Metabolic Activation and Carcinogenesis of Tobacco-Specific Nitrosamine N’-Nitrosonornicotine (NNN): A Density Function Theory and Molecular Docking Study



Abstract
1. Introduction
2. Materials and Methods
2.1. DFT Calculations
2.2. Molecular Docking
3. Results and Discussion
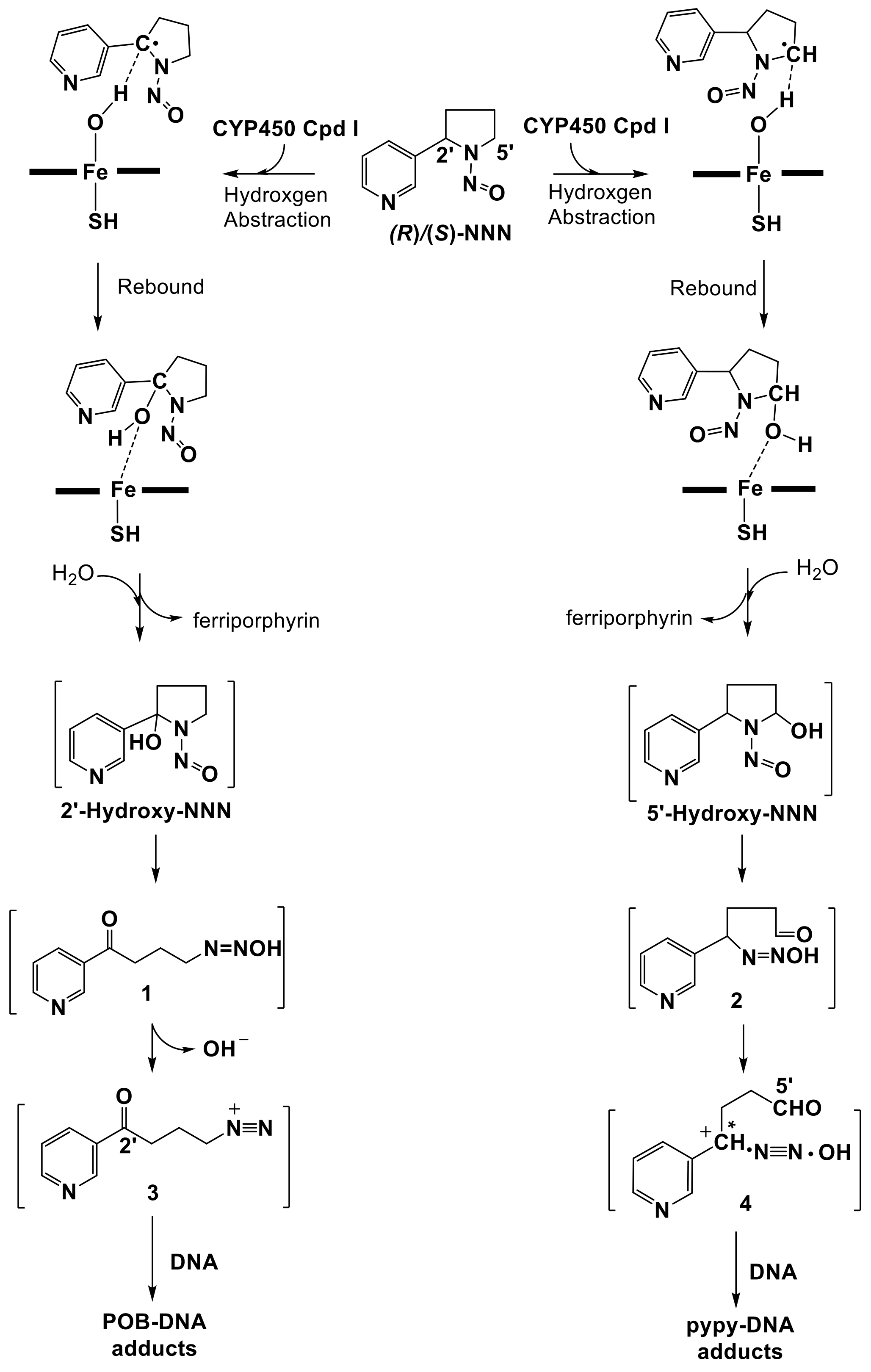
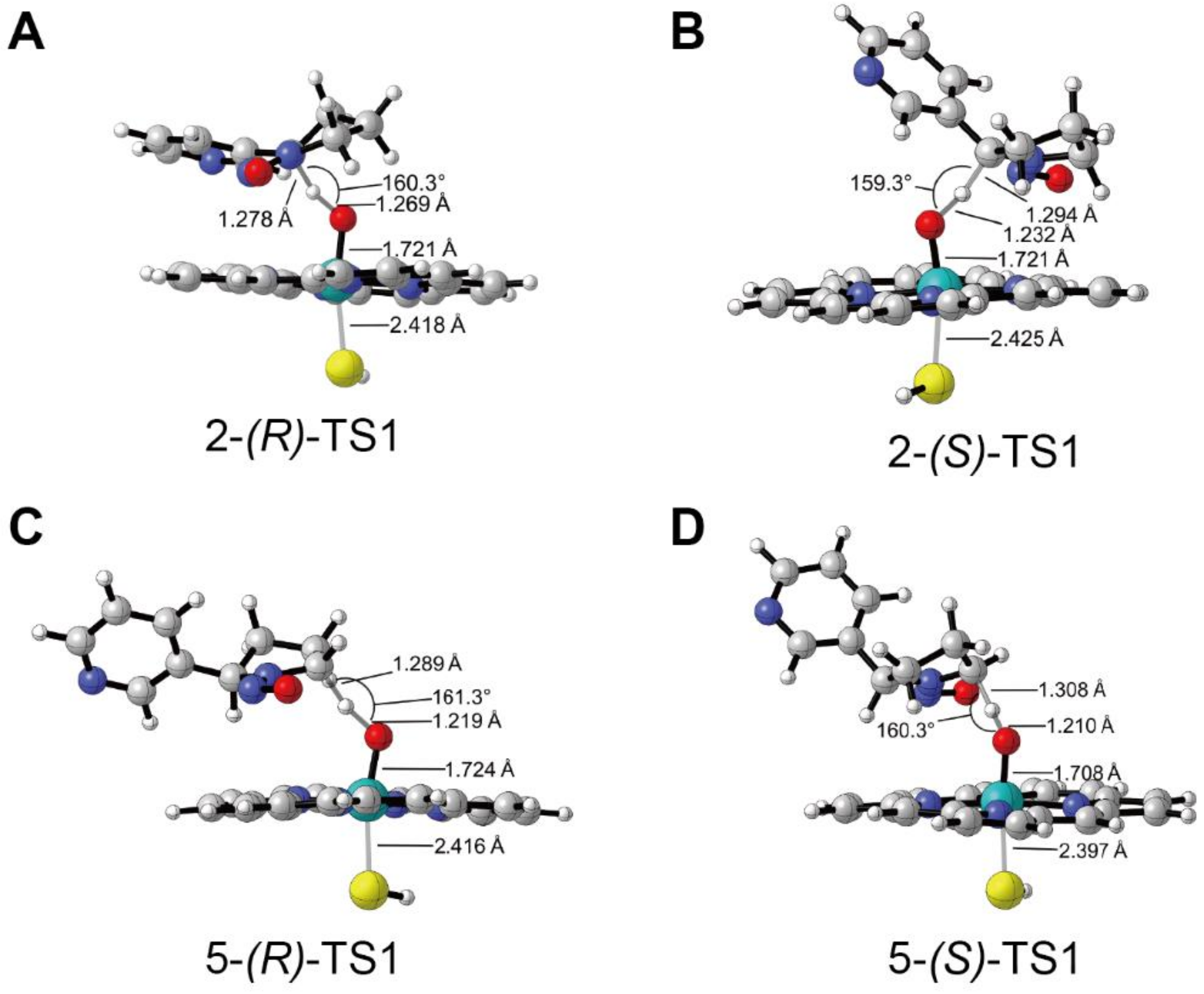
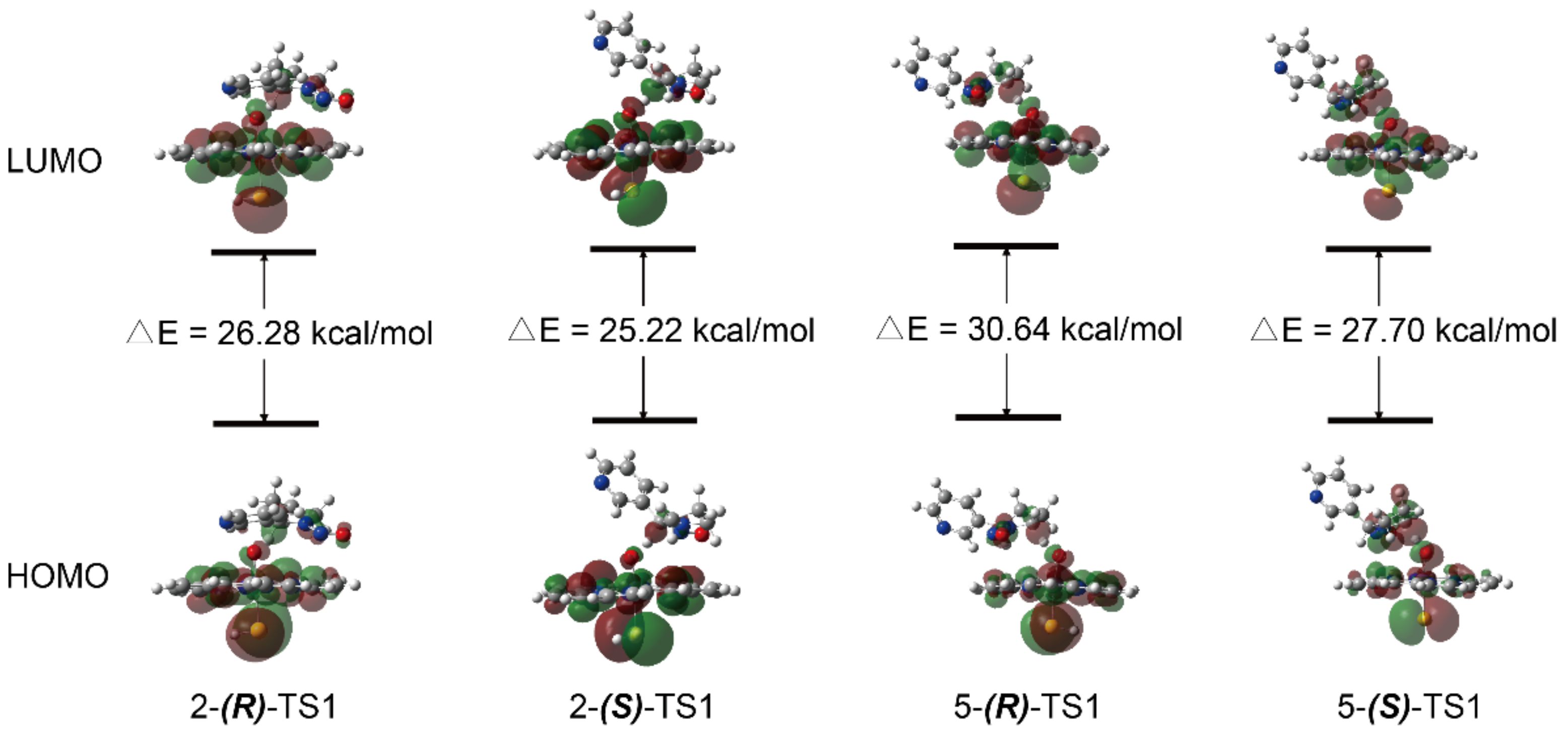
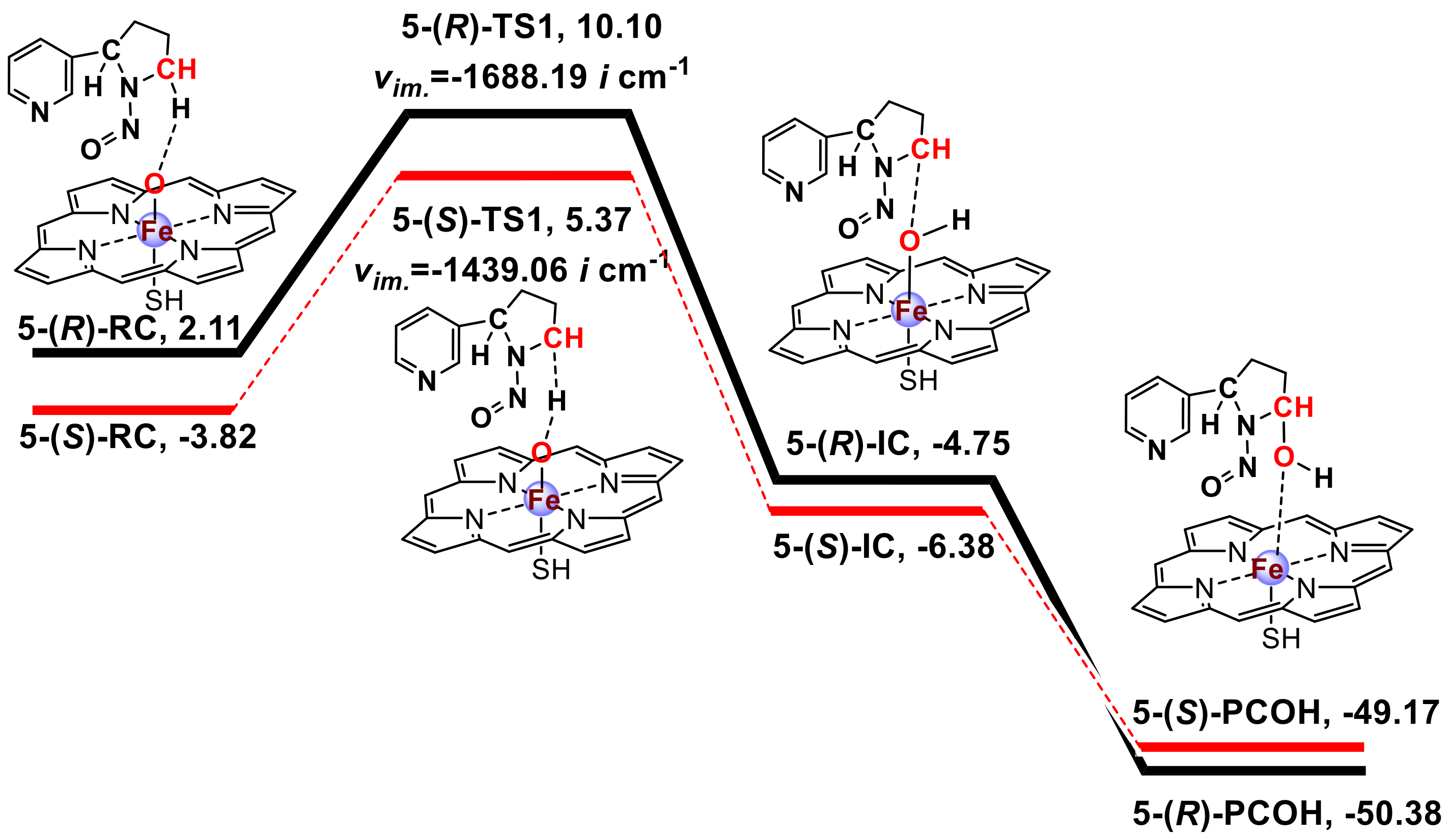
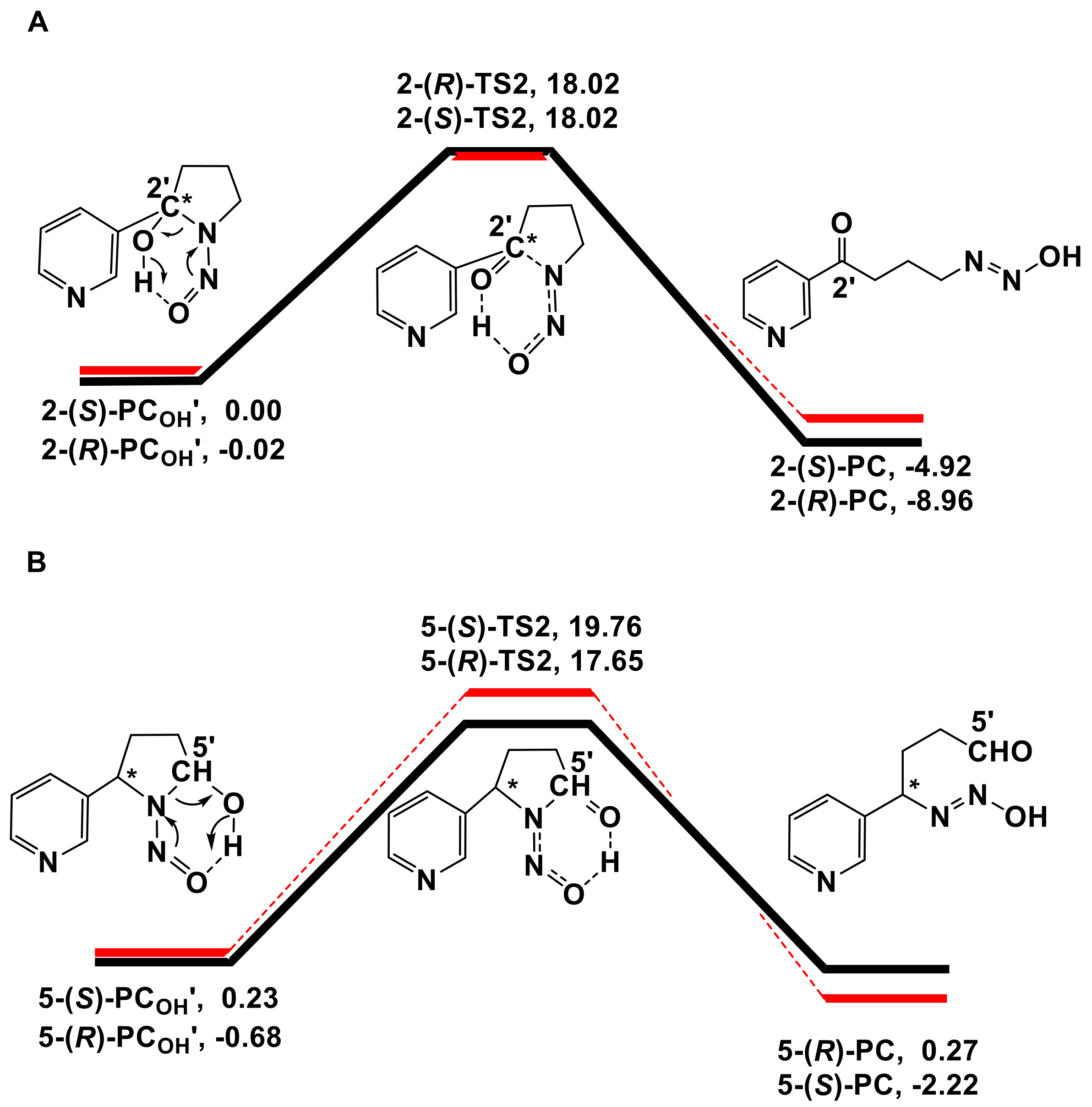
3.1. Metabolic Activation of (R)-NNN and (S)-NNN
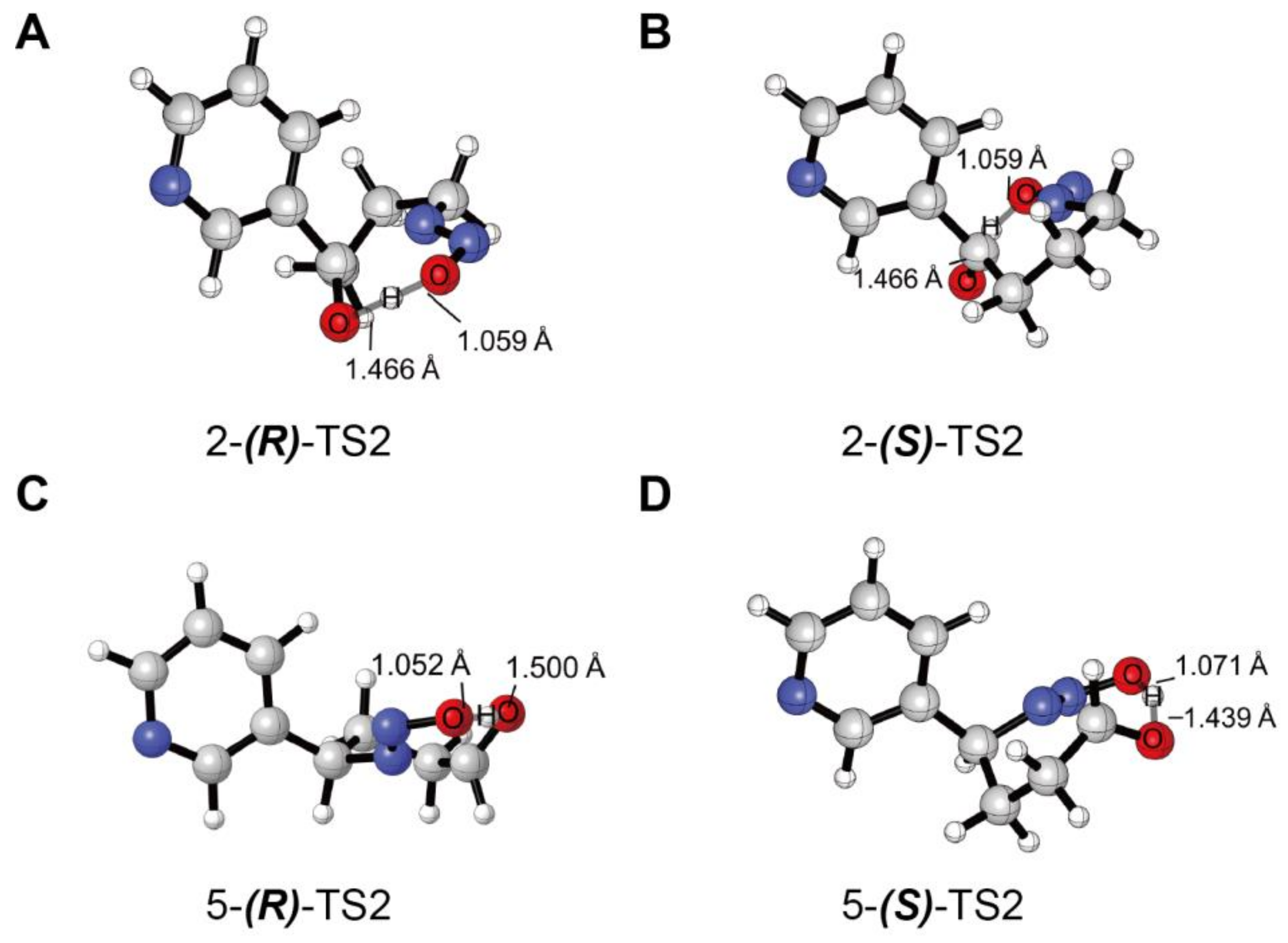
3.2. Decomposition of the Hydroxylation Products
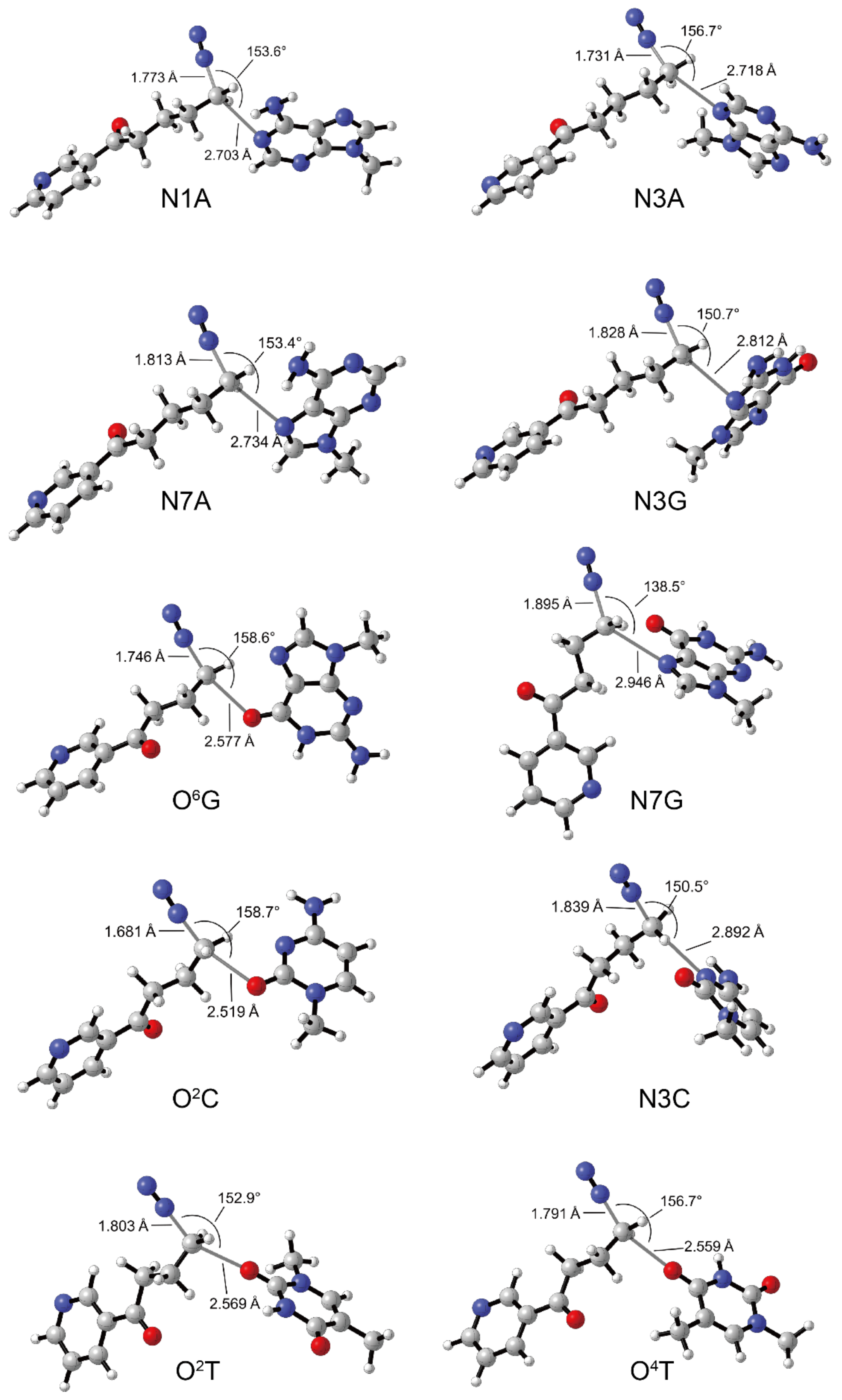
3.3. DNA Alkylation of 2′-Hydroxylation Products
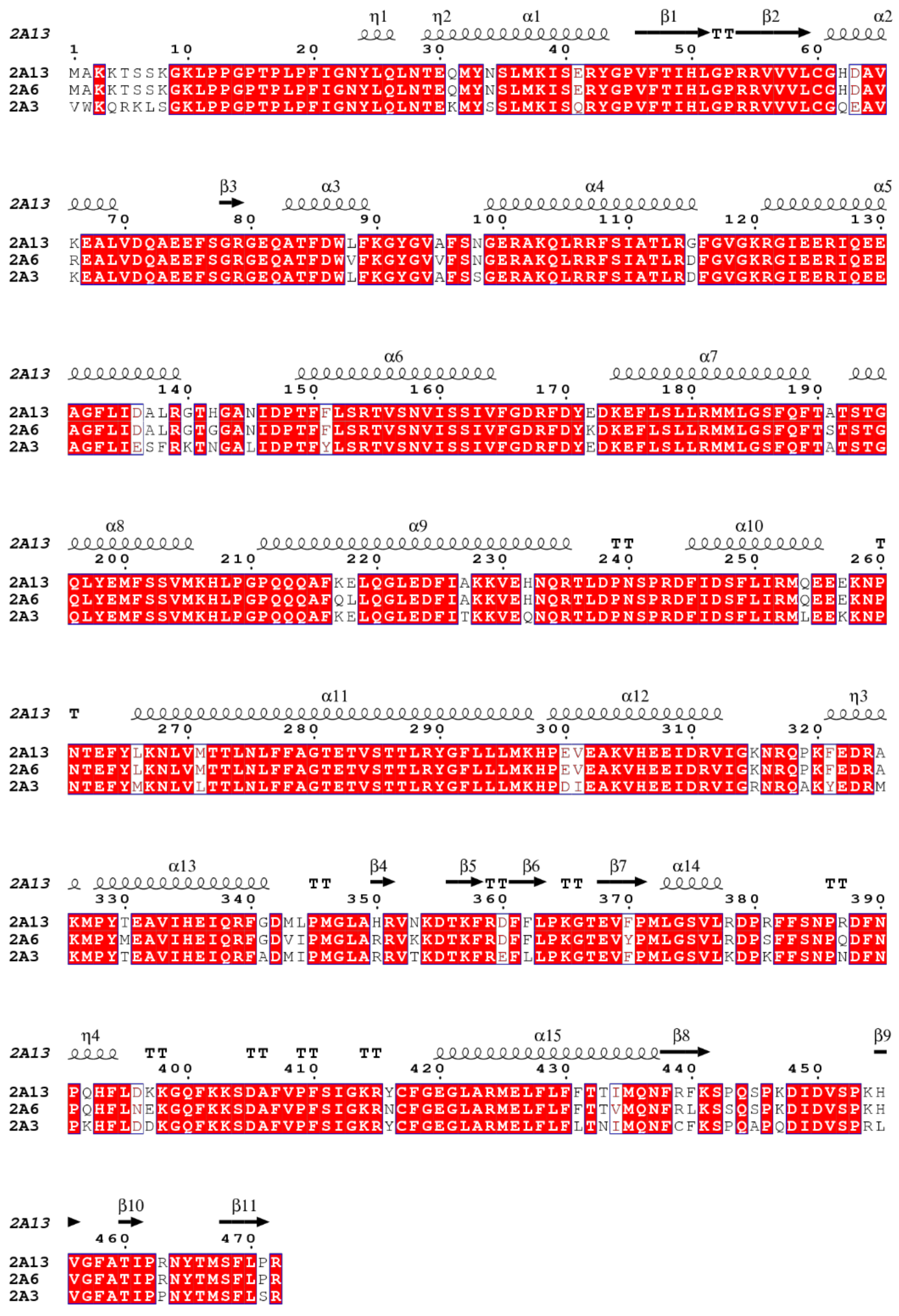
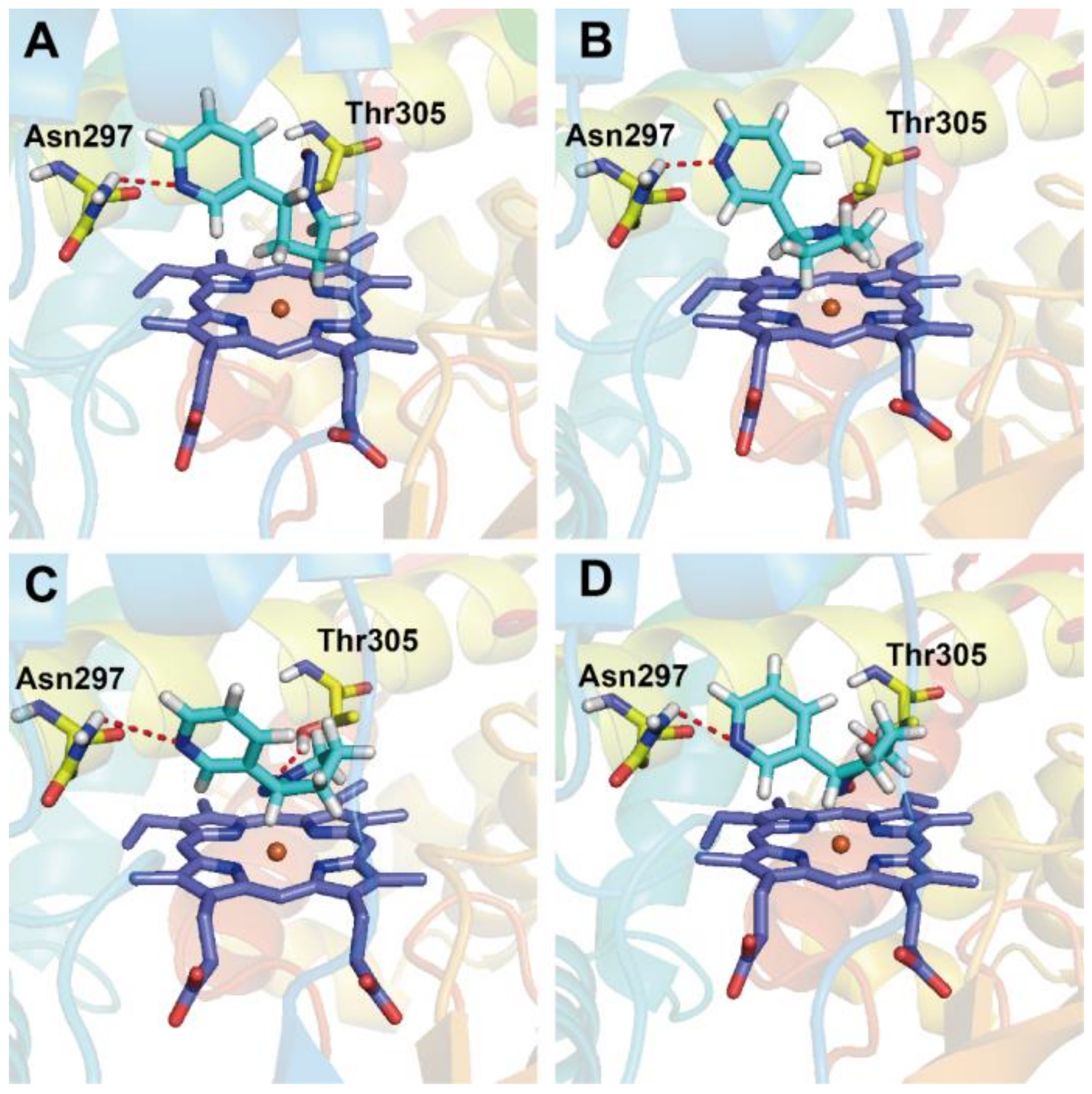
3.4. Docking Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- IARC. Smokeless tobacco and some tobacco-specific N-nitrosamines. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 2007. [Google Scholar]
- Stepanov, I.; Yershova, K.; Carmella, S.; Upadhyaya, P.; Hecht, S.S. Levels of (S)-N’-nitrosonornicotine in US tobacco products. Nicot. Tob. Res. 2013, 15, 1305–1310. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S. Biochemistry, Biology, and Carcinogenicity of tobacco-specific N-nitrosamines. Chem. Res. Toxicol. 1998, 11, 559–603. [Google Scholar] [CrossRef] [PubMed]
- Balbo, S.; James-Yi, S.; Johnson, C.S.; O’Sullivan, M.G.; Stepanov, I.; Wang, M.; Bandyopadhyay, D.; Kassie, F.; Carmella, S.; Upadhyaya, P.; et al. (S)-N’-nitrosonornicotine, a constituent of smokeless tobacco, is a powerful oral cavity carcinogen in rats. Carcinogenesis 2013, 34, 2178–2183. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.B.; Hecht, S.S.; Hoffmann, D. Metabolic alpha-hydroxylation of tobacco-specific carcinogen, N’-nitrosonornicotine. Cancer Res. 1978, 38, 3639–3645. [Google Scholar] [PubMed]
- Hecht, S.S.; Lin, D. Comparative mutagenicity of 4-(carbethoxynitrosamino)-4-(3-pyridyl) butanal and 4-(carbethoxynitrosamino)-1-(3-pyridyl)-1-butanone, model compounds for alpha-hydroxylation of n’-nitrosonornicotine. Carcinogenesis 1986, 7, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Lao, Y.; Yu, N.; Kassie, F.; Villalta, P.W.; Hecht, S.S. Analysis of pyridyloxobutyl DNA adducts in F344 rats chronically treated with (R)- and (S)-N’-nitrosonornicotine. Chem. Res. Toxicol. 2007, 20, 246–256. [Google Scholar] [CrossRef] [PubMed]
- McIntee, E.J.; Hecht, S.S. Metabolism of N’-nitrosonornicotine enantiomers by cultured rat esophagus and in vivo in rats. Chem. Res. Toxicol. 2000, 13, 192–199. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, M.; Villalta, P.W.; Lindgren, B.R.; Lao, Y.; Hecht, S.S. Quantitation of pyridyloxobutyl DNA adducts in nasal and oral mucosa of rats treated chronically with enantiomers of N’-nitrosonornicotine. Chem. Res. Toxicol. 2009, 22, 949–956. [Google Scholar] [CrossRef]
- Zhao, L.; Balbo, S.; Wang, M.; Upadhyaya, P.; Khariwala, S.S.; Villalta, P.W.; Hecht, S.S. Quantitation of pyridyloxobutyl-DNA adducts in tissues of rats treated chronically with (R)- or (S)-N’-nitrosonornicotine (NNN) in a carcinogenicity study. Chem. Res. Toxicol. 2013, 26, 1526–1535. [Google Scholar] [CrossRef]
- Upadhyaya, P.; Zimmerman, C.L.; Hecht, S.S. Metabolism and pharmacokinetics of N’-nitrosonornicotine in the patas monkey. Drug Metab. Dispos. 2002, 30, 1115–1122. [Google Scholar] [CrossRef]
- Patten, C.J.; Smith, T.J.; Friesen, M.J.; Tynes, R.E.; Yang, C.S.; Murphy, S.E. Evidence for cytochrome P450 2A6 and 3A4 as major catalysts for N’-nitrosonornicotine alpha-hydroxylation by human liver microsomes. Carcinogenesis 1997, 18, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Staretz, M.E.; Murphy, S.E.; Patten, C.J.; Nunes, M.G.; Koehl, W.; Amin, S.; Koenig, L.A.; Guengerich, F.P.; Hecht, S.S. Comparative metabolism of the tobacco-related carcinogens benzo[a]pyrene, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol, and N’-nitrosonornicotine in human hepatic microsomes. Drug Metab. Dispos. 1997, 25, 154–162. [Google Scholar] [PubMed]
- Wong, H.L.; Murphy, S.E.; Hecht, S.S. Cytochrome P450 2A-Catalyzed Metabolic activation of structurally similar carcinogenic nitrosamines: N’-nitrosonornicotine enantiomers, N-nitrosopiperidine, and N-nitrosopyrrolidine. Chem. Res. Toxicol. 2005, 18, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Castonguay, A.; Stoner, G.D.; Schut, H.; Hecht, S.S. Metabolism of tobacco-specific N-nitrosamines by cultured human-tissues. Proc. Natl. Acad. Sci. USA 1983, 80, 6694–6697. [Google Scholar] [CrossRef] [PubMed]
- Green, M.T. C-H bond activation in heme proteins: The role of thiolate ligation in cytochrome P450. Curr. Opin. Chem. Biol. 2009, 13, 84–88. [Google Scholar] [CrossRef]
- De Montellano Ortiz, P.R. Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev. 2010, 110, 932–948. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Cohen, S.; Wang, Y.; Chen, H.; Kumar, D.; Thiel, W. P450 Enzymes: Their structure, reactivity, and selectivity-modeled by QM/MM calculations. Chem. Rev. 2010, 110, 949–1017. [Google Scholar] [CrossRef]
- Ogliaro, F.; Harris, N.; Cohen, S.; Filatov, M.; de Visser, S.P.; Shaik, S. A model “rebound” mechanism of hydroxylation by cytochrome P450: Stepwise and effectively concerted pathways, and their reactivity patterns. J. Am. Chem. Soc. 2000, 122, 8977–8989. [Google Scholar] [CrossRef]
- De Visser, S.P.; Ogliaro, F.; Sharma, P.K.; Shaik, S. What factors affect the regioselectivity of oxidation by cytochrome P450? a DFT study of allylic hydroxylation and double bond epoxidation in a model reaction. J. Am. Chem. Soc. 2002, 124, 11809–11826. [Google Scholar] [CrossRef]
- Kamachi, T.; Yoshizawa, K. A theoretical study on the mechanism of camphor hydroxylation by compound I of cytochrome P450. J. Am. Chem. Soc. 2003, 125, 4652–4661. [Google Scholar] [CrossRef]
- Gelb, M.H.; Heimbrook, D.C.; Malkonen, P.; Sligar, S.G. Stereochemistry and deuterium-isotope effects in camphor hydroxylation by the cytochrome P450cam monoxygenase system. Biochemistry 1982, 21, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, M.; Shen, R.; Choi, S.Y.; Toy, P.H.; Hollenberg, P.F.; Vaz, A.; Coon, M.J. Cytochrome P450-catalyzed hydroxylation of mechanistic probes that distinguish between radicals and cations. Evidence for cationic but not for radical intermediates. J. Am. Chem. Soc. 2000, 122, 2677–2686. [Google Scholar] [CrossRef]
- Wang, Y.; Kumar, D.; Yang, C.; Han, K.; Shaik, S. Theoretical study of N-demethylation of substituted N,N-Dimethylanilines by cytochrome P450: The mechanistic significance of kinetic isotope effect profiles. J. Phys. Chem. B. 2007, 111, 7700–7710. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Nandi, S. Choices have consequences: The nexus between DNA repair pathways and genomic instability in cancer. Clin. Transl. Med. 2016, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Nandi, S. DNA damage response and cancer therapeutics through the lens of the Fanconi Anemia DNA repair pathway. Cell Commun. Signal. 2017, 15, 41. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Nandi, S. Synthetic lethality in DNA repair network: A novel avenue in targeted cancer therapy and combination therapeutics. IUBMB Life 2017, 69, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Nandi, S. Rare genetic diseases with defects in DNA repair: Opportunities and challenges in orphan drug development for targeted cancer therapy. Cancers 2018, 10, 298. [Google Scholar] [CrossRef]
- Sun, G.H.; Zhao, L.J.; Zhong, R.G. The induction and repair of DNA interstrand crosslinks and implications in cancer chemotherapy. Anti-Cancer Agents Med. Chem. 2016, 16, 221–246. [Google Scholar]
- Sun, G.H.; Zhao, L.J.; Zhong, R.G.; Peng, Y.Z. The specific role of O6-methylguanine-DNA methyltransferase inhibitors in cancer chemotherapy. Future Med. Chem. 2018, 10, 1971–1996. [Google Scholar] [CrossRef]
- Sun, G.H.; Fan, T.J.; Zhao, L.J.; Zhou, Y.; Zhong, R.G. The potential of combi-molecules with DNA-damaging function as anticancer agents. Future Med. Chem. 2017, 9, 403–435. [Google Scholar] [CrossRef]
- Sun, G.H.; Zhao, L.J.; Fan, T.J.; Li, S.S.; Zhong, R.G. Investigations on the effect of O6-benzylguanine on the formation of dG-dC interstrand cross-links induced by chloroethylnitrosoureas in human glioma cells using stable isotope dilution high-performance liquid chromatography electrospray ionization tandem mass spectrometry. Chem. Res. Toxicol. 2014, 27, 1253–1262. [Google Scholar] [PubMed]
- Zarth, A.T.; Upadhyaya, P.; Yang, J.; Hecht, S.S. DNA adduct formation from metabolic 5′-hydroxylation of the tobacco-specific carcinogen N′-nitrosonornicotine in human enzyme systems and in rats. Chem. Res. Toxicol. 2016, 29, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry III. the role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Shahrokh, K.; Orendt, A.; Yost, G.S.; Cheatham, T.E.I. Quantum mechanically derived amber-compatible heme parameters for various states of the cytochrome P450 catalytic cycle. J. Comput. Chem. 2012, 33, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Schlegel, H.B. Reaction-path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction-path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Miertus, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum—A direct utilization of ab initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Rendic, S.; Guengerich, F.P. Contributions of human enzymes in carcinogen metabolism. Chem. Res. Toxicol. 2012, 25, 1316–1383. [Google Scholar] [CrossRef]
- Murphy, S.E.; Isaac, I.S.; Ding, X.X.; McIntee, E.J. Specificity of cytochrome P450 2A3-catalyzed alpha-hydroxylation of N’-nitrosonornicotine enantiomers. Drug Metab. Dispos. 2000, 28, 1263–1266. [Google Scholar] [PubMed]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 2016, 86. [Google Scholar]
- Fiser, A.; Sali, A. ModLoop: Automated modeling of loops in protein structures. Bioinformatics 2003, 19, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.H.; Fan, T.J.; Zhang, N.; Ren, T.; Zhao, L.J.; Zhong, R.G. Identification of the structural features of guanine derivatives as MGMT inhibitors using 3D-QSAR modeling combined with molecular docking. Molecules 2016, 21, 823. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.H.; Zhang, N.; Zhao, L.J.; Fan, T.J.; Zhang, S.F.; Zhong, R.G. Synthesis and antitumor activity evaluation of a novel combi-nitrosourea prodrug: Designed to release a DNA cross-linking agent and an inhibitor of O6-alkylguanine-DNA alkyltransferase. Bioorg. Med. Chem. 2016, 24, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.A.; Szklarz, G.D.; Scott, E.E. Human cytochrome P450 1A1 structure and utility in understanding drug and xenobiotic metabolism. J. Biol. Chem. 2013, 288, 12932–12943. [Google Scholar] [CrossRef]
- DeVore, N.M.; Scott, E.E. Nicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone binding and access channel in human cytochrome P450 2A6 and 2A13 enzymes. J. Biol. Chem. 2012, 287, 26576–26585. [Google Scholar] [CrossRef]
- Porubsky, P.R.; Battaile, K.P.; Scott, E.E. human cytochrome P450 2E1 structures with fatty acid analogs reveal a previously unobserved binding mode. J. Biol. Chem. 2010, 285, 22282–22290. [Google Scholar] [CrossRef]
- Williams, P.A.; Cosme, J.; Vinkovic, D.M.; Ward, A.; Angove, H.C.; Day, P.J.; Vonrhein, C.; Tickle, I.J.; Jhoti, H. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science 2004, 305, 683–686. [Google Scholar] [CrossRef]
- De Visser, S.P.; Kumar, D.; Cohen, S.; Shacham, R.; Shaik, S. A predictive pattern of computed barriers for C-H hydroxylation by compound I of cytochrome P450. J. Am. Chem. Soc. 2004, 126, 8362–8363. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Schüürmann, G. Computational evidence for α-nitrosamino radical as initial metabolite for both the p450 dealkylation and denitrosation of carcinogenic nitrosamines. J. Phys. Chem. B 2012, 116, 903–912. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Ogliaro, F.; Shaik, S. How does ethene inactivate cytochrome P450 en route to its epoxidation? A density functional study. Angew. Chem. Int. Edit. 2001, 40, 2871–2874. [Google Scholar] [CrossRef]
- De Visser, S.P.; Ogliaro, F.; Harris, N.; Shaik, S. Multi-state epoxidation of ethene by cytochrome P450: A quantum chemical study. J. Am. Chem. Soc. 2001, 123, 3037–3047. [Google Scholar] [CrossRef]
- De Visser, S.P.; Kumar, D.; Shaik, S. How do aldehyde side products occur during alkene epoxidation by cytochrome P450? Theory reveals a state-specific multi-state scenario where the high-spin component leads to all side products. J. Inorg. Biochem. 2004, 98, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Hirao, H.; Kumar, D.; Thiel, W.; Shaik, S. Two states and two more in the mechanisms of hydroxylation and epoxidation by cytochrome P450. J. Am. Chem. Soc. 2005, 127, 13007–13018. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; de Visser, S.P.; Shaik, S. Multistate reactivity in styrene epoxidation by compound I of cytochrome P450: Mechanisms of products and side products formation. Chem-Eur. J. 2005, 11, 2825–2835. [Google Scholar] [CrossRef]
- Ma, G.C.; Yu, H.Y.; Xu, T.; Wei, X.X.; Chen, J.R.; Lin, H.J.; Schuurmann, G. Computational insight into the activation mechanism of carcinogenic N’-nitrosonornicotine (NNN) catalyzed by cytochrome P450. Environ. Sci. Technol. 2018, 52, 11838–11847. [Google Scholar] [CrossRef]
- Lonsdale, R.; Harvey, J.N.; Mulholland, A.J. Inclusion of dispersion effects significantly improves accuracy of calculated reaction barriers for cytochrome P450 catalyzed reactions. J. Phys. Chem. Lett. 2010, 1, 3232–3237. [Google Scholar] [CrossRef]
- Grimme, S. Density functional theory with london dispersion corrections. Wires. Comput. Mol. Sci. 2011, 1, 211–228. [Google Scholar] [CrossRef]
- Finneman, J.I.; Fishbein, J.C. Mechanisms of benzyl group-transfer in the decay of (E)-arylmethanediazoates and aryldiazomethanes in aqueous-solutions. J. Am. Chem. Soc. 1995, 117, 4228–4239. [Google Scholar] [CrossRef]
- Beranek, D.T. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat. Res. 1990, 231, 11. [Google Scholar] [CrossRef]
- Lewis, D.F.; Ioannides, C.; Parke, D.V. Cytochromes P450 and species differences in xenobiotic metabolism and activation of carcinogen. Environ. Health Perspect. 1998, 106, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.F.; Heilman, J.M.; Blans, P.; Fishbein, J.C. The structure of DNA dictates purine atom site selectivity in alkylation by primary diazonium ions. Chem. Res. Toxicol. 2005, 18, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Blans, P.; Fishbein, J.C. Determinants of selectivity in alkylation of nucleosides and DNA by secondary diazonium ions: Evidence for, and consequences of, a preassociation mechanism. Chem. Res. Toxicol. 2004, 17, 1531–1539. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Villalta, P.W.; Upadhyaya, P.; Hecht, S.S. Analysis of O6-[4-(3-Pyridyl)-4-oxobut-1-yl]-2′-deoxyguanosine and other DNA adducts in rats treated with enantiomeric or racemic N’-nitrosonornicotine. Chem. Res. Toxicol. 2016, 29, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Schlicht, K.E.; Berg, J.Z.; Murphy, S.E. Effect of CYP2A13 active site mutation N297A on metabolism of coumarin and tobacco-specific nitrosamines. Drug Metab. Dispos. 2009, 37, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shen, Z.H.; Shen, J.; Liu, G.X.; Li, W.H.; Tang, Y. Computational insights into the different catalytic activities of CYP2A13 and CYP2A6 on NNK. J. Mol. Graph. Model. 2011, 30, 1–9. [Google Scholar] [CrossRef] [PubMed]
- He, X.Y. Identification of critical amino acid residues of humanCYP2A13 for the metabolic activation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco-specific carcinogen. Drug Metab. Dispos. 2004, 32, 1516–1521. [Google Scholar] [CrossRef]
- Ding, X.; Kaminsky, L.S. Human extrahepatic cytochromes P450: Function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu. Rev. Pharmacol. 2003, 43, 149–173. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | ΔG ‡ (kcal/mol) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| N1A | N3A | N7A | N3G | O6G | N7G | N3C | O2C | O2T | O4T | |
| TS | 2.42 | 0.92 | 3.85 | 0.86 | 2.35 | 4.72 | 5.03 | 1.71 | 3.82 | 2.79 |
| CYP450 Enzymes | Scoring Function | |
|---|---|---|
| (R)-NNN | (S)-NNN | |
| Human 1A1-Chain A | 55.29 | 70.67 |
| Human 2A6-Chain A | 63.88 | 73.98 |
| Human 2A13-Chain A | 58.32 | 73.55 |
| Human 2E1-Chain A | 53.79 | 62.59 |
| Human 3A4 | 59.19 | 61.56 |
| Rat 2A3 | 47.33 | 53.83 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, T.; Sun, G.; Zhao, L.; Cui, X.; Zhong, R. Metabolic Activation and Carcinogenesis of Tobacco-Specific Nitrosamine N’-Nitrosonornicotine (NNN): A Density Function Theory and Molecular Docking Study. Int. J. Environ. Res. Public Health 2019, 16, 178. https://doi.org/10.3390/ijerph16020178
Fan T, Sun G, Zhao L, Cui X, Zhong R. Metabolic Activation and Carcinogenesis of Tobacco-Specific Nitrosamine N’-Nitrosonornicotine (NNN): A Density Function Theory and Molecular Docking Study. International Journal of Environmental Research and Public Health. 2019; 16(2):178. https://doi.org/10.3390/ijerph16020178
Chicago/Turabian StyleFan, Tengjiao, Guohui Sun, Lijiao Zhao, Xin Cui, and Rugang Zhong. 2019. "Metabolic Activation and Carcinogenesis of Tobacco-Specific Nitrosamine N’-Nitrosonornicotine (NNN): A Density Function Theory and Molecular Docking Study" International Journal of Environmental Research and Public Health 16, no. 2: 178. https://doi.org/10.3390/ijerph16020178
APA StyleFan, T., Sun, G., Zhao, L., Cui, X., & Zhong, R. (2019). Metabolic Activation and Carcinogenesis of Tobacco-Specific Nitrosamine N’-Nitrosonornicotine (NNN): A Density Function Theory and Molecular Docking Study. International Journal of Environmental Research and Public Health, 16(2), 178. https://doi.org/10.3390/ijerph16020178