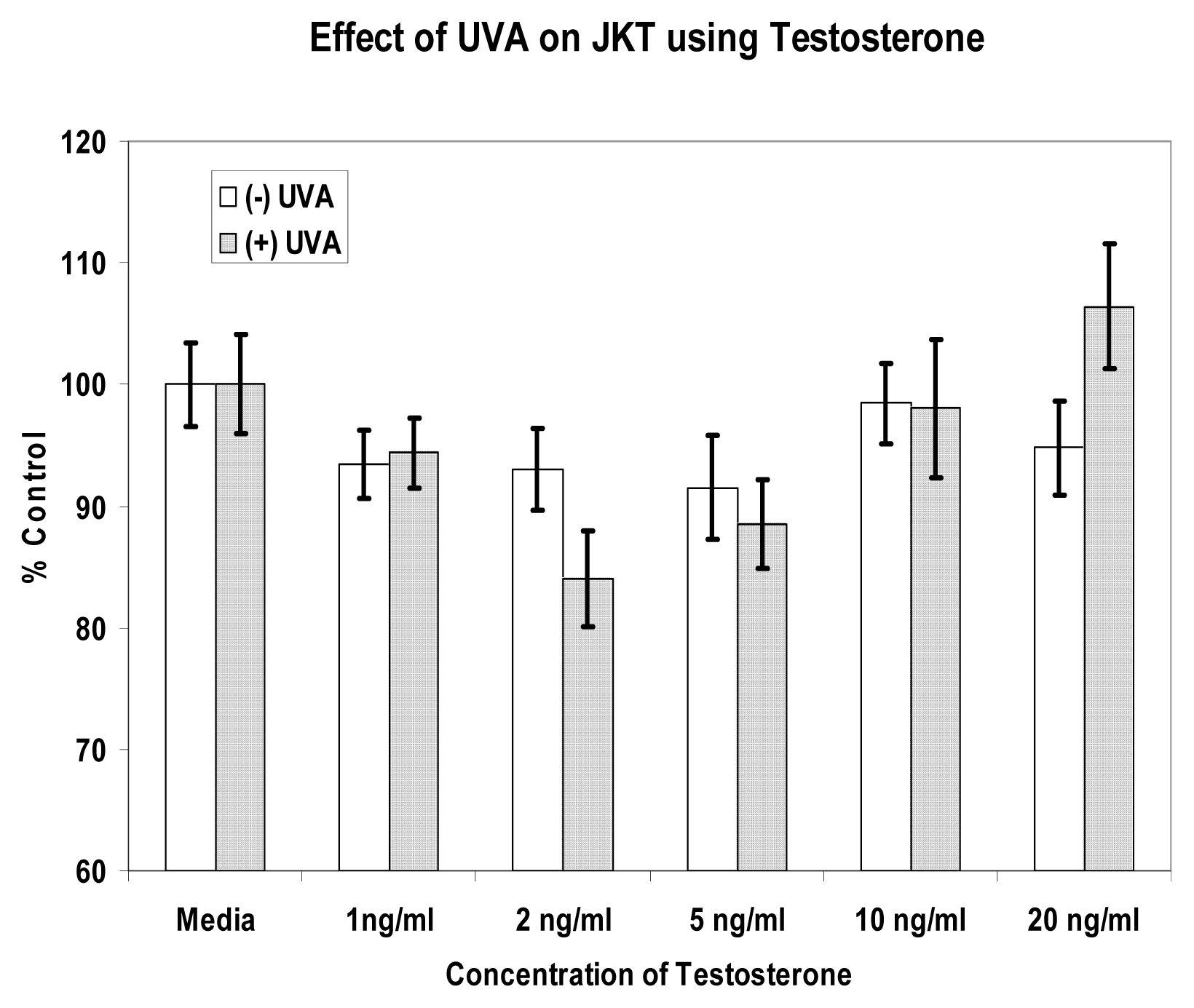
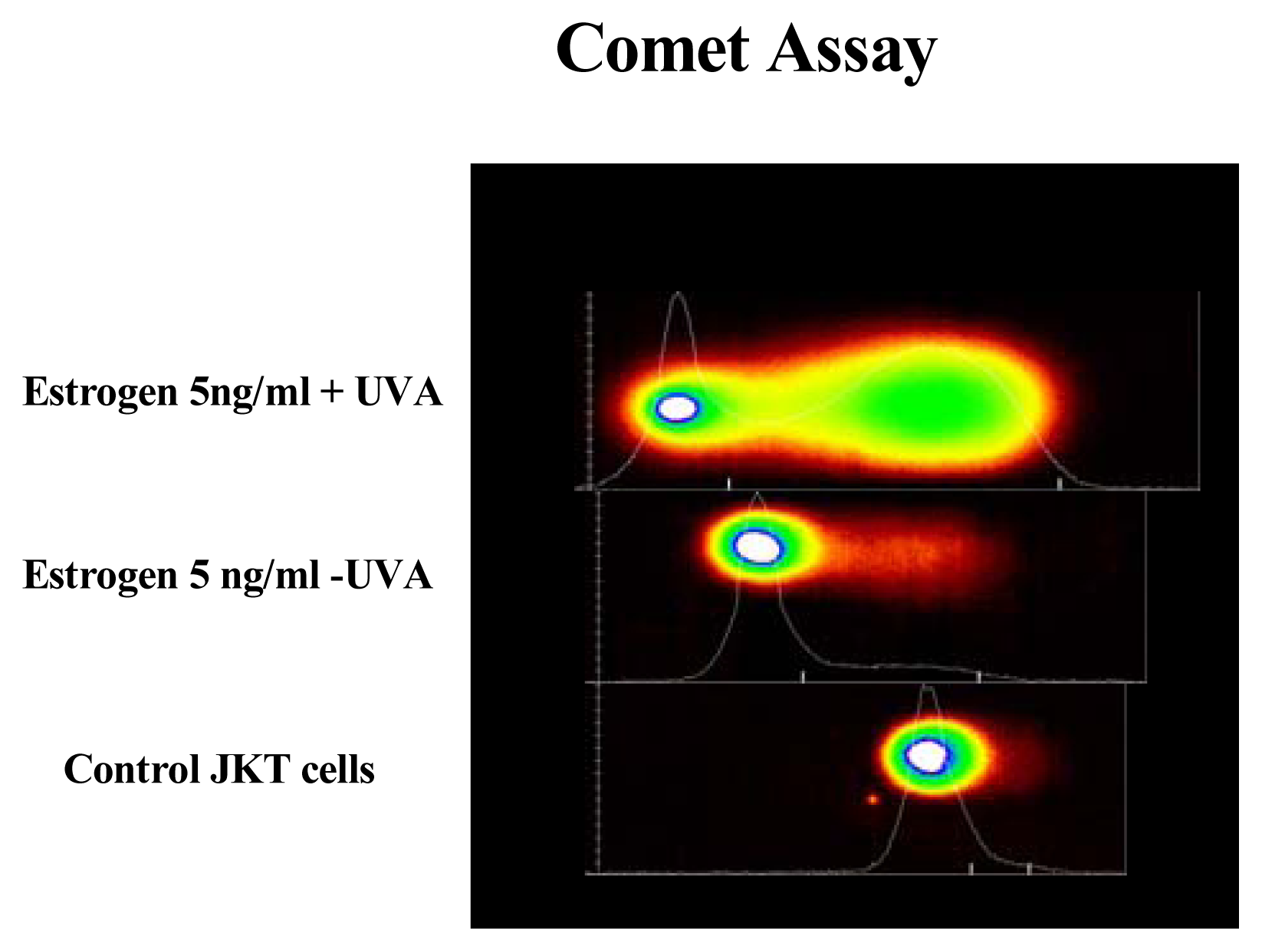
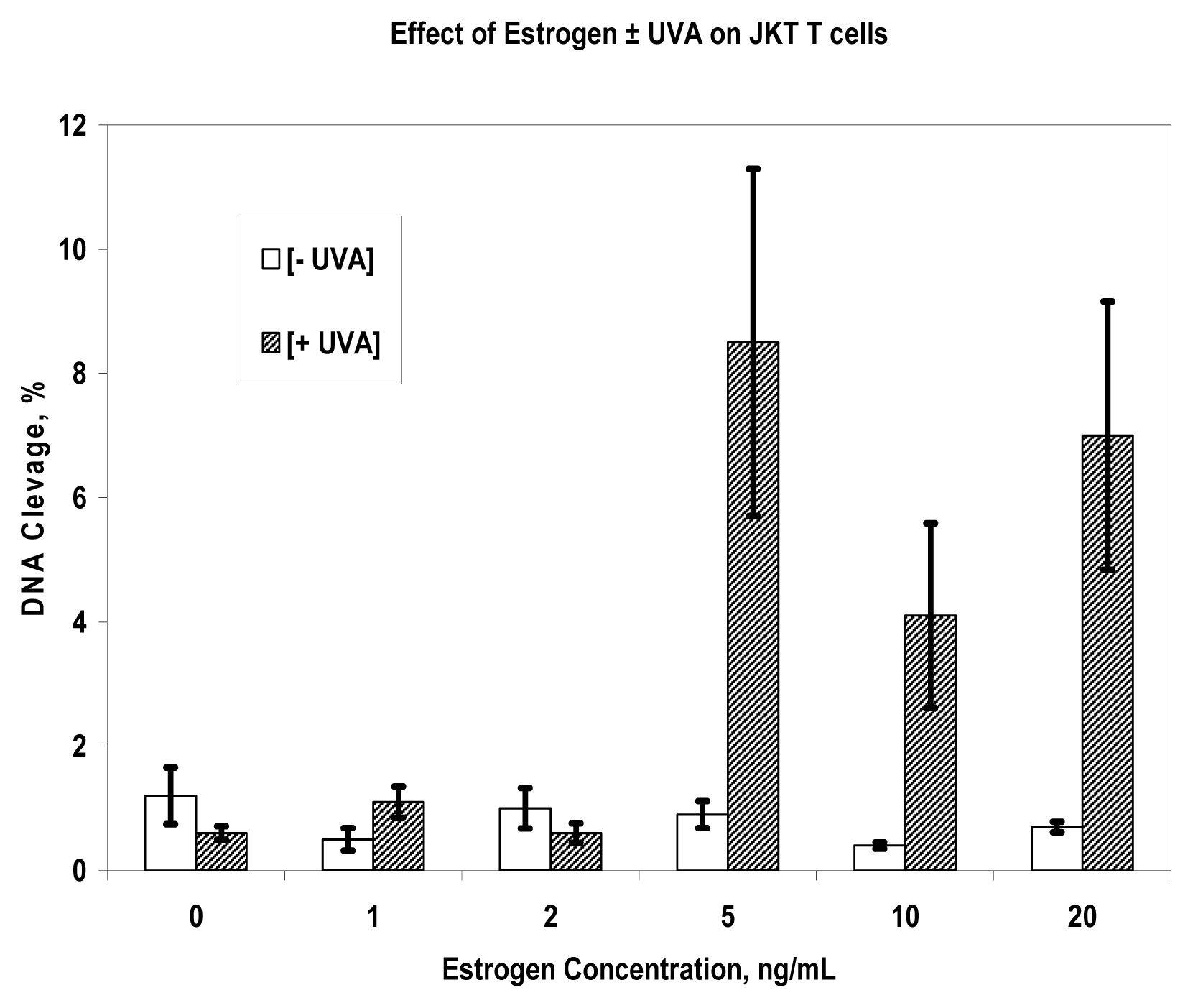
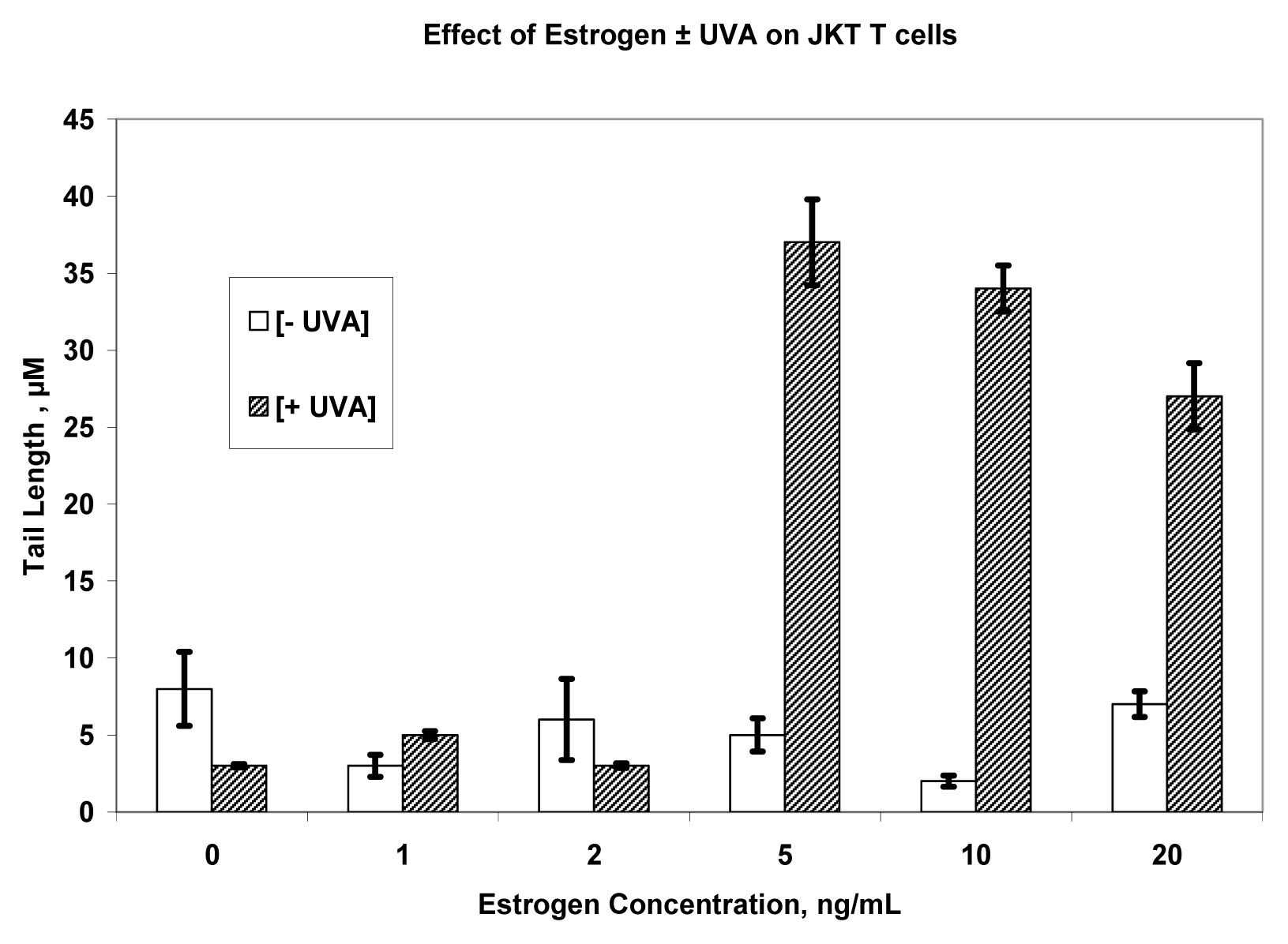
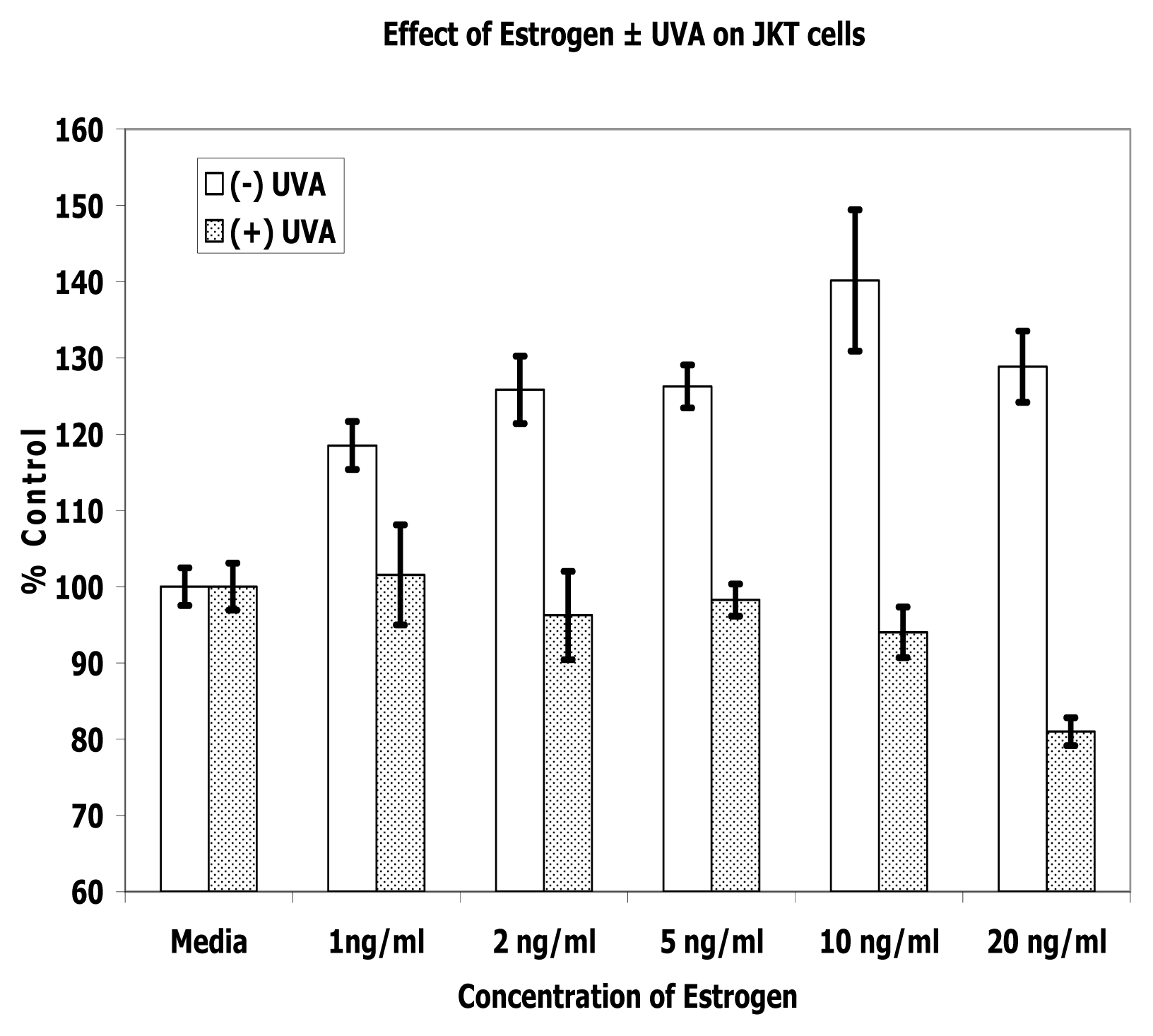
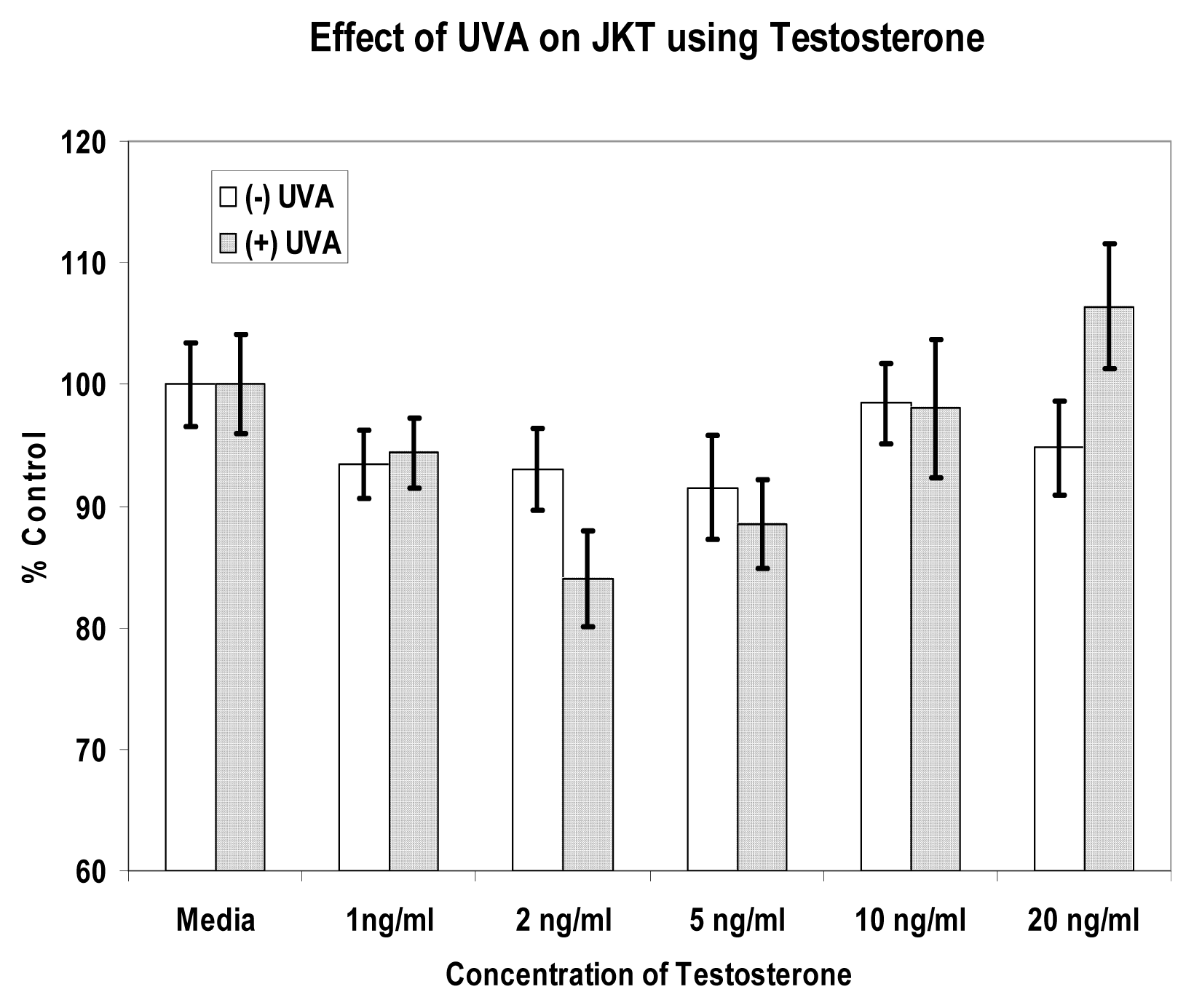
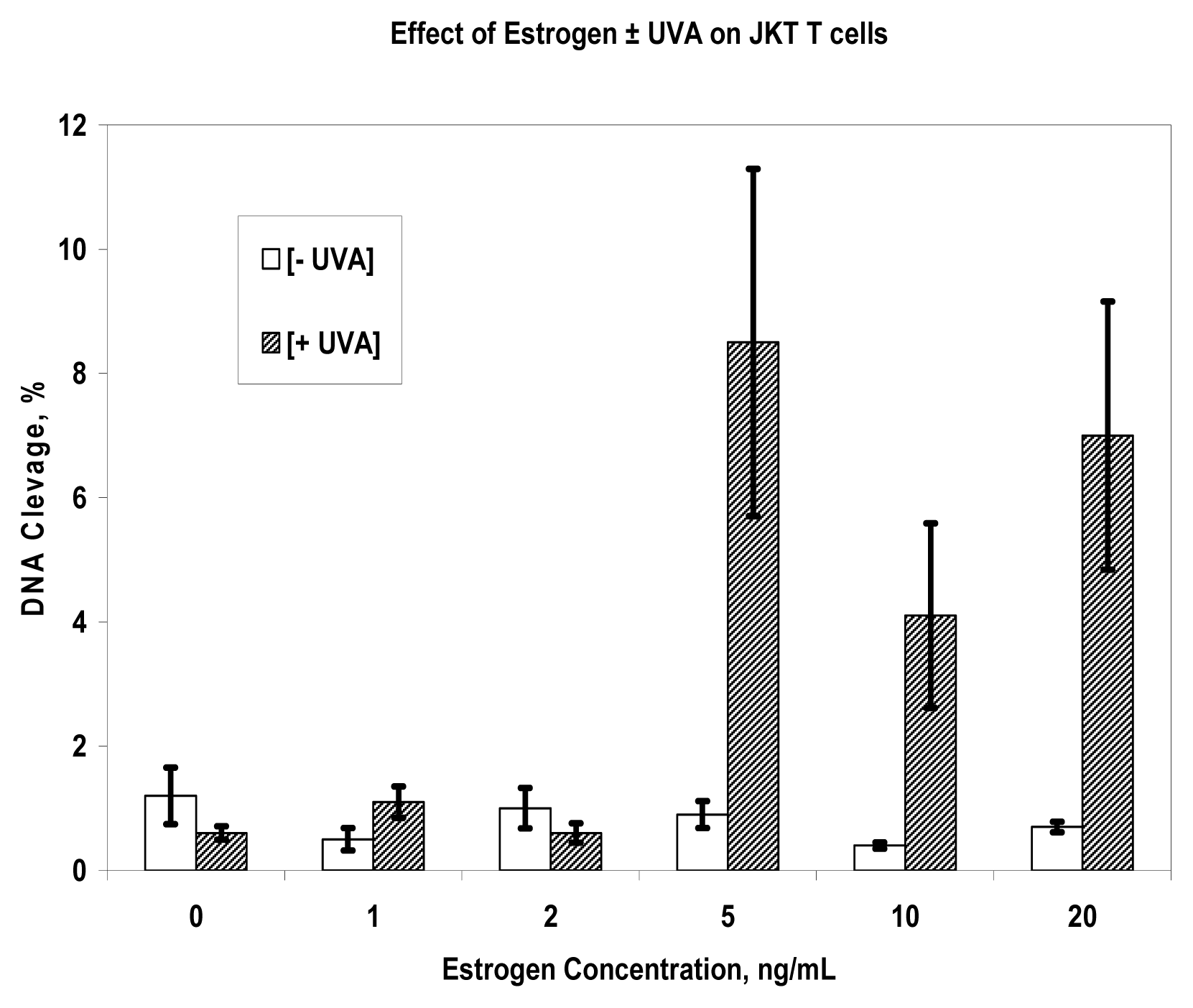
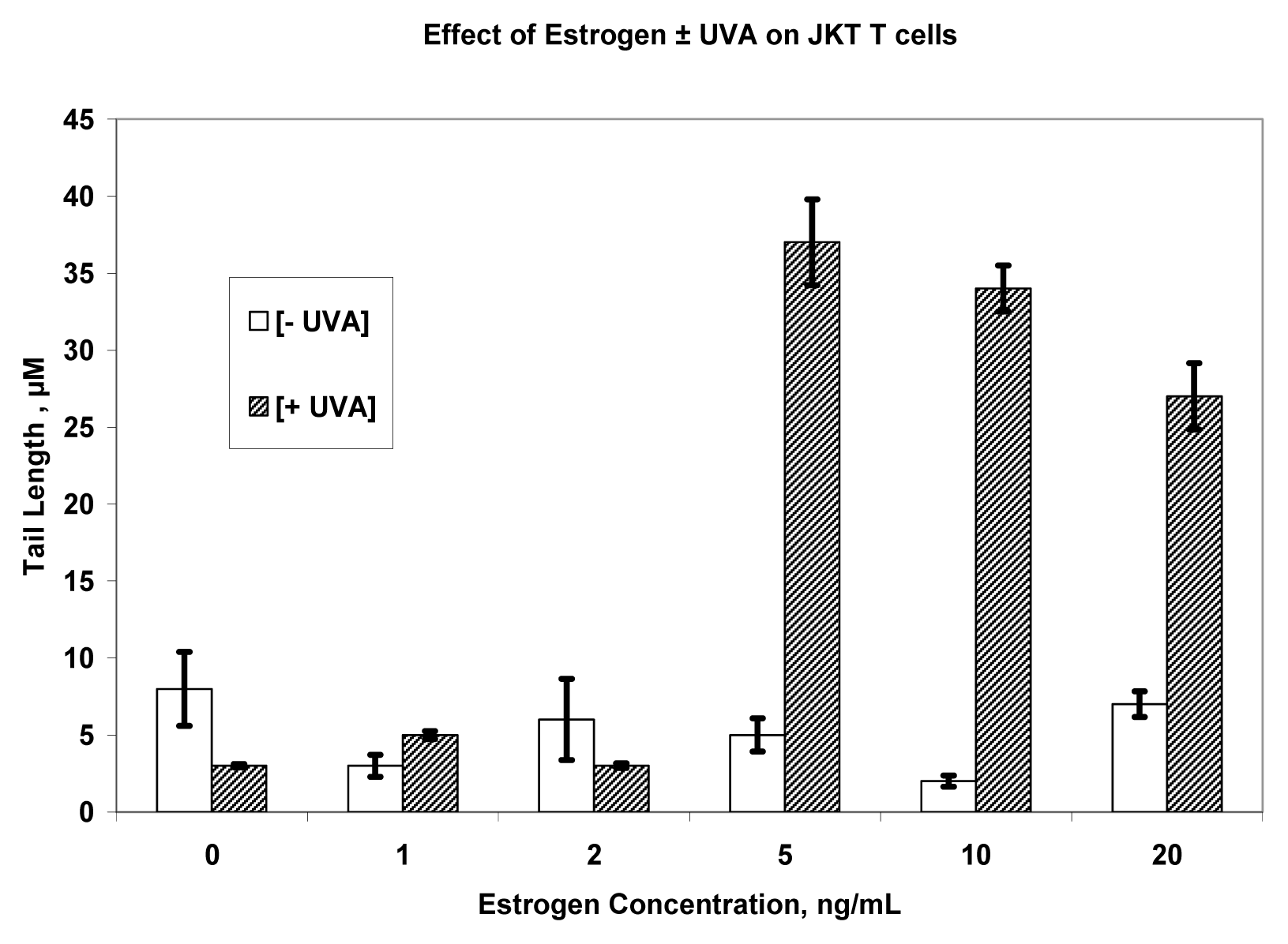
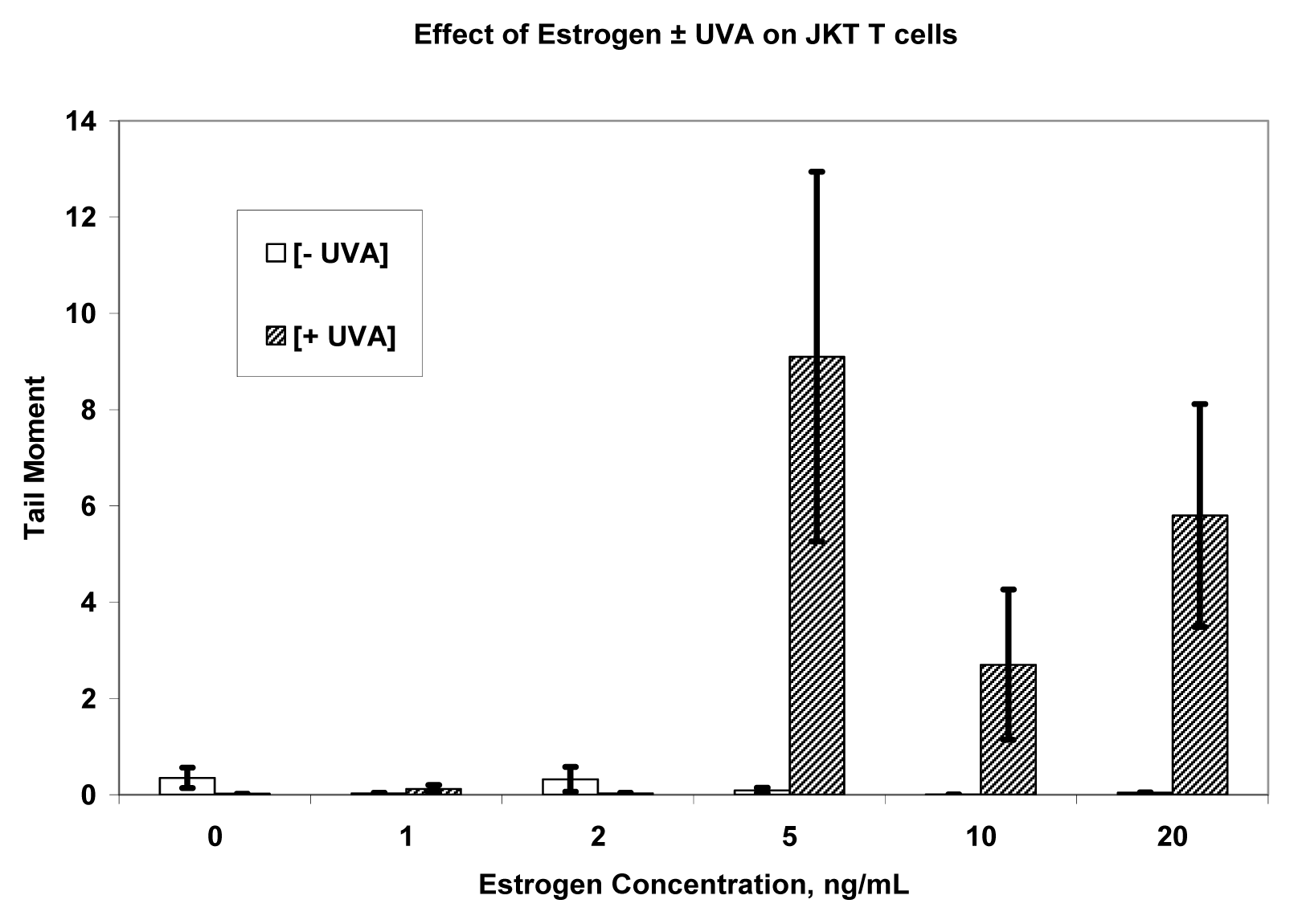
Results
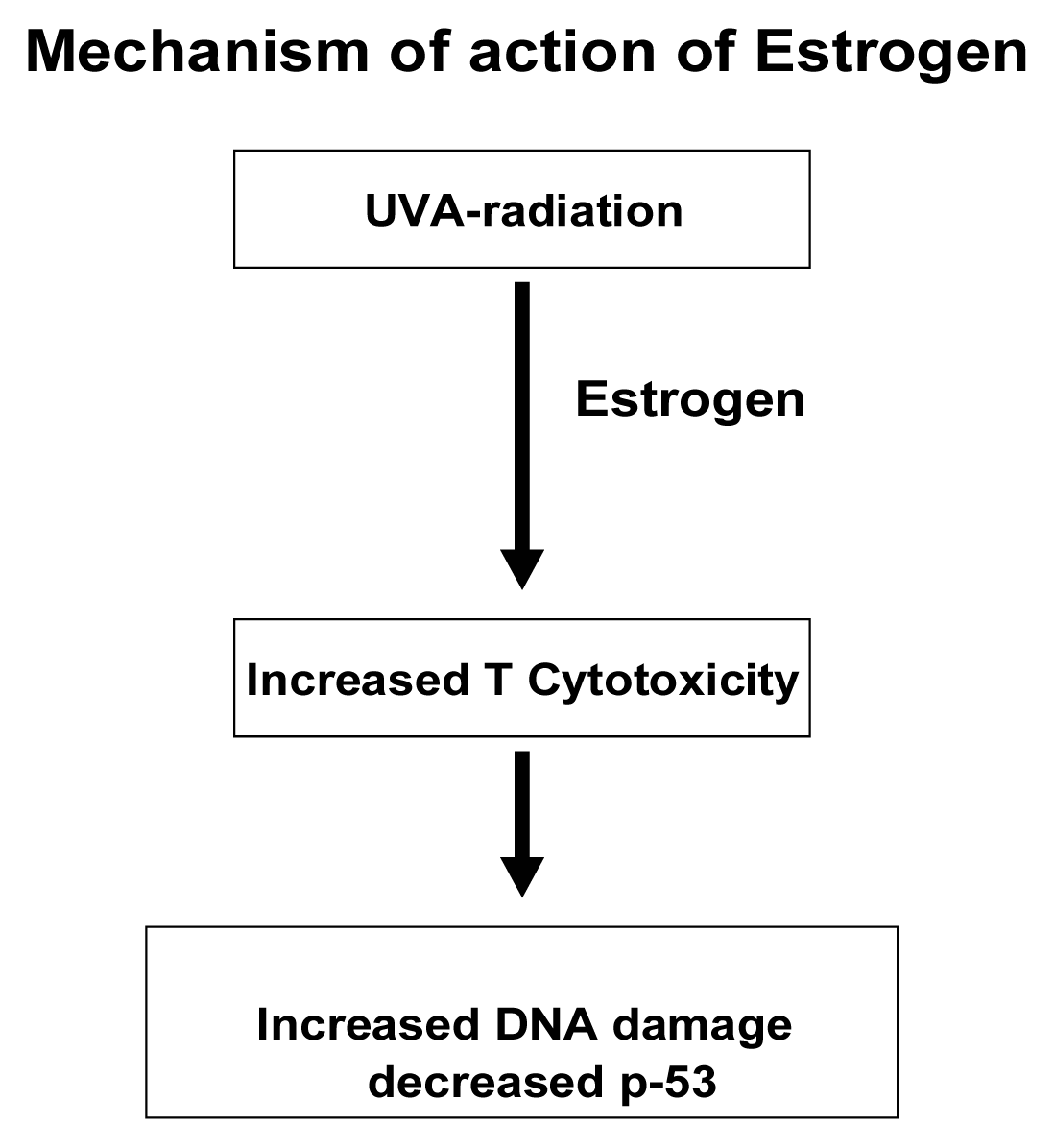
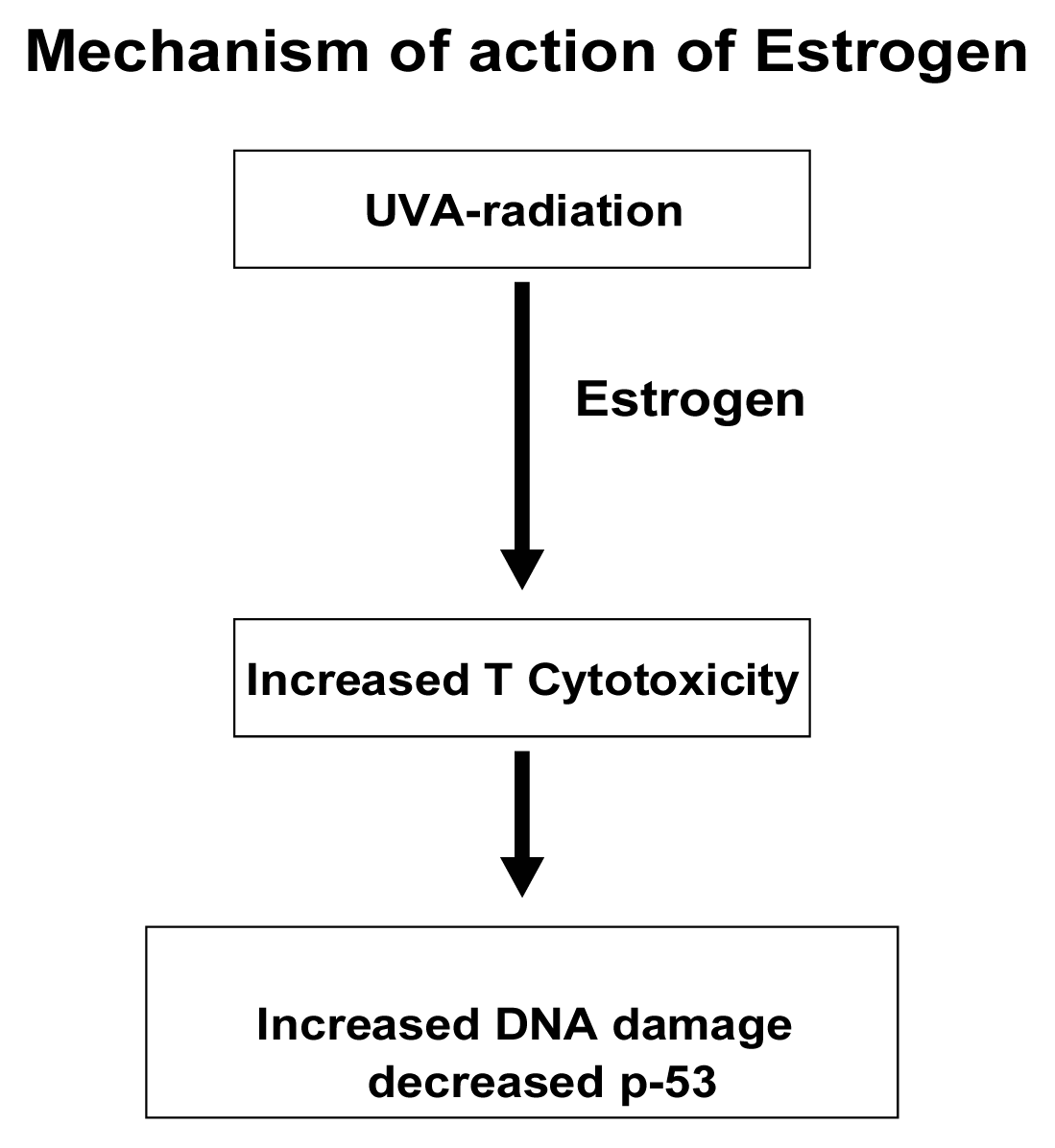
Discussion


References
- Matsumura, Y.; Ananthaswamy, H. N. Toxic effects of ultraviolet radiation on the skin. Toxicol Appl Pharmacol 2004, 195(3), 298–308. [Google Scholar]
- Young, A. R. Tanning devices--fast track to skin cancer? Pigment Cell Res 2004, 17, 2–9. [Google Scholar]
- Sunlight Ultraviolet Radiation, and the Skin. NIH Consensus Statement, Online May 8–10 1989, 7(8), 1–29.
- Aubin, F.; Mousson, C. Ultraviolet light-induced regulatory (suppressor) T cells: an approach for promoting induction of operational allograft tolerance? Transplantation 2004, 15(77), S29–31. [Google Scholar]
- Cutolo, M.; Sulli, A.; Capellino, S.; Villaggio, B.; Montagna, P.; Seriolo, B.; Straub, R. H. Sex hormones influence on the immune system: basic and clinical aspects in autoimmunity. Lupus 2004, 13(9), 635–8. [Google Scholar]
- Graham-Evans, B.; Tchounwou, P. B.; Cohly, H. Cytotoxicity and Proliferation Studies with Arsenic in Established Human Cell Lines: Keratinocytes, Melanocytes, Dendritic Cells, Dermal Fibroblasts, Microvascular Endothelial Cells, Monocytes and T-Cells. Int. J. Mol. Sci 2003, 4, 13–21. [Google Scholar]
- Jenkins, J. K.; Suwannaroj, S.; Elbourne, K. B.; Ndebele, K.; McMurray, R. W. 17-β estradiol alters Jurkat lymphocyte cell cycling and induces apoptosis through suppression of bcl-2 and cyclin A. Internat. Immunopharm 2001, 1, 1867–1911. [Google Scholar]
- Hu, S.; Ma, F.; Collado-Mesa, F.; Kirsner, R. S. Ultraviolet radiation and incidence of non-Hodgkin’s lymphoma among Hispanics in the United States. Cancer Epidemiol Biomarkers Prev 2004, 13, 59–64. [Google Scholar]
- Kanariou, M.; Petridou, E.; Vrachnou, E.; Trichopoulos, D. Lymphocyte alterations after prolonged sunlight exposure. Journal of Epidemiology and Biostatistics 2001, 6, 463–465. [Google Scholar]
- Godar, D. E. UVA1 radiation triggers two different final apoptotic pathways. J Invest Dermatol 1999, 112, 3–12. [Google Scholar]
- John, E. M.; Dreon, D. M.; Koo, J.; Schwartz, G. G. Residential sunlight exposure is associated with a decreased risk of prostate cancer. J Steroid Biochem Mol Biol 2004, 89–90, 549–52. [Google Scholar]
- Robsahm, T. E.; Tretli, S.; Dahlback, A.; Moan, J. Vitamin D3 from sunlight may improve the prognosis of breast-, colon- and prostate cancer (Norway). Cancer Causes Control 2004, 15, 149–58. [Google Scholar]
- Lehmann, B.; Sauter, W.; Knuschke, P.; Dressler, S.; Meurer, M. Demonstration of UVB-induced synthesis of 1 alpha, 25-dihydroxyvitamin D3 (calcitriol) in human skin by microdialysis. Arch. Dermatol. Res 2003, 295, 24–8. [Google Scholar]
- Grant, W. B. An estimate of premature cancer mortality in the U.S. due to inadequate doses of solar ultraviolet-B radiation. Cancer 2002, 94, 1867–75. [Google Scholar]
- Grant, W. B. An ecologic study of dietary and solar ultraviolet-B links to breast carcinoma mortality rates. Cancer 2002, 94, 272–81. [Google Scholar]
- Garland, C. F.; Garland, F. C. Do sunlight and vitamin D reduce the likelihood of colon cancer? Int J. Epidemiol 1980, 9, 227–31. [Google Scholar]
- Freifelder, D. Molecular Biology, 2nd edn; Jones and Bartlett: Boston, 1987; pp. 277–92. [Google Scholar]
- Lehmann, A. R.; Norris, P. G. DNA repair deficient photodermatoses. Semin. Dermatol 1990, 9, 55–62. [Google Scholar]
- Wang, L.; Cohly, H.; Yan, J.; Graham-Evans, B.; Hwang, H-M.; Yu, H. UVA light-induced toxic effects of 1-hydroxypyrene on human jurkat T-cells. Bull. Environ. Contam. Toxicol 2004, 72, 1240–1246. [Google Scholar]
- Wang, L.; Yan, J.; Wang, S.; Cohly, H.; Fu, P. P.; Hwang, H. M.; Yu, H. Phototoxicity and DNA damage induced by the cosmetic ingredient chemical azulene in human Jurkat T-cells. Mutat Res 2004, 562, 143–50. [Google Scholar]
- McMurray, R. W.; Ndebele, K.; Hardy, K. J.; Jenkins, J. K. 17-beta-estradiol suppresses IL-2 and IL-2 receptor. Cytokine 2001, 14, 324–33. [Google Scholar]
- Wyllie, A. H. The genetic regulation of apoptosis. Curr Opin Genet Dev 1995, 5, 97–104. [Google Scholar]
- Reed, J. C. Bcl-2 and the regulation of programmed cell death. J. Cell Biol 1994, 124, 1–6. [Google Scholar]
- Takei, S.; Redford, A.; Katayama, S.; Toyoda, H. Destabilization of tumor necrosis factor-alpha RNA by 5-alpha dihydrotestosterone in Jurkat cells. Life Sci 2000, 66, PL277–82. [Google Scholar]
- Wu, F. C. W. Endocrine aspects of anabolic steroids. Clin. Chem 1997, 43, 1289–1292. [Google Scholar]
- Dubey, R. K.; Jackson, E. K. Estrogen-induced cardiorenal protection: potential cellular, biochemical, and molecular mechanisms. Am J. Physiol Renal Physiol 2001, 280, F365–F388. [Google Scholar]
- Khodarev, N. N.; Sokolova, I. A.; Vaughan, A. T. Mechanisms of induction of apoptotic DNA fragmentation. Int. J. Radiat. Biol 1998, 73, 455–467. [Google Scholar]
- Khodarev, N. N.; Sokolova, I. A.; Vaughan, A. T. Association between DNA cleavage during apoptosis and regions of chromatin replication. J. Cell. Biochem 1998, 70, 604–615. [Google Scholar]
- Inohara, N.; Koseki, T.; Chen, S.; Wu, X.; Nunez, G. CIDE, a novel family of cell death activators with homology to the 45 kDa subunit of the DNA fragmentation factor. EMBO J 1998, 17, 2526–2533. [Google Scholar]
- Bedner, E.; Li, X.; Gorczyca, W.; Melamed, M. R.; Darzynkiewicz, Z. Analysis of apoptosis by laser scanning cytometry. Cytometry 1999, 35, 181–195. [Google Scholar]
- Liu, X.; Zou, H.; Slaughter, C.; Wang, X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell 1997, 89, 175–184. [Google Scholar]
- Tang, D.; Kidd, V. J. Cleavage of DFF-45/ICAD by multiple caspases is essential for its function during apoptosis. J. Biol. Chem 1998, 273, 28549–28552. [Google Scholar]
- Olive, P. L.; Banath, J. P. Sizing highly fragmented DNA in individual apoptotic cells using the comet assay and a DNA cross-linking agent. Exp. Cell Res 1995, 221, 19–26. [Google Scholar]
- Whitmore, S. E.; Morison, W.; Potten, C. S.; Chadwick, C. Tanning salon exposure and molecular alterations. J. Am Acad Dermatol 2001, 44, 775–80. [Google Scholar]
- Kastan, M. B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R. W. Participation of p53 protein in the cellular response to DNA damage. Cancer Res 1991, 51, 6304–6311. [Google Scholar]
- Kastan, M. B.; Zhan, Q.; El-Deiry, W. S.; Carrier, F.; Jacks, T.; Walsh, W. V.; Plunkett, B. S.; Vogelstein, B.; Fornace, A. J., Jr. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia telangiectasia. Cell 1992, 71, 587–597. [Google Scholar]
- Shaw, P.; Bovey, R.; Tardy, S.; Sahli, R.; Sordat, B.; Costa, J. Introduction of apoptosis by wild-type p53 in a human colon tumor-derived cell line. Proc Natl Acad Sci USA 1992, 89, 4495–4499. [Google Scholar]
- McMurray, R.; May, W. Sex Hormones and systemic lupus erythematosis: Review and meta-analysis. Arthritis and Rheumatism 2003, 48, 2100–2110. [Google Scholar]
- American Cancer Society. Cancer Prevention and early detection Facts and Figures. 2004. www.cancer.org/downloads/STT/CPED2004PWSecured.pdf. Last update 12-11-04.
© 2005 MDPI. All rights reserved.
Share and Cite
Cohly, H.H.P.; Graham-Evans, B.; Ndebele, K.; Jenkins, J.K.; McMurray, R.; Yan, J.; Yu, H.; Angel, M.F. Effect of Light Irradiation and Sex Hormones on Jurkat T Cells: 17β-Estradiol but Not Testosterone Enhances UVA-Induced Cytotoxicity in Jurkat Lymphocytes. Int. J. Environ. Res. Public Health 2005, 2, 156-163. https://doi.org/10.3390/ijerph2005010156
Cohly HHP, Graham-Evans B, Ndebele K, Jenkins JK, McMurray R, Yan J, Yu H, Angel MF. Effect of Light Irradiation and Sex Hormones on Jurkat T Cells: 17β-Estradiol but Not Testosterone Enhances UVA-Induced Cytotoxicity in Jurkat Lymphocytes. International Journal of Environmental Research and Public Health. 2005; 2(1):156-163. https://doi.org/10.3390/ijerph2005010156
Chicago/Turabian StyleCohly, Hari H.P., Barbara Graham-Evans, Kenneth Ndebele, John K. Jenkins, Robert McMurray, Jian Yan, Hongtao Yu, and Michael F. Angel. 2005. "Effect of Light Irradiation and Sex Hormones on Jurkat T Cells: 17β-Estradiol but Not Testosterone Enhances UVA-Induced Cytotoxicity in Jurkat Lymphocytes" International Journal of Environmental Research and Public Health 2, no. 1: 156-163. https://doi.org/10.3390/ijerph2005010156
APA StyleCohly, H. H. P., Graham-Evans, B., Ndebele, K., Jenkins, J. K., McMurray, R., Yan, J., Yu, H., & Angel, M. F. (2005). Effect of Light Irradiation and Sex Hormones on Jurkat T Cells: 17β-Estradiol but Not Testosterone Enhances UVA-Induced Cytotoxicity in Jurkat Lymphocytes. International Journal of Environmental Research and Public Health, 2(1), 156-163. https://doi.org/10.3390/ijerph2005010156