
Computer-Aided Multi-Epitope Vaccine Design against Enterobacter xiangfangensis

 ,
,  , , ,
, , , 

Abstract
:1. Introduction
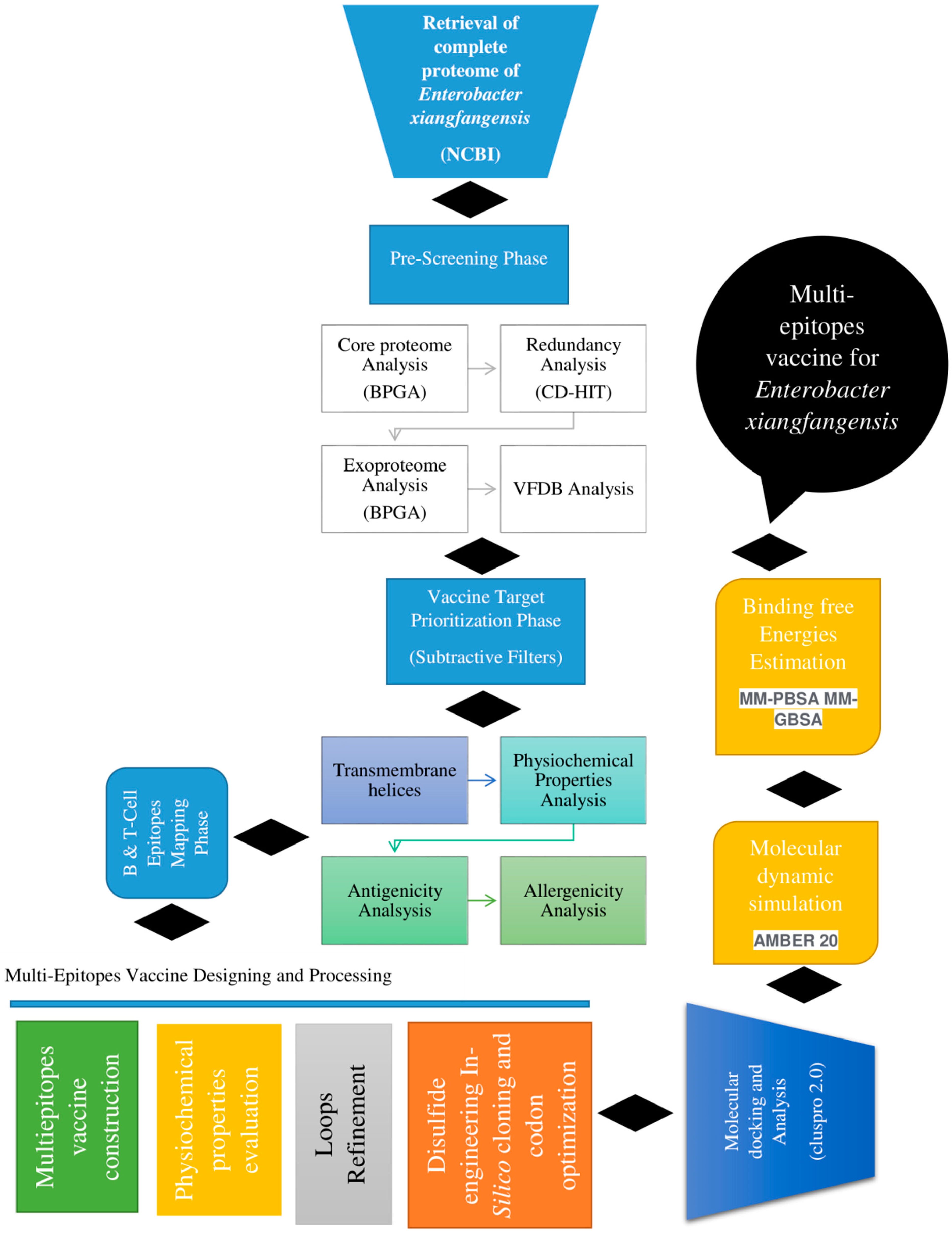
2. Research Methodology
2.1. Complete Proteome Retrieval and Bacterial Pan-Genome Analysis (BPGA)
2.2. Redundancy, Subcellular Localization and VFDB Analysis
2.3. Transmembrane Helices, Antigenicity, Allergenicity, Water Solubility and Physicochemical Property and Homology Analysis
2.4. Epitope Prediction and Prioritization Phase
2.5. Multi-Epitope-Based Vaccine Designing and Processing Phase
2.6. Structure Prediction and Loop Refinement
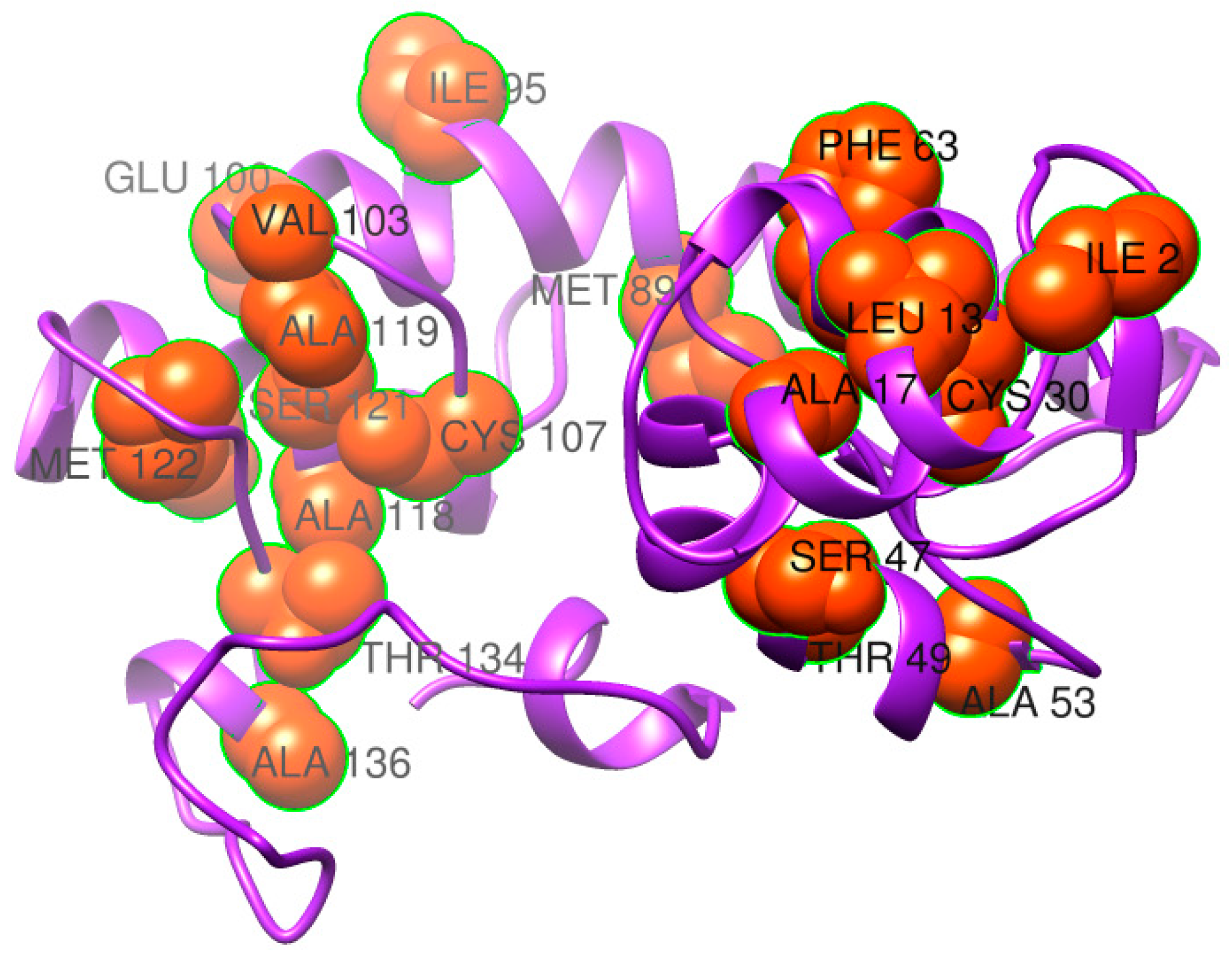
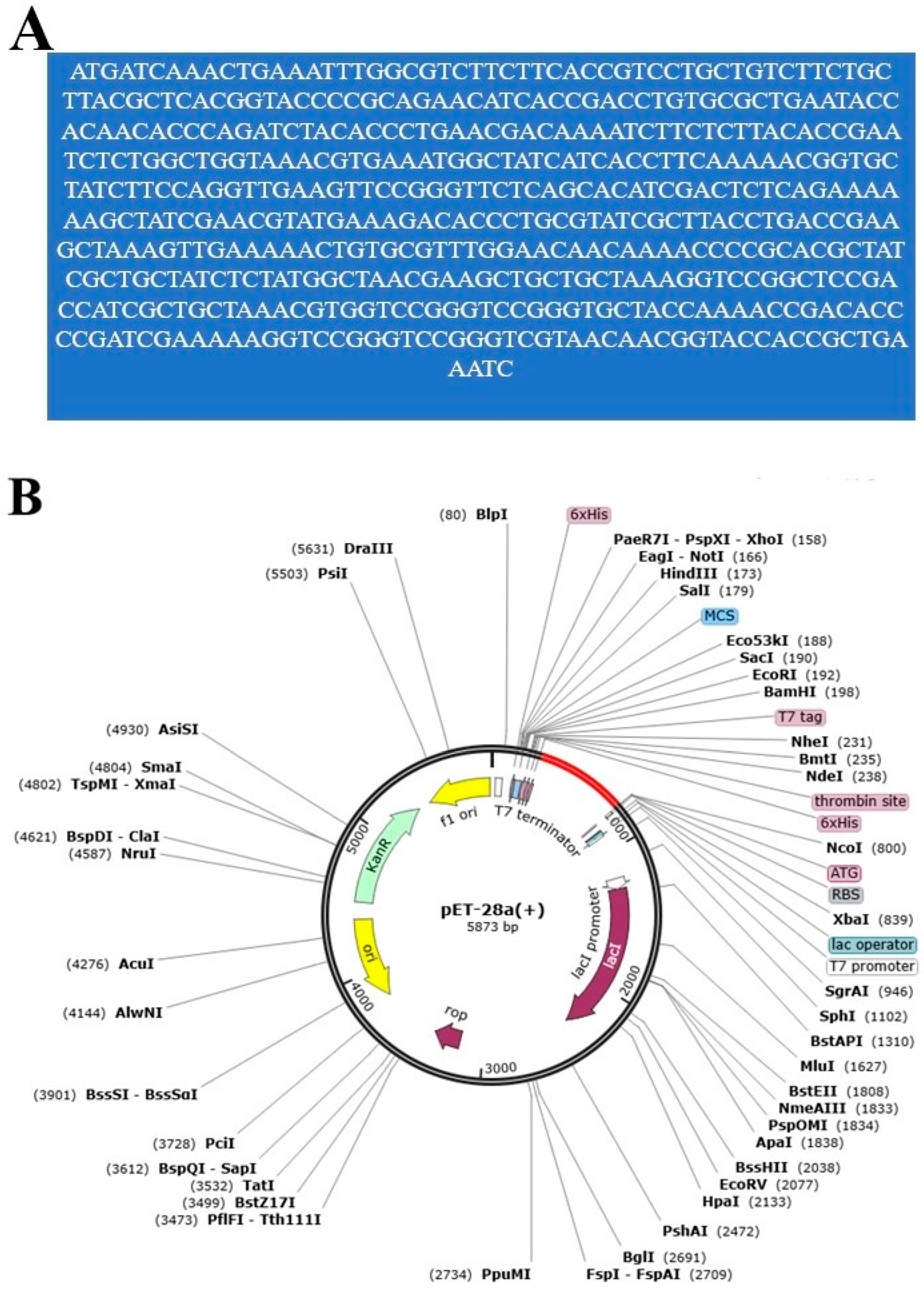
2.7. Disulfide Engineering and In Silico Codon Optimization Analysis
2.8. Secondary Structure, Solubility, Z-Score and Population Coverage Analysis
2.9. Molecular Docking
2.10. Molecular Dynamics Simulation
2.11. Immune Simulation
3. Results and Discussion
3.1. E. xiangfangensis Complete Proteome Retrieval and BPGA Analysis
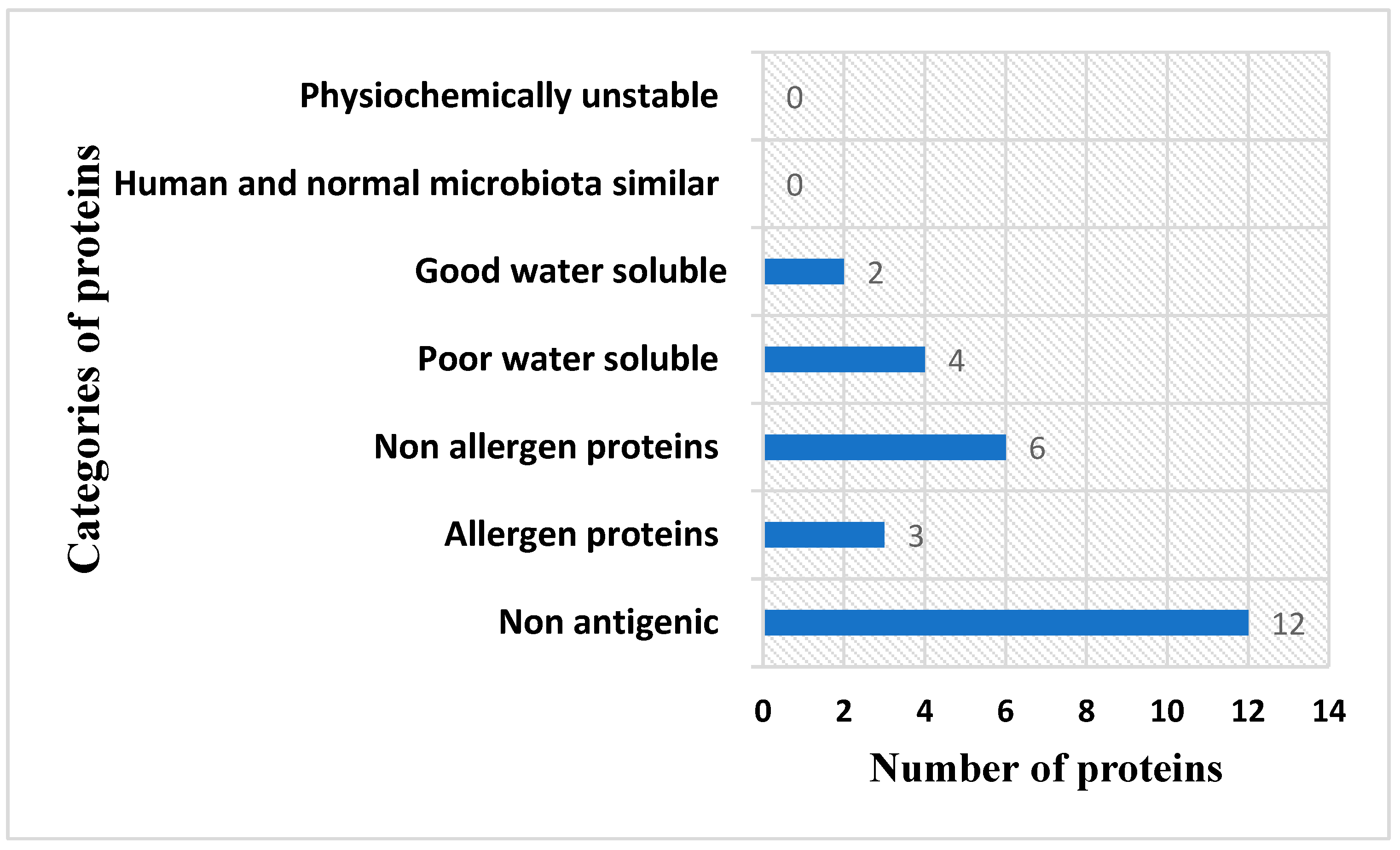
3.2. Epitope Mapping Phase
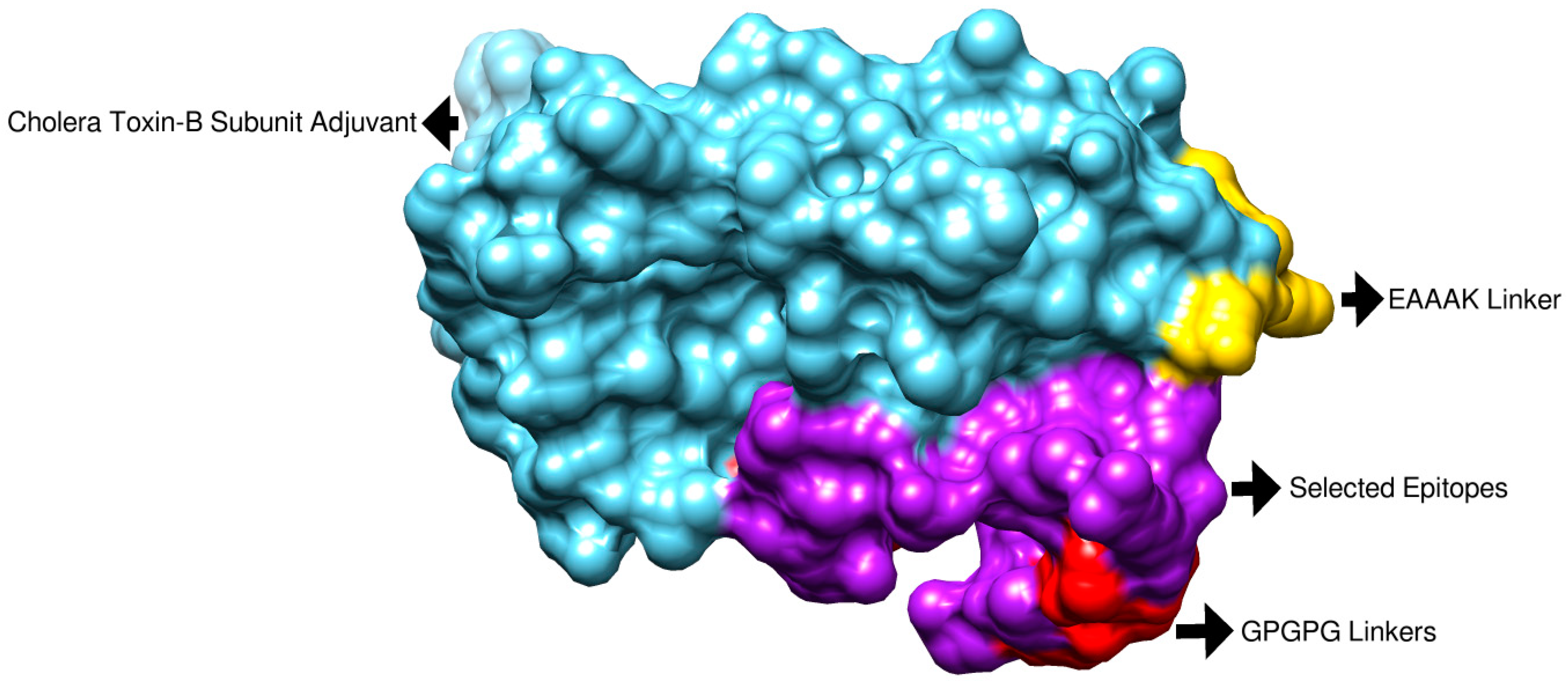
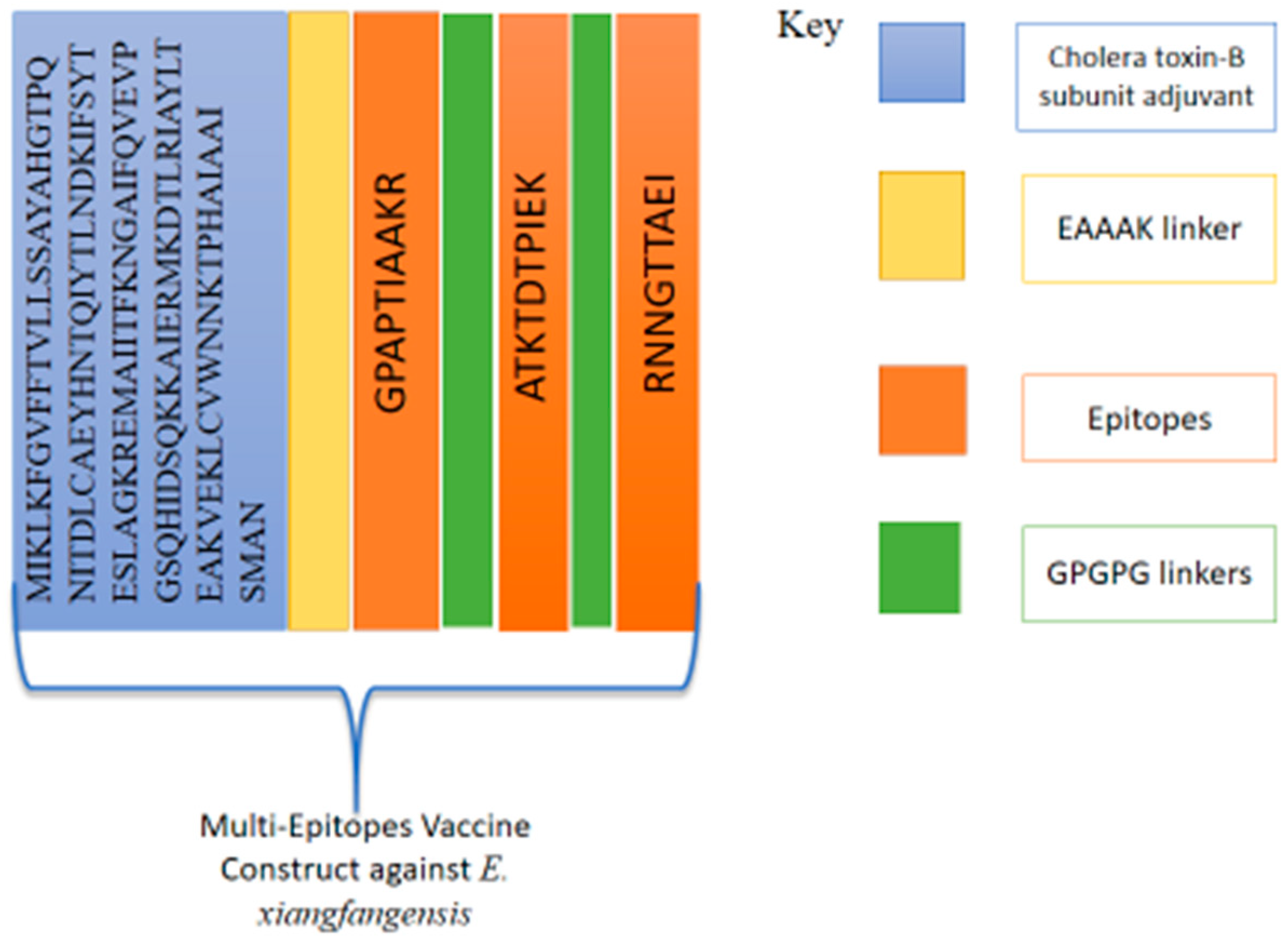
3.3. Physicochemical Property Evaluation and Vaccine Structure Prediction
3.4. Loop Refinement, Disulfide Engineering, In Silico Codon Optimization and Population Coverage Analysis
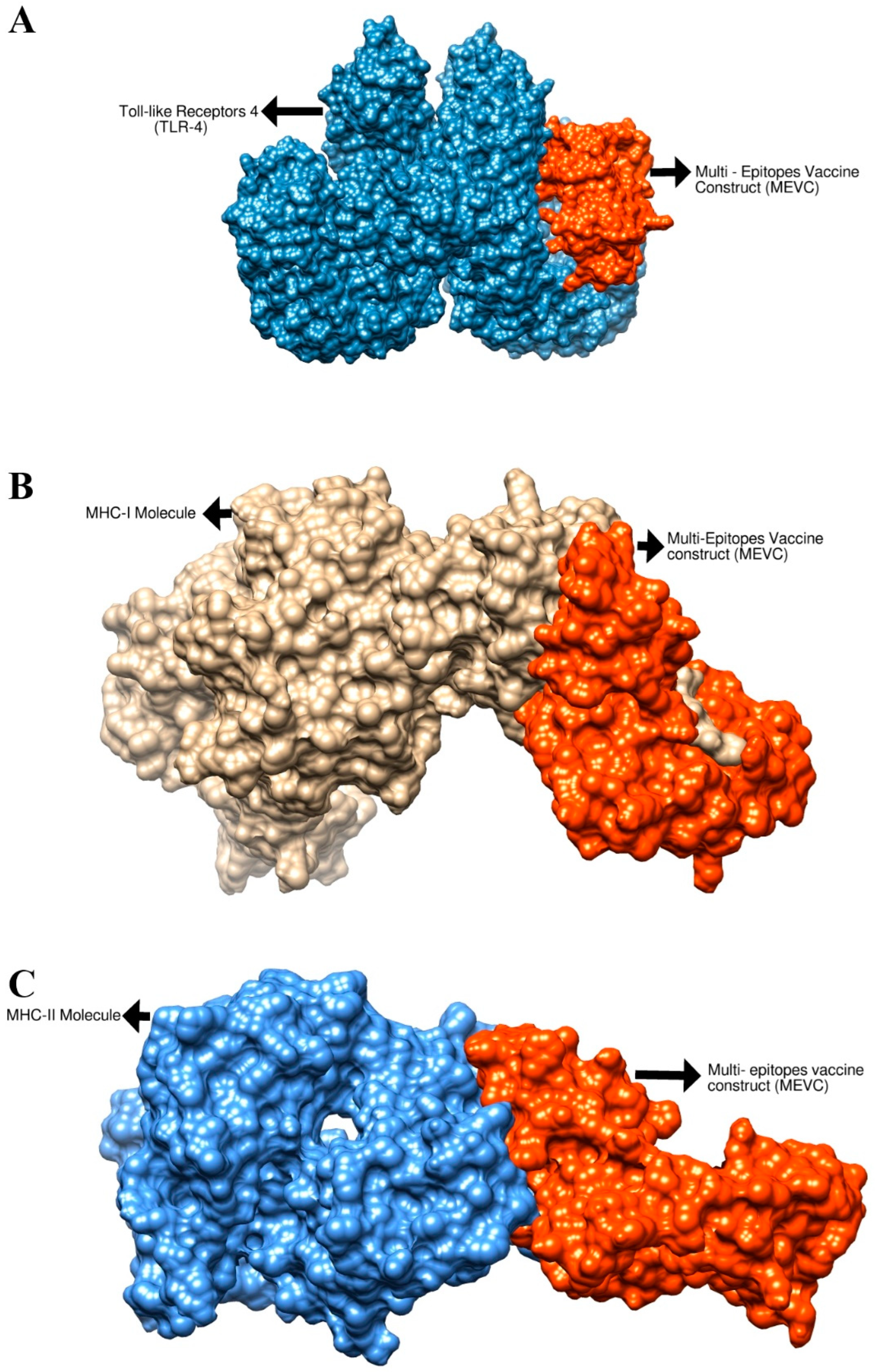
3.5. Molecular Docking Analysis
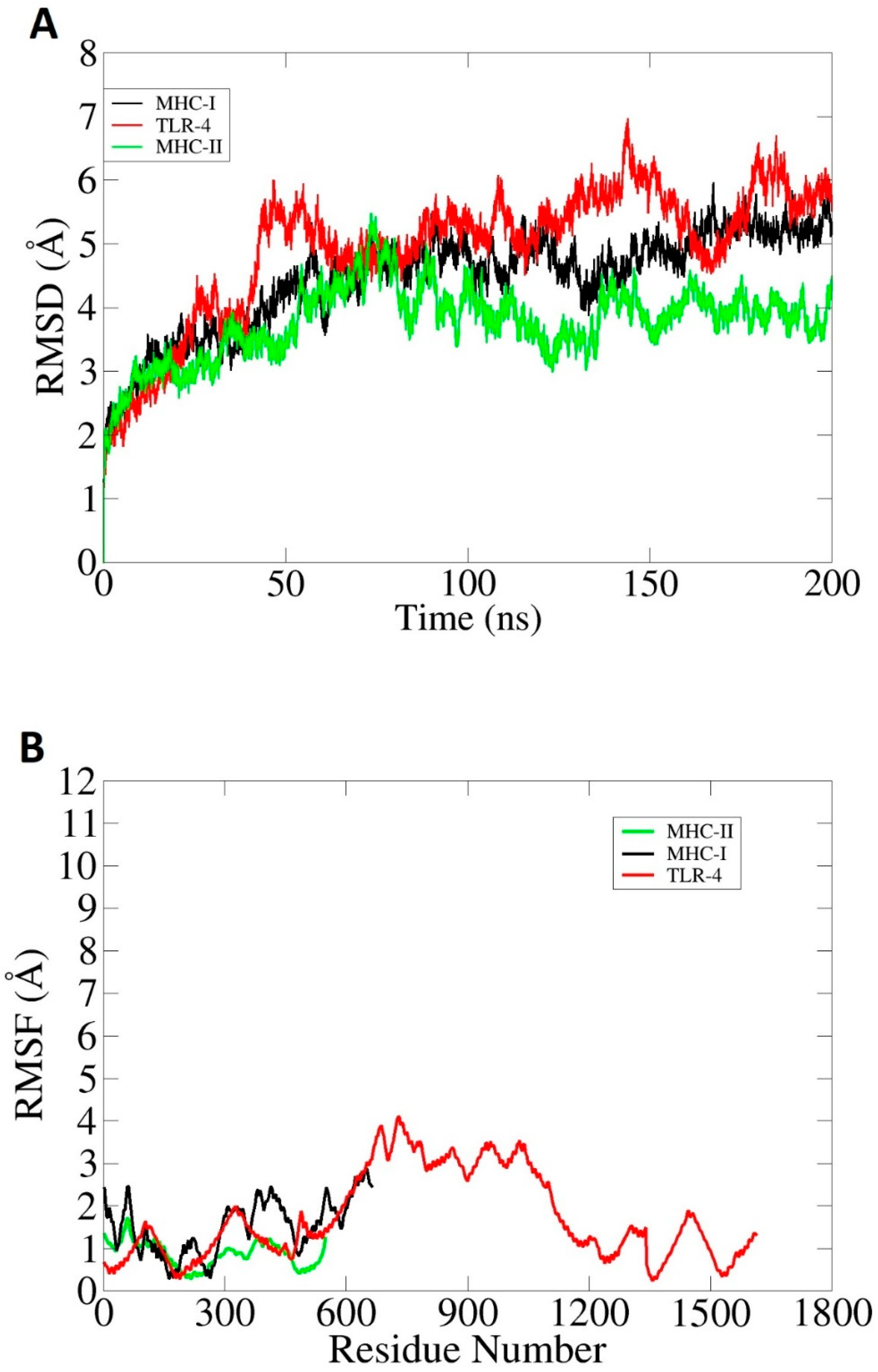
3.6. Molecular Dynamics Simulation (MDS) and Binding Free Energy Calculation
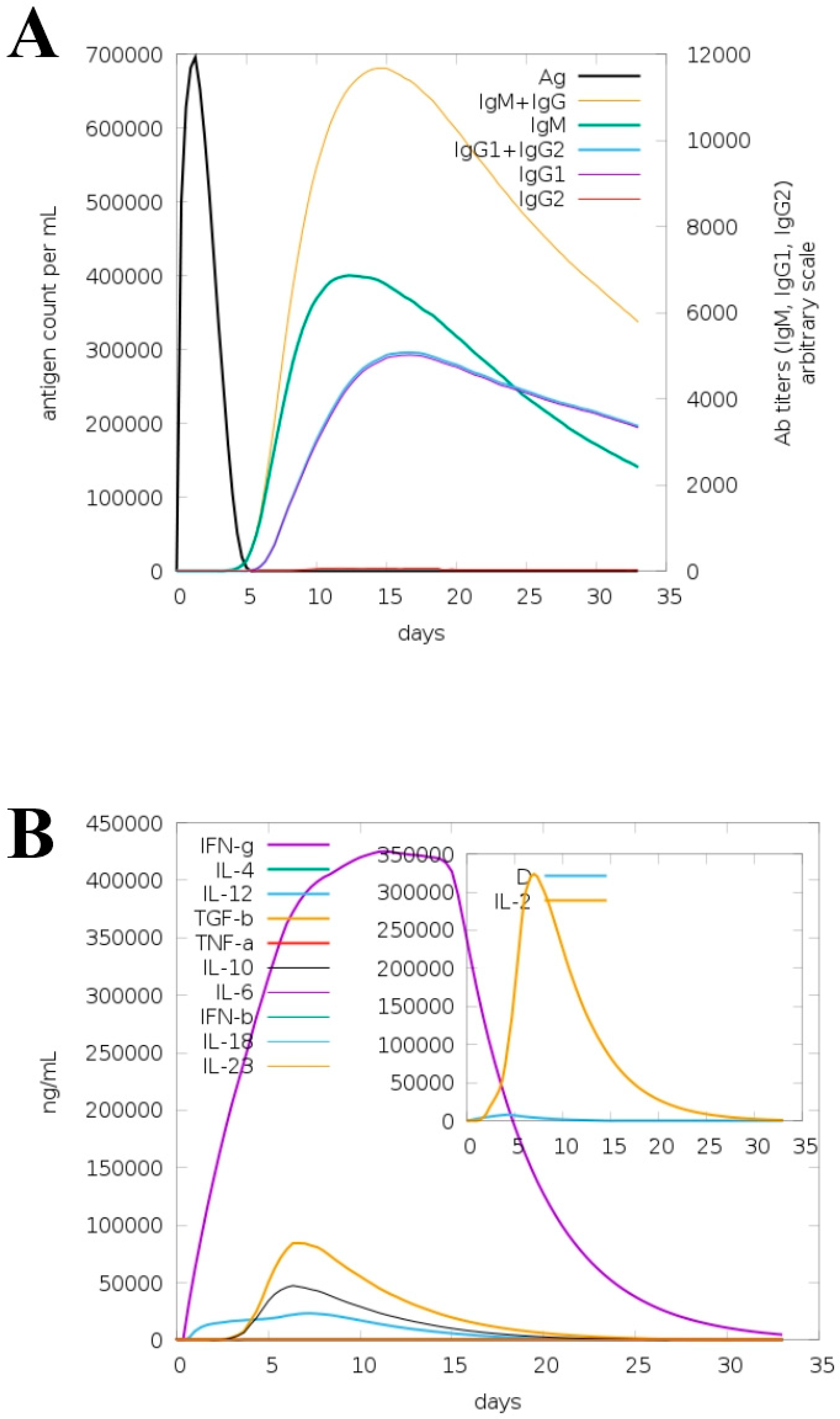
3.7. Immune Simulation for Model Vaccine
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hutchings, M.; Truman, A.; Wilkinson, B. Antibiotics: Past, Present and Future. Curr. Opin. Microbiol. 2019, 51, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, Research, and Development of New Antibiotics: The WHO Priority List of Antibiotic-Resistant Bacteria and Tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Ullah, A.; Ahmad, S.; Ismail, S.; Afsheen, Z.; Khurram, M.; Tahir ul Qamar, M.; AlSuhaymi, N.; Alsugoor, M.H.; Allemailem, K.S. Towards A Novel Multi-Epitopes Chimeric Vaccine for Simulating Strong Immune Responses and Protection against Morganella Morganii. Int. J. Environ. Res. Public Health 2021, 18, 10961. [Google Scholar] [CrossRef] [PubMed]
- Klemm, E.J.; Wong, V.K.; Dougan, G. Emergence of Dominant Multidrug-Resistant Bacterial Clades: Lessons from History and Whole-Genome Sequencing. Proc. Natl. Acad. Sci. USA 2018, 115, 12872–12877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacconelli, E.; Magrini, N.; Kahlmeter, G.; Singh, N. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics; World Health Organization: Geneva, Switzerland, 2017; Volume 27, pp. 318–327.
- Chokshi, A.; Sifri, Z.; Cennimo, D.; Horng, H. Global Contributors to Antibiotic Resistance. J. Glob. Infect. Dis. 2019, 11, 36. [Google Scholar] [PubMed]
- Abdulla, F.; Nain, Z.; Hossain, M.M.; Syed, S.B.; Khan, M.S.A.; Adhikari, U.K. A Comprehensive Screening of the Whole Proteome of Hantavirus and Designing a Multi-Epitope Subunit Vaccine for Cross-Protection against Hantavirus: Structural Vaccinology and Immunoinformatics Study. Microb. Pathog. 2021, 150, 104705. [Google Scholar] [CrossRef] [PubMed]
- Excler, J.L.; Saville, M.; Berkley, S.; Kim, J.H. Vaccine Development for Emerging Infectious Diseases. Nat. Med. 2021, 27, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Micoli, F.; Bagnoli, F.; Rappuoli, R.; Serruto, D. The Role of Vaccines in Combatting Antimicrobial Resistance. Nat. Rev. Microbiol. 2021, 19, 287–302. [Google Scholar] [CrossRef]
- Uhlemann, A.-C.; Annavajhala, M.; Gomez-Simmonds, A. Multidrug-Resistant Enterobacter Cloacae Complex Emerging as a Global, Diversifying Threat. Front. Microbiol. 2019, 10, 44. [Google Scholar]
- Gu, C.T.; Li, C.Y.; Yang, L.J.; Huo, G.C. Enterobacter xiangfangensis sp. Nov., Isolated from Chinese Traditional Sourdough, and Reclassification of Enterobacter Sacchari Zhu et Al. 2013 as Kosakonia sacchari comb. Nov. Int. J. Syst. Evol. Microbiol. 2014, 64, 2650–2656. [Google Scholar] [CrossRef] [Green Version]
- Peirano, G.; Matsumura, Y.; Adams, M.D.; Bradford, P.; Motyl, M.; Chen, L.; Kreiswirth, B.N.; Pitout, J.D.D. Genomic Epidemiology of Global Carbapenemase-Producing Enterobacter spp., 2008–2014. Emerg. Infect. Dis. 2018, 24, 1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismail, S.; Ahmad, S.; Azam, S.S. Vaccinomics to Design a Novel Single Chimeric Subunit Vaccine for Broad-Spectrum Immunological Applications Targeting Nosocomial Enterobacteriaceae Pathogens. Eur. J. Pharm. Sci. 2020, 146, 105258. [Google Scholar] [CrossRef] [PubMed]
- Rappuoli, R. Reverse Vaccinology. Curr. Opin. Microbiol. 2000, 3, 445–450. [Google Scholar] [CrossRef]
- Sette, A.; Rappuoli, R. Reverse Vaccinology: Developing Vaccines in the Era of Genomics. Immunity 2010, 33, 530–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA-an Ultra-Fast Pan-Genome Analysis Pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asad, Y.; Ahmad, S.; Rungrotmongkol, T.; Ranaghan, K.E.; Azam, S.S. Immuno-Informatics Driven Proteome-Wide Investigation Revealed Novel Peptide-Based Vaccine Targets against Emerging Multiple Drug Resistant Providencia Stuartii. J. Mol. Graph. Model. 2018, 80, 238–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emes, R.D.; Yang, Z. Duplicated Paralogous Genes Subject to Positive Selection in the Genome of Trypanosoma Brucei. PLoS ONE 2008, 3, e2295. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A Web Server for Clustering and Comparing Biological Sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Gul, S.; Ahmad, S.; Ullah, A.; Ismail, S.; Khurram, M.; ul Qamar, M.T.; Hakami, A.R.; Alkhathami, A.G.; Alrumaihi, F.; Allemailem, K.S. Designing a Recombinant Vaccine against Providencia Rettgeri Using Immunoinformatics Approach. Vaccines 2022, 10, 189. [Google Scholar] [CrossRef]
- Yu, N.Y.; Wagner, J.R.; Laird, M.R.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved Protein Subcellular Localization Prediction with Refined Localization Subcategories and Predictive Capabilities for All Prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A Reference Database for Bacterial Virulence Factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naz, A.; Awan, F.M.; Obaid, A.; Muhammad, S.A.; Paracha, R.Z.; Ahmad, J.; Ali, A. Identification of Putative Vaccine Candidates against Helicobacter Pylori Exploiting Exoproteome and Secretome: A Reverse Vaccinology Based Approach. Infect. Genet. Evol. 2015, 32, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L.L. Predicting Transmembrane Protein Topology with a Hidden Markov Model: Application to Complete Genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.; Azam, S.S. A Novel Approach of Virulome Based Reverse Vaccinology for Exploring and Validating Peptide-Based Vaccine Candidates against the Most Troublesome Nosocomial Pathogen: Acinetobacter Baumannii. J. Mol. Graph. Model. 2018, 83, 1–11. [Google Scholar] [CrossRef]
- Ismail, S.; Shahid, F.; Khan, A.; Bhatti, S.; Ahmad, S.; Naz, A.; Almatroudi, A.; ul Qamar, M.T. Pan-Vaccinomics Approach Towards a Universal Vaccine Candidate Against WHO Priority Pathogens to Address Growing Global Antibiotic Resistance. Comput. Biol. Med. 2021, 136, 104705. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A Server for Prediction of Protective Antigens, Tumour Antigens and Subunit Vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v. 2—A Server for in Silico Prediction of Allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef]
- ProtParam, E. ExPASy-ProtParam Tool. 2017. Available online: https://web.expasy.org/protparam/ (accessed on 15 March 2022).
- Bibi, S.; Ullah, I.; Zhu, B.; Adnan, M.; Liaqat, R.; Kong, W.-B.; Niu, S. In Silico Analysis of Epitope-Based Vaccine Candidate against Tuberculosis Using Reverse Vaccinology. Sci. Rep. 2021, 11, 1249. [Google Scholar] [CrossRef]
- Ahmad, S.; Ranaghan, K.E.; Azam, S.S. Combating Tigecycline Resistant Acinetobacter Baumannii: A Leap Forward towards Multi-Epitope Based Vaccine Discovery. Eur. J. Pharm. Sci. 2019, 132, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Blast, N. Basic Local Alignment Search Tool. National Library of Medicine, National Institutes of Health. 2015. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 15 March 2022).
- Vita, R.; Overton, J.A.; Greenbaum, J.A.; Ponomarenko, J.; Clark, J.D.; Cantrell, J.R.; Wheeler, D.K.; Gabbard, J.L.; Hix, D.; Sette, A.; et al. The Immune Epitope Database (IEDB) 3.0. Nucleic Acids Res. 2014, 43, D405–D412. [Google Scholar] [CrossRef]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving Sequence-Based B-Cell Epitope Prediction Using Conformational Epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dar, H.A.; Ismail, S.; Waheed, Y.; Ahmad, S.; Jamil, Z.; Aziz, H.; Hetta, H.F.; Muhammad, K. Designing a Multi-Epitope Vaccine against Mycobacteroides Abscessus by Pangenome-Reverse Vaccinology. Sci. Rep. 2021, 11, 11197. [Google Scholar] [CrossRef] [PubMed]
- Tahir ul Qamar, M.; Ahmad, S.; Fatima, I.; Ahmad, F.; Shahid, F.; Naz, A.; Abbasi, S.W.; Khan, A.; Mirza, M.U.; Ashfaq, U.A.; et al. Designing Multi-Epitope Vaccine against Staphylococcus Aureus by Employing Subtractive Proteomics, Reverse Vaccinology and Immuno-Informatics Approaches. Comput. Biol. Med. 2021, 132, 104389. [Google Scholar] [CrossRef]
- Dorosti, H.; Eslami, M.; Negahdaripour, M.; Ghoshoon, M.B.; Gholami, A.; Heidari, R.; Dehshahri, A.; Erfani, N.; Nezafat, N.; Ghasemi, Y. Vaccinomics Approach for Developing Multi-Epitope Peptide Pneumococcal Vaccine. J. Biomol. Struct. Dyn. 2019, 37, 3524–3535. [Google Scholar] [CrossRef] [PubMed]
- Omoniyi, A.A.; Adebisi, S.S.; Musa, S.A.; Nzalak, J.O.; Danborno, B.; Bauchi, Z.M.; Badmus, I.T.; Olatomide, O.D.; Oladimeji, O.J.; Nyengaard, J.R. Immunoinformatics Analysis and In-Silico Design of Multi-Epitopes Vaccine against Lassa Virus. ResherchSquare 2021. preprint. [Google Scholar]
- Baseer, S.; Ahmad, S.; Ranaghan, K.E.; Azam, S.S. Towards a Peptide-Based Vaccine against Shigella Sonnei: A Subtractive Reverse Vaccinology Based Approach. Biologicals 2017, 50, 87–99. [Google Scholar] [CrossRef] [Green Version]
- Baldauf, K.J.; Royal, J.M.; Hamorsky, K.T.; Matoba, N. Cholera Toxin B: One Subunit with Many Pharmaceutical Applications. Toxins 2015, 7, 974–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alharbi, M.; Alshammari, A.; Alasmari, A.F.; Alharbi, S.M.; ul Qamar, M.; Ullah, A.; Ahmad, S.; Irfan, M.; Khalil, A.A.K. Designing of a Recombinant Multi-Epitopes Based Vaccine against Enterococcus Mundtii Using Bioinformatics and Immunoinformatics Approaches. Int. J. Environ. Res. Public Health 2022, 19, 3729. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A Protein Structure and Structural Feature Prediction Server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef] [Green Version]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein Structure Refinement Driven by Side-Chain Repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [Green Version]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A Novel Tool to Adapt Codon Usage of a Target Gene to Its Potential Expression Host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural Summaries of PDB Entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Hebditch, M.; Carballo-Amador, M.A.; Charonis, S.; Curtis, R.; Warwicker, J. Protein–Sol: A Web Tool for Predicting Protein Solubility from Sequence. Bioinformatics 2017, 33, 3098–3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiederstein, M.; Sippl, M.J. ProSA-Web: Interactive Web Service for the Recognition of Errors in Three-Dimensional Structures of Proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, N.; Panda, P.K.; Shah, K.; Sukla, L.B.; Chaubey, P. Population Coverage Analysis of T-Cell Epitopes of Neisseria Meningitidis Serogroup B from Iron Acquisition Proteins for Vaccine Design. Bioinformation 2011, 6, 255. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Lim-Wilby, M. Molecular Docking. In Molecular Modeling of Proteins; Springer: Berlin/Heidelberg, Germany, 2008; pp. 365–382. Available online: https://link.springer.com/protocol/10.1007/978-1-59745-177-2_19 (accessed on 15 April 2022).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottarella, S.E.; Beglov, D.; Beglova, N.; Nugent, M.A.; Kozakov, D.; Vajda, S. Docking Server for the Identification of Heparin Binding Sites on Proteins. J. Chem. Inf. Model. 2014, 54, 2068–2078. [Google Scholar] [CrossRef]
- Moradi, A.; Ranjbar, Z.; Guo, L.; Javadpour, S.; Ramezanzadeh, B. Molecular Dynamic (MD) Simulation and Electrochemical Assessments of the Satureja Hortensis Extract for the Construction of Effective Zinc-Based Protective Film on Carbon Steel. J. Mol. Liq. 2021, 338, 116606. [Google Scholar] [CrossRef]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.; Cheatham, T.; Cruzeiro, V.W.D.; Darden, T.; Duke, R.E.; Giambasu, G.; et al. Amber 2020; University of California Press: San Francisco, CA, USA, 2020. [Google Scholar]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Antechamber: An Accessory Software Package for Molecular Mechanical Calculations. J. Am. Chem. Soc 2001, 222, U403. [Google Scholar]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. The FF14SB Force Field. Amber 2014, 14, 29–31. [Google Scholar]
- Roe, D.R.; Cheatham III, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Turner, P.J. XMGRACE, Version 5.1.; Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology: Beaverton, OR, USA, 2005. [Google Scholar]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Castiglione, F.; Bernaschi, M. C-Immsim: Playing with the Immune Response. In Proceedings of the 16th International Symposium on Mathematical Theory of Networks and Systems (MTNS2004), Leuven, Belgium, 5–9 July 2004. [Google Scholar]
- Hassan, A.; Naz, A.; Obaid, A.; Paracha, R.Z.; Naz, K.; Awan, F.M.; Muhmmad, S.A.; Janjua, H.A.; Ahmad, J.; Ali, A. Pangenome and Immuno-Proteomics Analysis of Acinetobacter Baumannii Strains Revealed the Core Peptide Vaccine Targets. BMC Genom. 2016, 17, 732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanober, G.; Ahmad, S.; Azam, S.S. Identification of Plausible Drug Targets by Investigating the Druggable Genome of MDR Staphylococcus Epidermidis. Gene Rep. 2017, 7, 147–153. [Google Scholar] [CrossRef]
- Barh, D.; Barve, N.; Gupta, K.; Chandra, S.; Jain, N.; Tiwari, S.; Leon-Sicairos, N.; Canizalez-Roman, A.; dos Santos, A.R.; Hassan, S.S.; et al. Exoproteome and Secretome Derived Broad Spectrum Novel Drug and Vaccine Candidates in Vibrio Cholerae Targeted by Piper Betel Derived Compounds. PLoS ONE 2013, 8, e52773. [Google Scholar] [CrossRef]
- Ali, A.; Naz, A.; Soares, S.C.; Bakhtiar, M.; Tiwari, S.; Hassan, S.S.; Hanan, F.; Ramos, R.; Pereira, U.; Barh, D.; et al. Pan-Genome Analysis of Human Gastric Pathogen H. Pylori: Comparative Genomics and Pathogenomics Approaches to Identify Regions Associated with Pathogenicity and Prediction of Potential Core Therapeutic Targets. Biomed. Res. Int. 2015, 2015, 139580. [Google Scholar] [CrossRef] [Green Version]
- Dombkowski, A.A.; Sultana, K.Z.; Craig, D.B. Protein Disulfide Engineering. FEBS Lett. 2014, 588, 206–212. [Google Scholar] [CrossRef] [Green Version]
- Zahroh, H.; Ma’rup, A.; Tambunan, U.S.F.; Parikesit, A.A. Immunoinformatics Approach in Designing Epitopebased Vaccine against Meningitis-Inducing Bacteria (Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae Type B). Drug Target Insights 2016, 10, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro Web Server for Protein–Protein Docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Carugo, O. How Root-Mean-Square Distance (Rmsd) Values Depend on the Resolution of Protein Structures That Are Compared. J. Appl. Crystallogr. 2003, 36, 125–128. [Google Scholar] [CrossRef]
- Knapp, B.; Lederer, N.; Omasits, U.; Schreiner, W. VmdICE: A Plug-in for Rapid Evaluation of Molecular Dynamics Simulations Using VMD. J. Comput. Chem. 2010, 31, 2868–2873. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vaccine Candidate Protein | B-Cell Peptide | Antigenicity Score |
|---|---|---|
| core/255/1/Org1_Gene3420 (ferrichrome porin (FhuA)) | AAETPKKEETITVTAAPAAQESAWGPAPTIAAKRTATATKTDTPIEKTPQSISVVTREEMDMKQPGT | 0.78 |
| PTTEPLKEIQFKMGTDNLWQTGFD | 0.53 | |
| LPREGTVVPYYDANGKAHKLPTDFNEGDEDNKISRR | 0.98 | |
| NDTFTVRQNLRYTK | 0.45 | |
| TSAFNRNNGTTAEINDQAF | 0.62 | |
| FEPLSGTTQGGKPFD | 0.42 | |
| TADPANPTSGFSVQG | 0.52 | |
| NTVTYYSSASPKAYESFNV | 0.85 | |
| core/4064/1/Org1_Gene715 (peptidoglycan-associated lipoprotein (Pal)) | SNKNASNDQSGEGMMGAGTGMDANGNGNMSSEEQARLQMQQLQQNNIVYFDLDKYDIRS | 0.42 |
| DERGTPEYNISL | 0.40 | |
| SYGKEKPAVLGHDEAAYSKN | 0.63 | |
| SNKNASNDQSGEGMMGAGTGMDANGNGNMSSEEQARLQMQQLQQNNIVYFDLDKYDIRS | 0.71 |
| Model | RMSD | MolProbity | Clash Score | Poor Rotamers | Rama Favored | GALAXY Energy |
|---|---|---|---|---|---|---|
| Initial | 0.000 | 4.112 | 237.6 | 7.9 | 88.0 | 32,580.03 |
| Model 1 | 1.055 | 1.757 | 5.5 | 0.0 | 92.8 | −2764.21 |
| Model 2 | 0.945 | 1.785 | 6.9 | 0.0 | 94.0 | −2752.33 |
| Model 3 | 1.115 | 1.884 | 9.0 | 0.0 | 94.0 | −2750.91 |
| Model 4 | 2.568 | 1.683 | 4.5 | 0.7 | 92.8 | −2737.98 |
| Model 5 | 1.068 | 1.857 | 7.3 | 0.0 | 92.8 | −2734.97 |
| Model 6 | 2.477 | 1.709 | 5.2 | 0.7 | 93.4 | −2729.57 |
| Model 7 | 1.017 | 1.779 | 5.2 | 0.0 | 91.6 | −2718.37 |
| Model 8 | 2.229 | 1.679 | 4.2 | 0.0 | 92.2 | −2718.16 |
| Model 9 | 0.933 | 1.870 | 8.7 | 0.7 | 94.0 | −2717.94 |
| Model 10 | 2.437 | 1.683 | 4.5 | 0.7 | 92.8 | −2716.26 |
| Complex | Interacting Residues |
|---|---|
| Vaccine–MHC-I Complex | Asp66, Ala173, Asn312, Ala74, Phe40, Ala68, Val119, Gly121, Thr80, Glu35, Gln34, Arg4, Asp17, Val44, Asn60, Arg105, Val44, Asn103, Pro187, Arg72, Glu17, Val42, Ile31, Phe122 |
| Vaccine–MHC-II Complex | Asn42, Lys41, Gly43, Glu44, Arg45, Lys94, Ala14, Arg97, Glu16, Asp98, Arg273, Tyr257, Leu272, Thr258, Arg219, Asp223, Gln222, Pro232, Phe208, Gln224, Lys19, Thr240, Ser61, Gly239, Val231 Arg202, His192 |
| Vaccine–TLR-4 Complex | Asn526, Leu25, Asp502, Lys477, Glu272, Tyr451, Phe573, Val605, Ser582, Ile450, Gln578, Val548, Ser552, Leu548 |
| Energy Parameter | TLR-4–Vaccine Complex | MHC-I–Vaccine Complex | MHC-II–Vaccine Complex |
|---|---|---|---|
| MM-GBSA | |||
| VDWAALS | −190.74 | −180.60 | −168.55 |
| EEL | −90.23 | −54.87 | −60.97 |
| Delta G gas | −280.97 | −235.47 | −229.52 |
| Delta G solv | 55.00 | 53.48 | 52.00 |
| Delta Total | −225.97 | −181.99 | 177.52 |
| MM-PBSA | |||
| VDWAALS | −190.74 | −180.60 | −168.55 |
| EEL | −90.23 | −54.87 | −60.97 |
| Delta G gas | −280.97 | −235.47 | −229.52 |
| Delta G solv | 48.99 | 45.57 | 52.09 |
| Delta Total | −231.98 | −189.9 | −177.43 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshammari, A.; Alharbi, M.; Alghamdi, A.; Alharbi, S.A.; Ashfaq, U.A.; Tahir ul Qamar, M.; Ullah, A.; Irfan, M.; Khan, A.; Ahmad, S. Computer-Aided Multi-Epitope Vaccine Design against Enterobacter xiangfangensis. Int. J. Environ. Res. Public Health 2022, 19, 7723. https://doi.org/10.3390/ijerph19137723
Alshammari A, Alharbi M, Alghamdi A, Alharbi SA, Ashfaq UA, Tahir ul Qamar M, Ullah A, Irfan M, Khan A, Ahmad S. Computer-Aided Multi-Epitope Vaccine Design against Enterobacter xiangfangensis. International Journal of Environmental Research and Public Health. 2022; 19(13):7723. https://doi.org/10.3390/ijerph19137723
Chicago/Turabian StyleAlshammari, Abdulrahman, Metab Alharbi, Abdullah Alghamdi, Saif Ali Alharbi, Usman Ali Ashfaq, Muhammad Tahir ul Qamar, Asad Ullah, Muhammad Irfan, Amjad Khan, and Sajjad Ahmad. 2022. "Computer-Aided Multi-Epitope Vaccine Design against Enterobacter xiangfangensis" International Journal of Environmental Research and Public Health 19, no. 13: 7723. https://doi.org/10.3390/ijerph19137723