Single Core Genome Sequencing for Detection of both Borrelia burgdorferi Sensu Lato and Relapsing Fever Borrelia Species

Abstract
:1. Introduction
2. Materials and Methods
2.1. Search for PCR Primers for Borrelial Core Genome Amplification
2.2. Sources of Borrelial Genomic DNAs
- (1)
- Frozen pure cultures of Borrelia burgdorferi sensu stricto strain B31 (ATCC 53210) and Borrelia coriaceae (ATCC 43381) in liquid media purchased from ATCC;
- (2)
- Venous blood specimens collected from U.S. patients submitted by licensed physicians for “Lyme bacteria” tests as part of routine patient management and 25 blind-coded simulated patient blood specimens from New York State (NYS) Department of Health (DOH) for a Borrelia burgdorferi proficiency test; the patients’ informed consent was not required because it is implied that informed consent was previously given for the scope of the treatment and because the patients’ identities were not revealed; ethical approval was not required because it is considered that this was not research but clinical/laboratory practice [41].
- (3)
- Blind-coded archived serum samples derived from patients with and without “Lyme disease” from the CDC under two Material Transfer Agreements (MTA No. NCEZID-R137154-00 and NCEZID-R147284-00);
- (4)
- Engorged Ixodes scapularis ticks removed from humans living in the United States and submitted for Lyme testing as part of preventive patient care; and
- (5)
- Unfed, questing Ixodes ricinus nymphs collected from free public access areas in Ireland by “flagging” in the counties of Kerry, Waterford, Galway, Wicklow, and Donegal (John Eoin Healy). Acquiring tick samples in areas of free public access in the Republic of Ireland for the purpose of laboratory analysis does not require approval from any state authorities or governmental agencies.
2.3. DNA Extractions
2.4. PCR Primers
- M1 (forward): 5′-ACGATGCACACTTGGTGTTAA-3′ and
- M2 (reverse): 5′-TCCGACTTATCACCGGCAGTC-3′
- Primary PCR Forward Bg1 primer: 5′- GACGTTAATTTATGAATAAGC-3′
- Primary PCR Reverse Bg6 primer: 5′- TTAACACCAAGTGTGCATCGT-3′
- Heminested PCR Forward Bg5 primer: 5′- CGGGATTATTGGGCGTAAAGGGTGAG-3′
- Heminested PCR Reverse Bg6 primer: 5′- TTAACACCAAGTGTGCATCGT-3′
2.5. PCR Conditions
2.6. Sanger Sequencing
3. Results
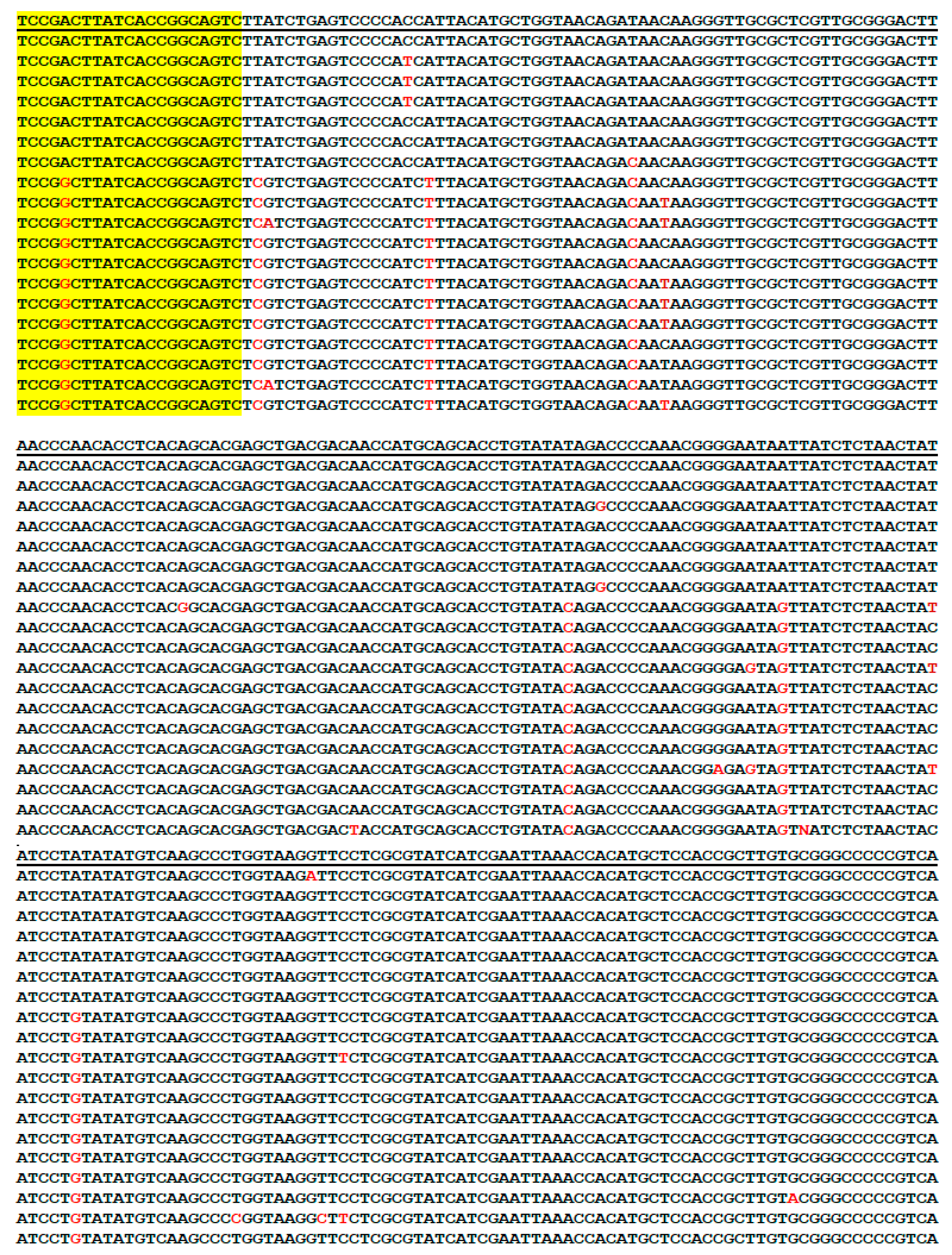
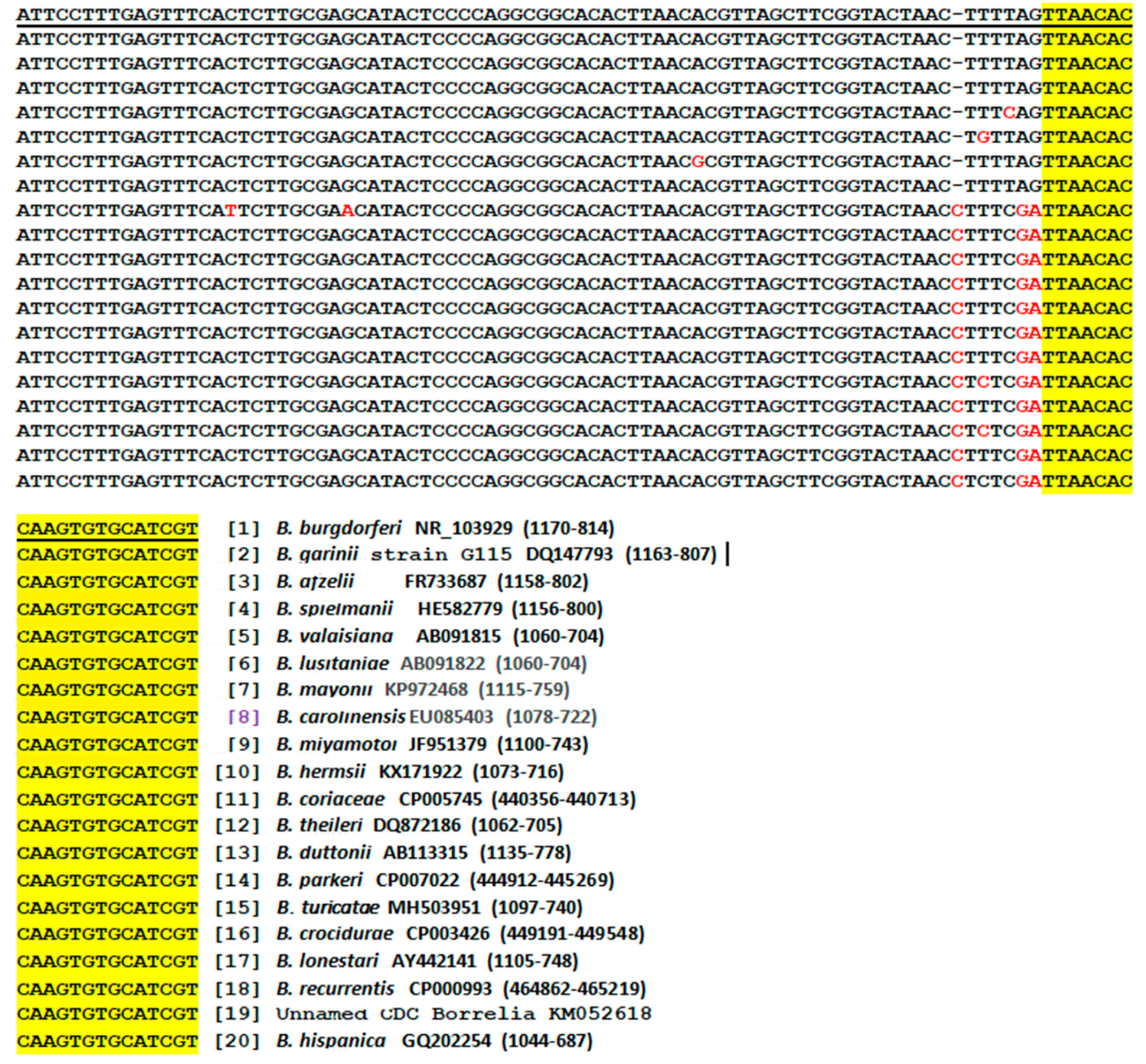
3.1. Genus-Specific PCR Primers for Metagenomic DNA Detection of Borrelia
3.2. Routine Detection of B. burgdorferi Sensu Lato
3.3. Routine Detection of B. garinii Bernie Strain
3.4. Routine Detection of Borrelia valaisiana
3.5. Routine Detection of B. miyamotoi
3.6. Routine Detection of Mixed Infection by B. burgdorferi and B. miyamotoi
3.7. Routine Detection of Other Relapsing Fever Borreliae
3.8. Supplementary Heminested PCR/Sequencing for Further Borrelial Speciation
3.9. Bg5/Bg6 Primer Sequencing for Distinction between B. burgdorferi and B. garinii
3.10. Potential Pitfalls in Metagenomic DNA Diagnosis of Lyme Borreliosis
3.11. Selection of the Borrelial 16S rRNA Gene Target Region for Routine Detection
4. Discussion
4.1. The Need for a Reliable Test for all Pathogenic Borreliae in Blood Samples
4.2. Sequencing of one PCR Amplicon for Detection of all Pathogenic Borreliae
4.3. Participation of Hospital Laboratories in Endemic Areas for Timely Diagnosis of Spirochetemia
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Steere, A.C.; Strle, F.; Wormser, G.P.; Hu, L.T.; Branda, J.A.; Hovius, J.W.; Li, X.; Mead, P.S. Lyme borreliosis. Nat. Rev. Dis. Primers 2016, 2, 16090. [Google Scholar] [CrossRef] [PubMed]
- Rudenko, N.; Golovchenko, M.; Grubhoffer, L.; Oliver, J.H., Jr. Updates on Borrelia burgdorferi sensu lato complex with respect to public health. Ticks Tick Borne Dis. 2011, 2, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Bunikis, J.; Tsao, J.; Garpmo, U.; Berglund, J.; Fish, D.; Barbour, A.G. Typing of Borrelia relapsing fever group strains. Emerg. Infect. Dis. 2004, 10, 1661–1664. [Google Scholar] [CrossRef] [PubMed]
- Rijpkema, S.G.; Tazelaar, D.J.; Molkenboer, M.J.; Noordhoek, G.T.; Plantinga, G.; Schouls, L.M.; Schellekens, J.F. Detection of Borrelia afzelii, Borrelia burgdorferi sensu stricto, Borrelia garinii and group VS116 by PCR in skin biopsies of patients with erythema migrans and acrodermatitis chronica atrophicans. Clin. Microbiol. Infect. 1997, 3, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Crowder, C.D.; Carolan, H.E.; Rounds, M.A.; Honig, V.; Mothes, B.; Haag, H.; Nolte, O.; Luft, B.J.; Grubhoffer, L.; Ecker, D.J.; et al. Prevalence of Borrelia miyamotoi in Ixodes ticks in Europe and the United States. Emerg. Infect. Dis. 2014, 20, 1678–1682. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Wilcox, S.; Mankoff, J.; Stricker, R.B. Severity of chronic Lyme disease compared to other chronic conditions: A quality of life survey. PeerJ 2014, 2, e322. [Google Scholar] [CrossRef]
- Feder, H.M., Jr.; Johnson, B.J.; O’Connell, S.; Shapiro, E.D.; Steere, A.C.; Wormser, G.P.; Ad Hoc International Lyme Disease Group; Agger, W.A.; Artsob, H.; Auwaerter, P.; et al. A critical appraisal of “chronic Lyme disease”. N. Engl. J. Med. 2007, 357, 1422–1430. [Google Scholar] [CrossRef]
- Magnarelli, L.A.; Anderson, J.F.; Johnson, R.C.; Nadelman, R.B.; Wormser, G.P. Comparison of different strains of Borrelia burgdorferi sensu lato used as antigens in enzyme-linked immunosorbent assays. J. Clin. Microbiol. 1994, 32, 1154–1158. [Google Scholar]
- Cutler, S.J.; Wright, D.J. Predictive value of serology in diagnosing Lyme borreliosis. J. Clin. Pathol. 1994, 47, 344–349. [Google Scholar] [CrossRef]
- Hinckley, A.F.; Connally, N.P.; Meek, J.I.; Johnson, B.J.; Kemperman, M.M.; Feldman, K.A.; White, J.L.; Mead, P.S. Lyme disease testing by large commercial laboratories in the United States. Clin. Infect. Dis. 2014, 59, 676–681. [Google Scholar] [CrossRef]
- CDC. Tickborne Diseases of the United States. Borrelia miyamotoi Disease. Available online: https://www.cdc.gov/ticks/tickbornediseases/borrelia-miyamotoi.html (accessed on 26 January 2019).
- Platonov, A.E.; Karan, L.S.; Kolyasnikova, N.M.; Makhneva, N.A.; Toporkova, M.G.; Maleev, V.V.; Fish, D.; Krause, P.J. Humans infected with relapsing fever spirochete Borrelia miyamotoi, Russia. Emerg. Infect. Dis. 2011, 17, 1816–1823. [Google Scholar] [CrossRef]
- CDC. Tickborne Diseases of the United States. Tickborne Relapsing Fever. Available online: https://www.cdc.gov/ticks/tickbornediseases/tbrf.html (accessed on 26 January 2019).
- Lee, S.H.; Vigliotti, J.S.; Vigliotti, V.S.; Jones, W.; Shearer, D.M. Detection of borreliae in archived sera from patients with clinically suspect Lyme disease. Int. J. Mol. Sci. 2014, 15, 4284–4298. [Google Scholar] [CrossRef]
- CDC. Southern Tick–Associated Rash Illness. Available online: https://www.cdc.gov/stari/symptoms/index.html (accessed on 26 January 2019).
- Barbour, A.G.; Restrepo, B.I. Antigenic variation in vector-borne pathogens. Emerg. Infect. Dis. 2000, 6, 449–457. [Google Scholar] [CrossRef]
- Theisen, M.; Frederiksen, B.; Lebech, A.M.; Vuust, J.; Hansen, K. Polymorphism in ospC gene of Borrelia burgdorferi and immunoreactivity of OspC protein: Implications for taxonomy and for use of OspC protein as a diagnostic antigen. J. Clin. Microbiol. 1993, 31, 2570–2576. [Google Scholar]
- Wang, I.N.; Dykhuizen, D.E.; Qiu, W.; Dunn, J.J.; Bosler, E.M.; Luft, B.J. Genetic diversity of ospC in a local population of Borrelia burgdorferi sensu stricto. Genetics 1999, 151, 15–30. [Google Scholar]
- Kannangara, D.W.; Patel, P. Report of Non-Lyme, Erythema Migrans Rashes from New Jersey with a Review of Possible Role of Tick Salivary Toxins. Vector Borne Zoonotic Dis. 2018, 18, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Grogg, K.L.; Edwards, W.D.; Wright, A.J.; Schwenk, N.M. Death from inappropriate therapy for Lyme disease. Clin. Infect. Dis. 2000, 31, 1107–1109. [Google Scholar] [CrossRef] [PubMed]
- Molloy, P.J.; Persing, D.H.; Berardi, V.P. False-positive results of PCR testing for Lyme disease [letter]. Clin. Infect. Dis. 2001, 33, 412–413. [Google Scholar] [CrossRef]
- Schutzer, S.E.; Body, B.A.; Boyle, J.; Branson, B.M.; Dattwyler, R.J.; Fikrig, E.; Gerald, N.J.; Gomes-Solecki, M.; Kintrup, M.; Ledizet, M.; et al. Direct Diagnostic Tests for Lyme Disease. Clin. Infect. Dis. 2019, 68, 1052–1057. [Google Scholar] [CrossRef] [PubMed]
- Matthijs, G.; Souche, E.; Alders, M.; Corveleyn, A.; Eck, S.; Feenstra, I.; Race, V.; Sistermans, E.; Sturm, M.; Weiss, M.; et al. Guidelines for diagnostic next-generation sequencing. Eur. J. Hum. Genet. 2016, 24, 2–5. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov Identifier: NCT03505879. Next Generation Sequencing Detection of Lyme Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT03505879 (accessed on 26 January 2019).
- Segerman, B. The genetic integrity of bacterial species: The core genome and the accessory genome, two different stories. Front Cell Infect. Microbiol. 2012, 2, 116. [Google Scholar] [CrossRef]
- Alcaraz, L.D.; Moreno-Hagelsieb, G.; Eguiarte, L.E.; Souza, V.; Herrera-Estrella, L.; Olmedo, G. Understanding the evolutionary relationships and major traits of Bacillus through comparative genomics. BMC Genom. 2010, 11, 332. [Google Scholar] [CrossRef]
- Barajas, H.R.; Romero, M.F.; Martínez-Sánchez, S.; Alcaraz, L.D. Global genomic similarity and core genome sequence diversity of the Streptococcus genus as a toolkit to identify closely related bacterial species in complex environments. PeerJ 2019, 6, e6233. [Google Scholar] [CrossRef]
- Segata, N.; Huttenhower, C. Toward an efficient method of identifying core genes for evolutionary and functional microbial phylogenies. PLoS ONE 2011, 6, e24704. [Google Scholar] [CrossRef]
- Diatta, G.; Souidi, Y.; Granjon, L.; Arnathau, C.; Durand, P.; Chauvancy, G.; Mané, Y.; Sarih, M.; Belghyti, D.; Renaud, F.; et al. Epidemiology of tick-borne borreliosis in Morocco. PLoS Negl. Trop. Dis. 2012, 6, e1810. [Google Scholar] [CrossRef]
- Deurenberg, R.H.; Bathoorn, E.; Chlebowicz, M.A.; Couto, N.; Ferdous, M.; García-Cobos, S.; Kooistra-Smid, A.M.; Raangs, E.C.; Rosema, S.; Veloo, A.C.; et al. Application of next generation sequencing in clinical microbiology and infection prevention. J. Biotechnol. 2017, 243, 16–24. [Google Scholar] [CrossRef]
- Tringe, S.G.; Rubin, E.M. Metagenomics: DNA sequencing of environmental samples. Nat. Rev. Genet. 2005, 6, 805–814. [Google Scholar] [CrossRef] [Green Version]
- Greninger, A.L. The challenge of diagnostic metagenomics. Expert Rev. Mol. Diagn. 2018, 18, 605–615. [Google Scholar] [CrossRef]
- Janda, J.M.; Abbott, S.L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: Pluses, perils, and pitfalls. J. Clin. Microbiol. 2007, 45, 2761–2764. [Google Scholar] [CrossRef]
- Hakovirta, J.R.; Prezioso, S.; Hodge, D.; Pillai, S.P.; Weigel, L.M. Identification and Analysis of Informative Single Nucleotide Polymorphisms in 16S rRNA Gene Sequences of the Bacillus cereus Group. J. Clin. Microbiol. 2016, 54, 2749–2756. [Google Scholar] [CrossRef] [Green Version]
- Peacock, S. Health care: Bring microbial sequencing to hospitals. Nature 2014, 509, 557–559. [Google Scholar] [CrossRef]
- Feria-Arroyo, T.P.; Castro-Arellano, I.; Gordillo-Perez, G.; Cavazos, A.L.; Vargas-Sandoval, M.; Grover, A.; Torres, J.; Medina, R.F.; de León, A.A.; Esteve-Gassent, M.D. Implications of climate change on the distribution of the tick vector Ixodes scapularis and risk for Lyme disease in the Texas-Mexico transboundary region. Parasit Vectors 2014, 7, 199. [Google Scholar] [CrossRef]
- Norris, S.J.; Barbour, A.G.; Fish, D.; Diuk-Wasser, M.A. Response to Esteve-Gassent et al.: flaB sequences obtained from Texas PCR products are identical to the positive control strain Borrelia burgdorferi B31. Parasit Vectors 2015, 8, 310. [Google Scholar] [CrossRef]
- NCBI. Pubmed Nucleotide. Available online: https://www.ncbi.nlm.nih.gov/nucleotide (accessed on 26 January 2019).
- Dieffenbach, C.W.; Lowe, T.M.; Dveksler, G.S. General concepts for PCR primer design. PCR Methods Appl. 1993, 3, S30–S37. [Google Scholar] [CrossRef]
- Chakravorty, S.; Helb, D.; Burday, M.; Connell, N.; Alland, D. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 2007, 69, 330–339. [Google Scholar] [CrossRef]
- Borovecki, A.; Mlinaric, A.; Horvat, M.; Supak Smolcic, V. Informed consent and ethics committee approval in laboratory medicine. Biochem. Med. 2018, 28, 373–382. [Google Scholar] [CrossRef]
- Benson, D.A.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2000, 28, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Vigliotti, J.S.; Vigliotti, V.S.; Jones, W.; Moorcroft, T.A.; Lantsman, K. DNA Sequencing Diagnosis of Off-Season Spirochetemia with Low Bacterial Density in Borrelia burgdorferi and Borrelia miyamotoi Infections. Int. J. Mol. Sci. 2014, 15, 11364–11386. [Google Scholar] [CrossRef]
- Wormser, G.P.; Bittker, S.; Cooper, D.; Nowakowski, J.; Nadelman, R.B.; Pavia, C. Yield of large-volume blood cultures in patients with early Lyme disease. J. Infect. Dis. 2001, 184, 1070–1072. [Google Scholar] [CrossRef]
- Schwartz, J.J.; Gazumyan, A.; Schwartz, I. rRNA gene organization in the Lyme disease spirochete, Borrelia burgdorferi. J. Bacteriol. 1992, 174, 3757–3765. [Google Scholar] [CrossRef]
- Lee, S.H.; Vigliotti, V.S.; Vigliotti, J.S.; Jones, W.; Pappu, S. Increased sensitivity and specificity of Borrelia burgdorferi 16S ribosomal DNA detection. Am. J. Clin. Pathol. 2010, 133, 569–576. [Google Scholar] [CrossRef]
- Rintala, A.; Pietilä, S.; Munukka, E.; Eerola, E.; Pursiheimo, J.P.; Laiho, A.; Pekkala, S.; Huovinen, P. Gut Microbiota Analysis Results Are Highly Dependent on the 16S rRNA Gene Target Region, Whereas the Impact of DNA Extraction Is Minor. J. Biomol. Tech. 2017, 28, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Steere, A.C.; Malawista, S.E.; Snydman, D.R.; Shope, R.E.; Andiman, W.A.; Ross, M.R.; Steele, F.M. Lyme arthritis: An epidemic of oligoarticular arthritis in children and adults in three connecticut communities. Arthritis Rheum. 1977, 20, 7–17. [Google Scholar] [CrossRef]
- Burgdorfer, W.; Barbour, A.G.; Hayes, S.F.; Benach, J.L.; Grunwaldt, E.; Davis, J.P. Lyme disease-a tick-borne spirochetosis? Science 1982, 216, 1317–1319. [Google Scholar] [CrossRef] [Green Version]
- Barbour, A.G.; Bunikis, J.; Travinsky, B.; Hoen, A.G.; Diuk-Wasser, M.A.; Fish, D.; Tsao, J.I. Niche partitioning of Borrelia burgdorferi and Borrelia miyamotoi in the same tick vector and mammalian reservoir species. Am. J. Trop Med. Hyg. 2009, 81, 1120–1131. [Google Scholar] [CrossRef]
- Liveris, D.; Varde, S.; Iyer, R.; Koenig, S.; Bittker, S.; Cooper, D.; McKenna, D.; Nowakowski, J.; Nadelman, R.B.; Wormser, G.P.; et al. Genetic diversity of Borrelia burgdorferi in lyme disease patients as determined by culture versus direct PCR with clinical specimens. J. Clin. Microbiol. 1999, 37, 565–569. [Google Scholar]
- Nowrouzian, F.L.; Adlerberth, I.; Wold, A.E. High frequency of false-positive signals in a real-time PCR-based “Plus/Minus” assay. Apmis 2009, 117, 68–72. [Google Scholar] [CrossRef]
- Ruiz-Villalba, A.; van Pelt-Verkuil, E.; Gunst, Q.D.; Ruijter, J.M.; van den Hoff, M.J. Amplification of nonspecific products in quantitative polymerase chain reactions (qPCR). Biomol. Detect. Quantif. 2017, 14, 7–18. [Google Scholar] [CrossRef]
- Liveris, D.; Schwartz, I.; McKenna, D.; Nowakowski, J.; Nadelman, R.; Demarco, J.; Iyer, R.; Bittker, S.; Cooper, D.; Holmgren, D.; et al. Comparison of five diagnostic modalities for direct detection of Borrelia burgdorferi in patients with early Lyme disease. Diagn. Microbiol. Infect. Dis. 2012, 73, 243–245. [Google Scholar] [CrossRef]
- Liveris, D.; Schwartz, I.; McKenna, D.; Nowakowski, J.; Nadelman, R.B.; DeMarco, J.; Iyer, R.; Cox, M.E.; Holmgren, D.; Wormser, G.P. Quantitation of cell-associated borrelial DNA in the blood of Lyme disease patients with erythema migrans. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 791–795. [Google Scholar] [CrossRef]
- Nowakowski, J.; Schwartz, I.; Liveris, D.; Wang, G.; Aguero-Rosenfeld, M.E.; Girao, G.; McKenna, D.; Nadelman, R.B.; Cavaliere, L.F.; Wormser, G.P.; Lyme Disease Study Group. Laboratory diagnostic techniques for patients with early Lyme disease associated with erythema migrans: A comparison of different techniques. Clin. Infect. Dis. 2001, 33, 2023–2027. [Google Scholar] [CrossRef]
- Corinaldesi, C.; Danovaro, R.; Dell’Anno, A. Simultaneous recovery of extracellular and intracellular DNA suitable for molecular studies from marine sediments. Appl. Env. Microbiol. 2005, 71, 46–50. [Google Scholar] [CrossRef]
- Persing, D.H.; Rutledge, B.J.; Rys, P.N.; Podzorski, D.S.; Mitchell, P.D.; Reed, K.D.; Liu, B.; Fikrig, E.; Malawista, S.E. Target imbalance: Disparity of Borrelia burgdorferi genetic material in synovial fluid from Lyme arthritis patients. J. Infect. Dis. 1994, 169, 668–672. [Google Scholar] [CrossRef]
- Jenkins, C.; Ling, C.L.; Ciesielczuk, H.L.; Lockwood, J.; Hopkins, S.; McHugh, T.D.; Gillespie, S.H.; Kibbler, C.C. Detection and identification of bacteria in clinical samples by 16S rRNA gene sequencing: Comparison of two different approaches in clinical practice. J. Med. Microbiol. 2012, 61, 483–488. [Google Scholar] [CrossRef]
- Aguero-Rosenfeld, M.E.; Wang, G.; Schwartz, I.; Wormser, G.P. Diagnosis of lyme borreliosis. Clin. Microbiol. Rev. 2005, 18, 484–509. [Google Scholar] [CrossRef]
- Cogswell, F.B.; Bantar, C.E.; Hughes, T.G.; Gu, Y.; Philipp, M.T. Host DNA can interfere with detection of Borrelia burgdorferi in skin biopsy specimens by PCR. J. Clin. Microbiol. 1996, 34, 980–982. [Google Scholar] [PubMed]
- Kwok, S.; Chang, S.Y.; Sninsky, J.J.; Wang, A. A guide to the design and use of mismatched and degenerate primers. PCR Methods Appl. 1994, 3, S39–S47. [Google Scholar] [CrossRef] [PubMed]
- Ryu, K.H.; Choi, S.H.; Lee, J.S. Restriction primers as short as 6-mers for PCR amplification of bacterial and plant genomic DNA and plant viral RNA. Mol. Biotechnol. 2000, 14, 1–3. [Google Scholar] [CrossRef]
- Kalle, E.; Kubista, M.; Rensing, C. Multi-template polymerase chain reaction. Biomol. Detect. Quantif. 2014, 2, 11–29. [Google Scholar] [CrossRef] [Green Version]
- Hong, G.; Lee, S.H.; Ge, S.; Zhou, S. A Novel Low Temperature PCR Assured High-Fidelity DNA Amplification. Int. J. Mol. Sci. 2013, 14, 12853–12862. [Google Scholar] [CrossRef] [Green Version]
- Maganga, G.D.; Kapetshi, J.; Berthet, N.; Kebela Ilunga, B.; Kabange, F.; Mbala Kingebeni, P.; Mondonge, V.; Muyembe, J.J.; Bertherat, E.; Briand, S.; et al. Ebola virus disease in the Democratic Republic of Congo. N. Engl. J. Med. 2014, 371, 2083–2091. [Google Scholar] [CrossRef] [PubMed]
- Cabral, G.B.; Ferreira, J.L.P.; Souza, R.P.; Cunha, M.S.; Luchs, A.; Figueiredo, C.A.; Brígido, L.F.M. Simple protocol for population (Sanger) sequencing for Zika virus genomic regions. Mem. Inst. Oswaldo Cruz 2018, 113, 38–44. [Google Scholar] [CrossRef]
- CDC. Ebola Virus Disease Case Definition for Reporting in EU. Available online: https://ecdc.europa.eu/en/ebola-virus-disease-case-definition-reporting-eu (accessed on 26 January 2019).
- CDC. Lyme Disease (Borrelia burgdorferi) 2017 Case Definition. Laboratory Criteria for Diagnosis. Available online: https://wwwn.cdc.gov/nndss/conditions/lyme-disease/case-definition/2017/ (accessed on 26 January 2019).
- CDC. Typhoid & Paratyphoid Fever. Available online: https://wwwnc.cdc.gov/travel/yellowbook/2018/infectious-diseases-related-to-travel/typhoid-paratyphoid-fever (accessed on 26 January 2019).
- Gentilini, M.; Bricaire, F. Chronic Lyme disease: A scam that should be condemned! Available online: https://doi.org/10.1016/j.medmal.2019.01.001 (accessed on 26 January 2019).
- Larsson, C.; Bergström, S. A novel and simple method for laboratory diagnosis of relapsing fever borreliosis. Open Microbiol. J. 2008, 2, 10–12. [Google Scholar] [PubMed]
- Dorward, D.W.; Fischer, E.R.; Brooks, D.M. Invasion and cytopathic killing of human lymphocytes by spirochetes causing Lyme disease. Clin. Infect. Dis. 1997, 25 (Suppl. 1), S2–S8. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Vigliotti, V.S.; Vigliotti, J.S.; Jones, W.; Williams, J.; Walshon, J. Early Lyme disease with spirochetemia—Diagnosed by DNA sequencing. BMC Res. Notes 2010, 3, 273. [Google Scholar] [CrossRef]
- Karan, L.; Makenov, M.; Kolyasnikova, N.; Stukolova, O.; Toporkova, M.; Olenkova, O. Dynamics of Spirochetemia and Early PCR Detection of Borrelia miyamotoi. Emerg. Infect. Dis. 2018, 24, 860–867. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
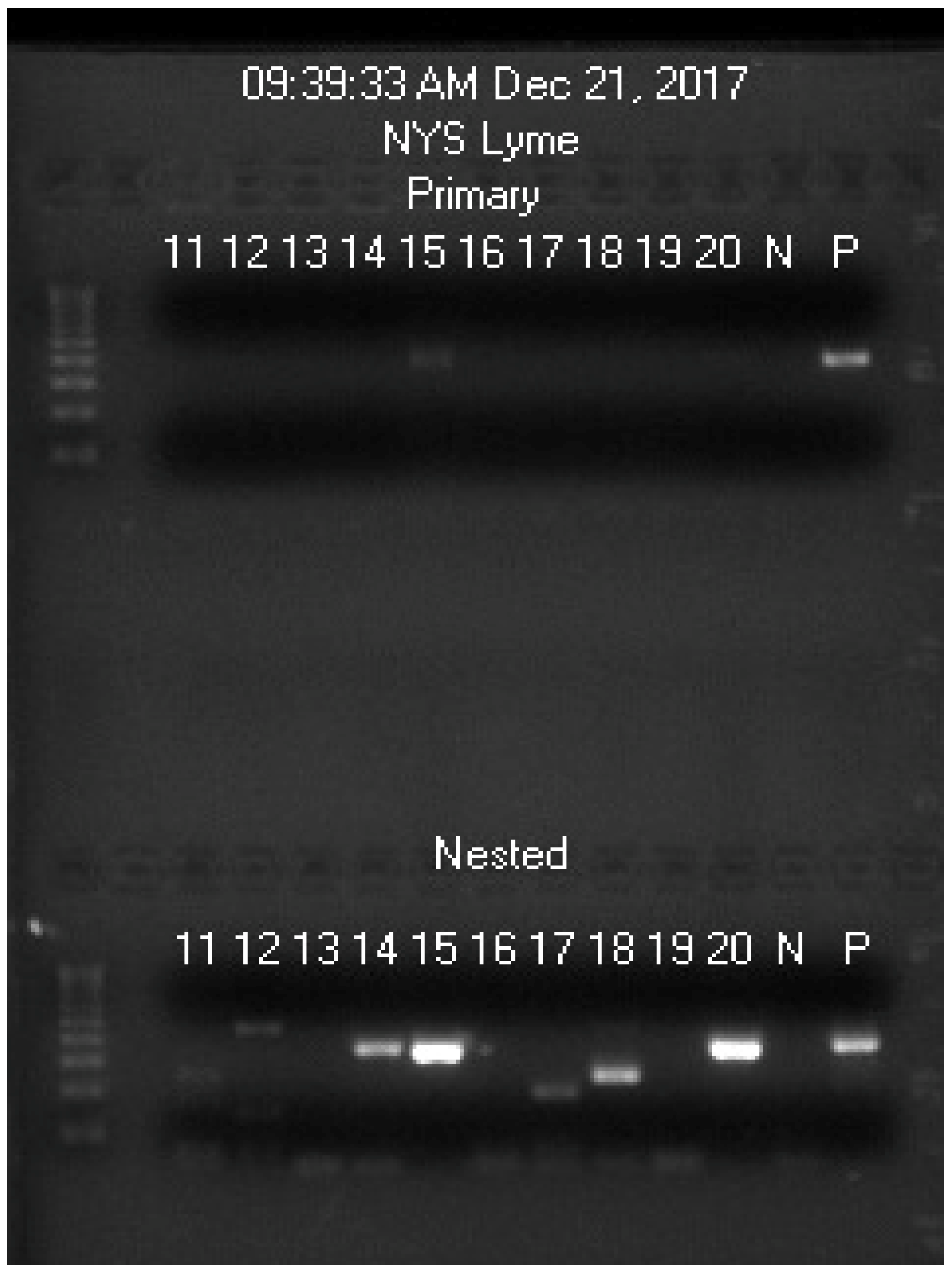
| Blood Sample Code No. | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 |
|---|---|---|---|---|---|---|---|---|---|---|
| Spiked with Bb culture | N | N | N | N | Bb | N | N | N | N | Bb |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.H.; Healy, J.E.; Lambert, J.S. Single Core Genome Sequencing for Detection of both Borrelia burgdorferi Sensu Lato and Relapsing Fever Borrelia Species. Int. J. Environ. Res. Public Health 2019, 16, 1779. https://doi.org/10.3390/ijerph16101779
Lee SH, Healy JE, Lambert JS. Single Core Genome Sequencing for Detection of both Borrelia burgdorferi Sensu Lato and Relapsing Fever Borrelia Species. International Journal of Environmental Research and Public Health. 2019; 16(10):1779. https://doi.org/10.3390/ijerph16101779
Chicago/Turabian StyleLee, Sin Hang, John Eoin Healy, and John S Lambert. 2019. "Single Core Genome Sequencing for Detection of both Borrelia burgdorferi Sensu Lato and Relapsing Fever Borrelia Species" International Journal of Environmental Research and Public Health 16, no. 10: 1779. https://doi.org/10.3390/ijerph16101779