
Experimental Data Extraction and in Silico Prediction of the Estrogenic Activity of Renewable Replacements for Bisphenol A

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
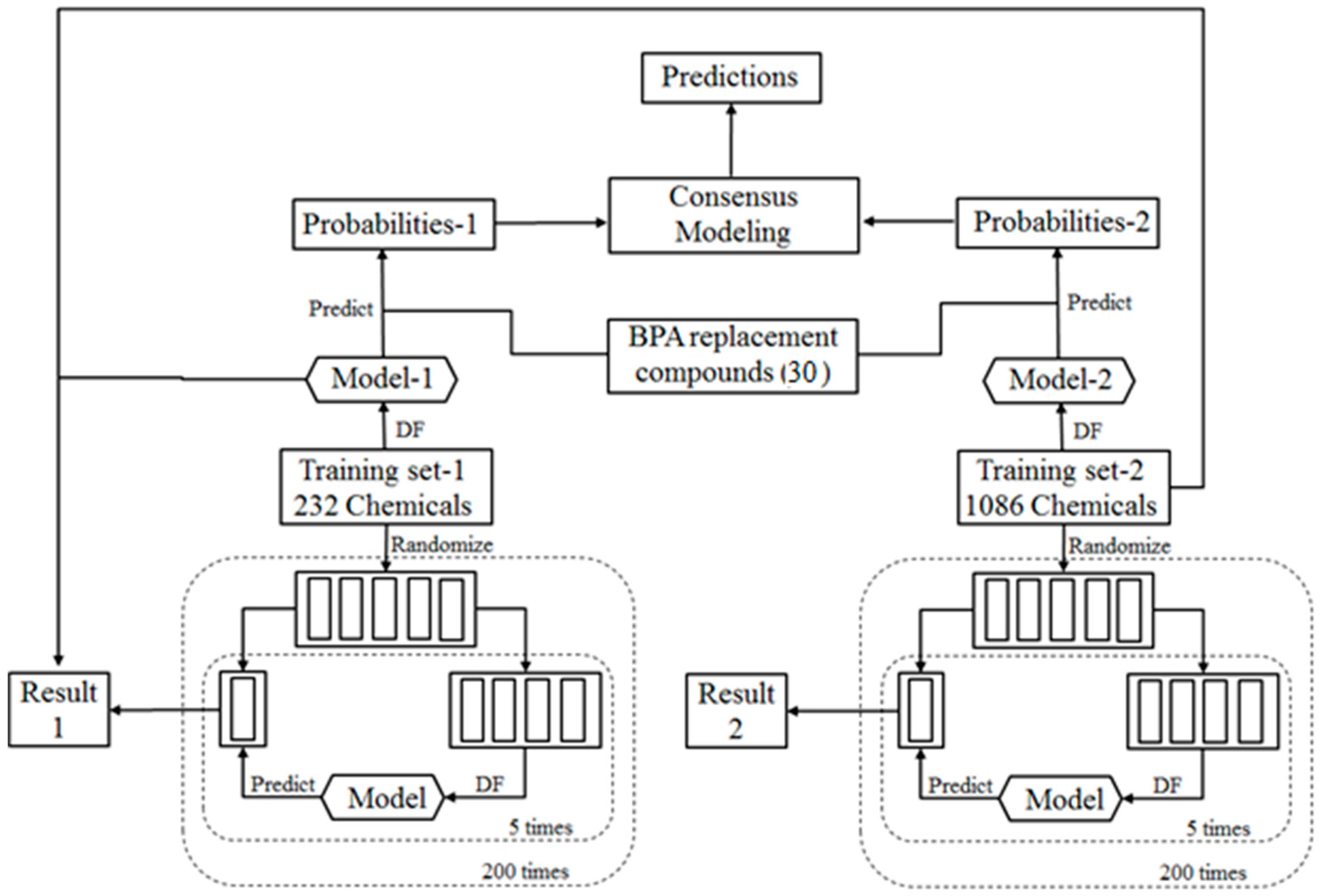
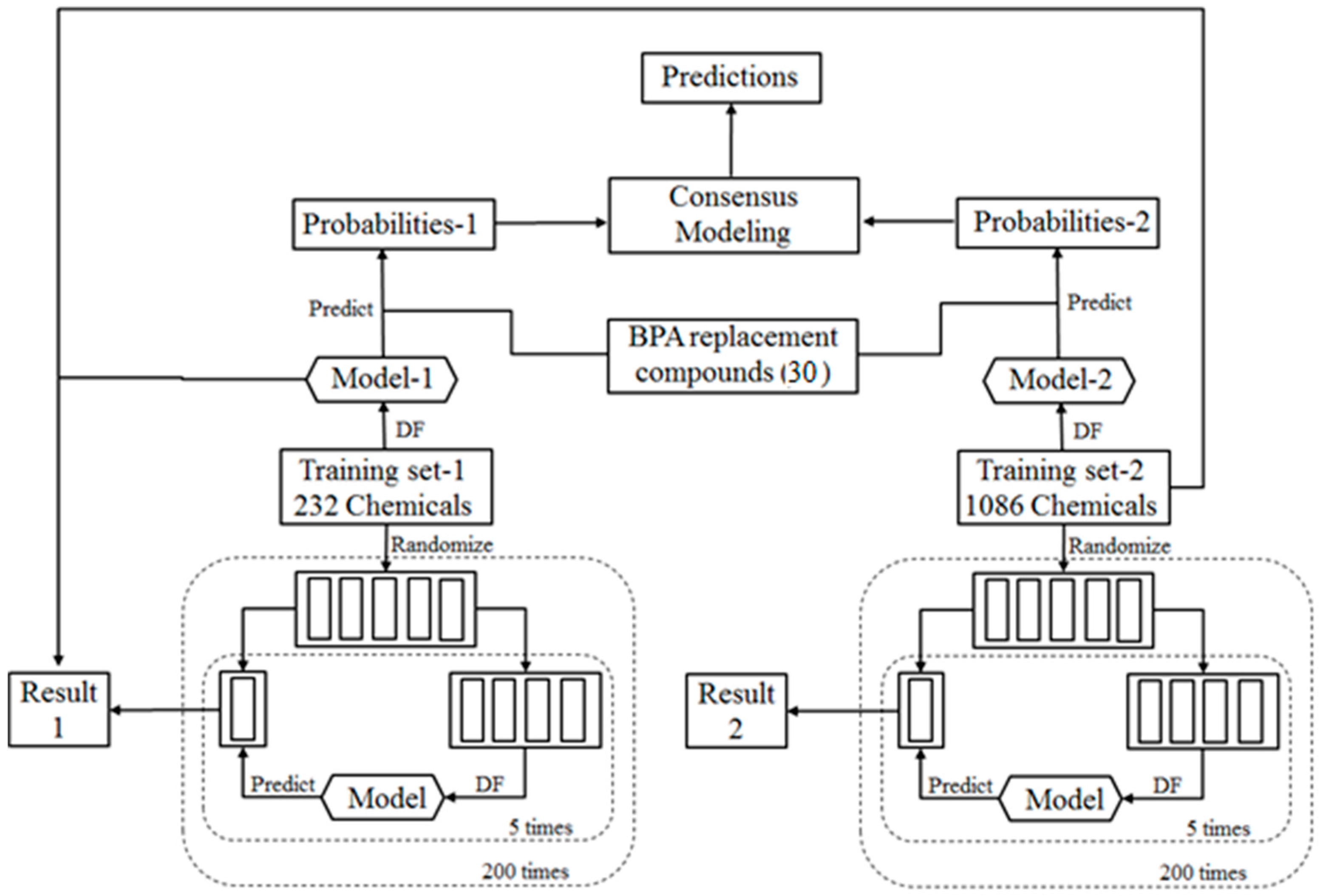
2.1. Study Design
2.2. Training Data Sets
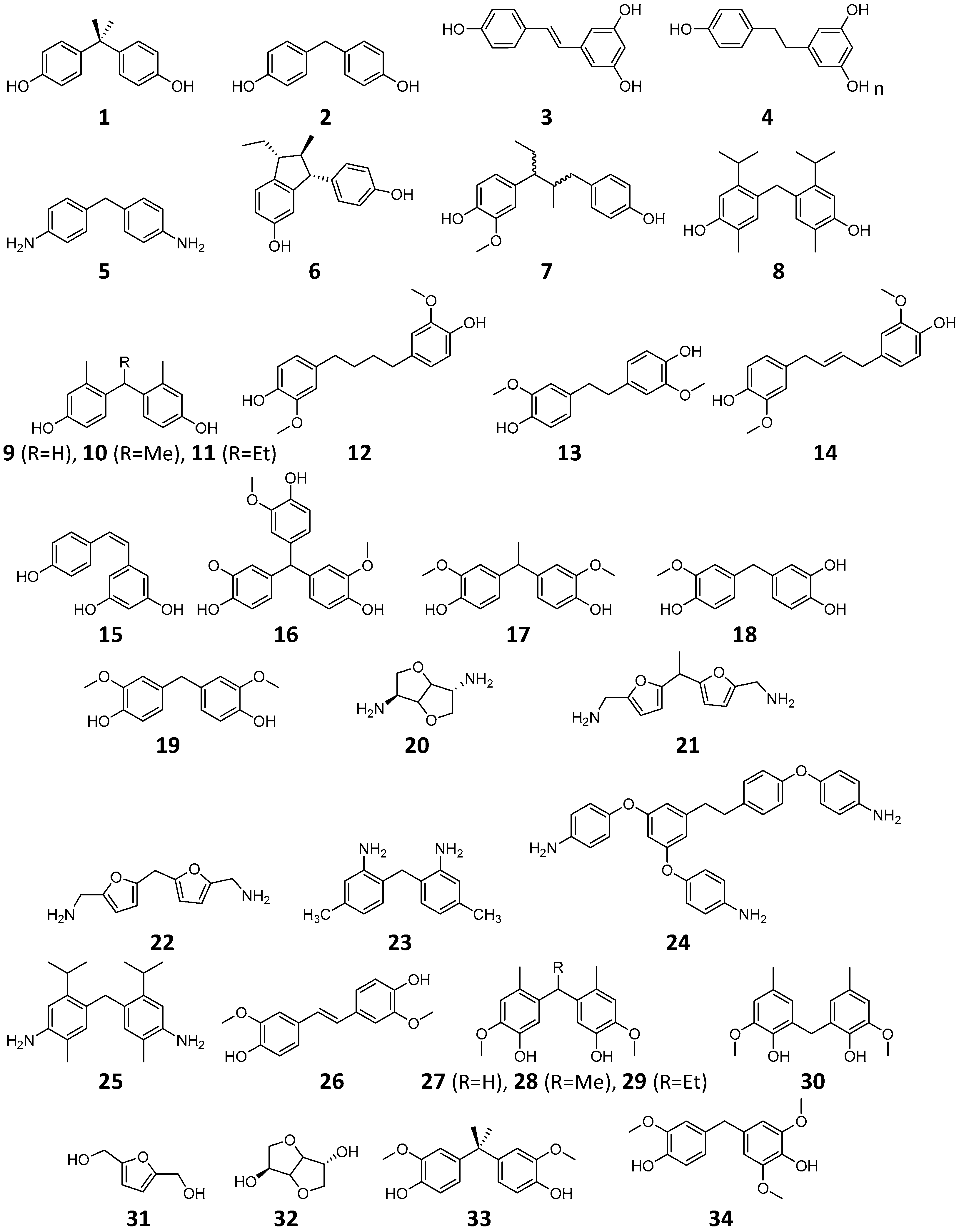
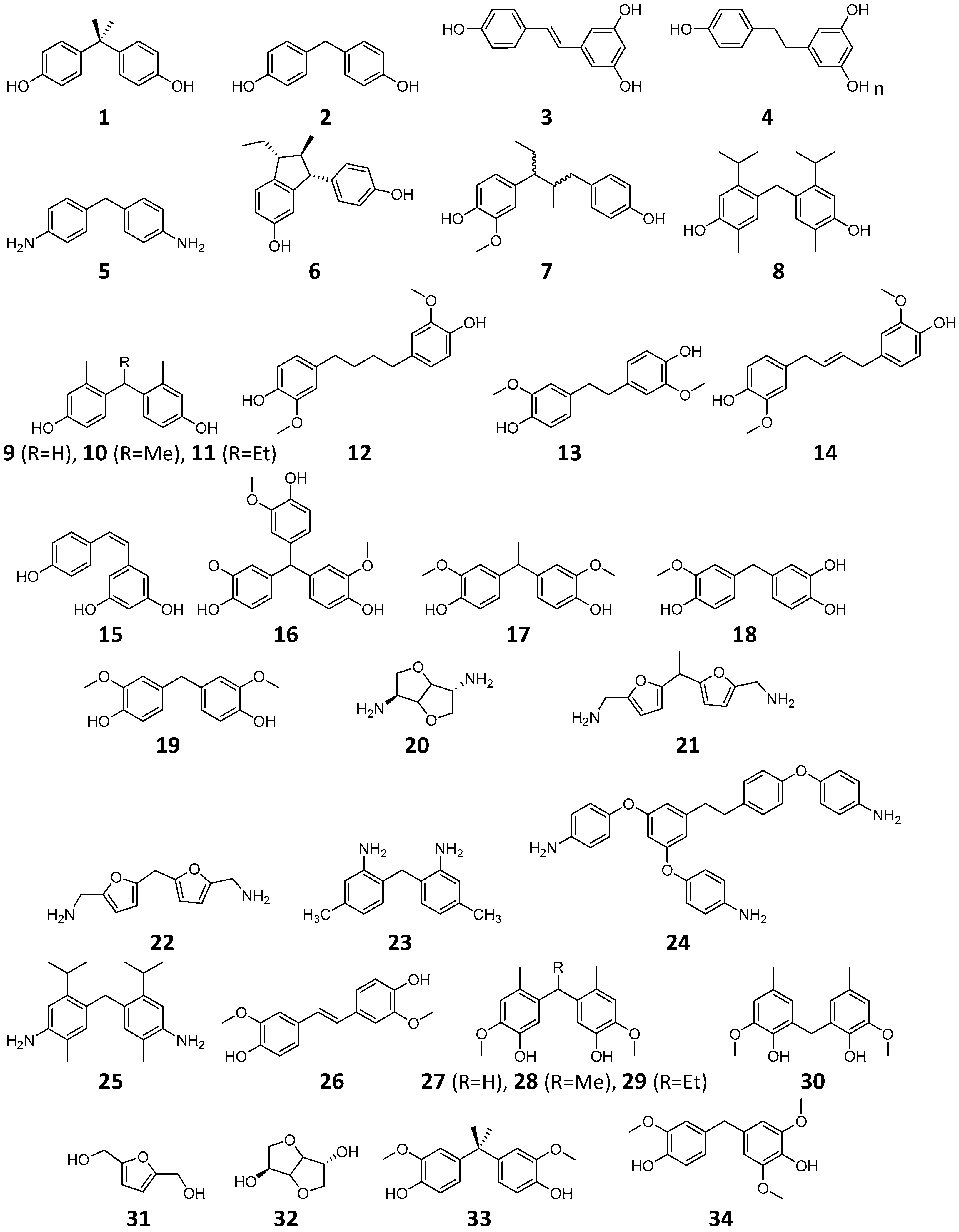
2.3. BPA Replacement Compounds
2.4. Molecular Descriptors
2.5. Individual Models
2.6. Prediction Performance
2.7. Cross Validations
2.8. External Validations
2.9. Consensus Modeling
2.10. Prediction Confidence
2.11. Quantitative Prediction
3. Results
3.1. Experimental Estrogenic Data
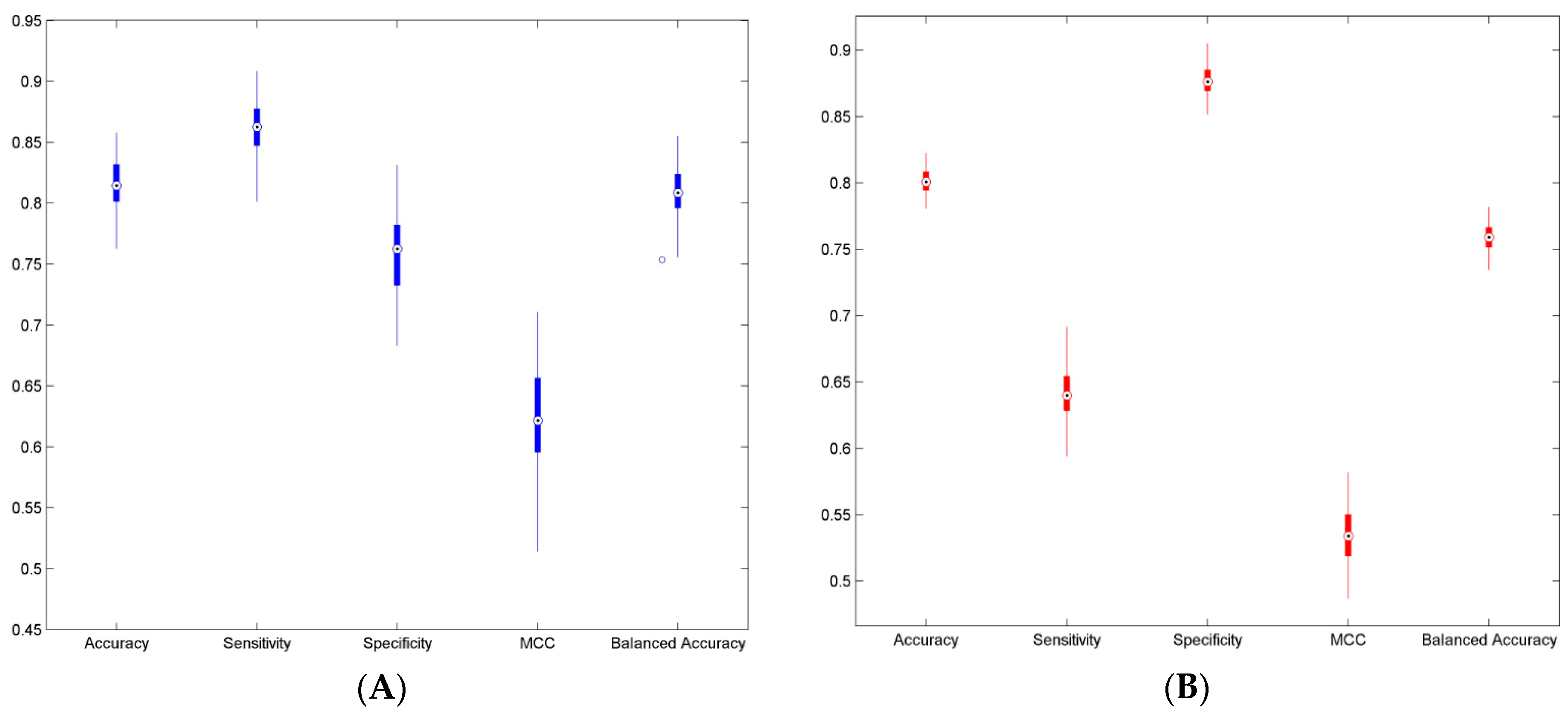
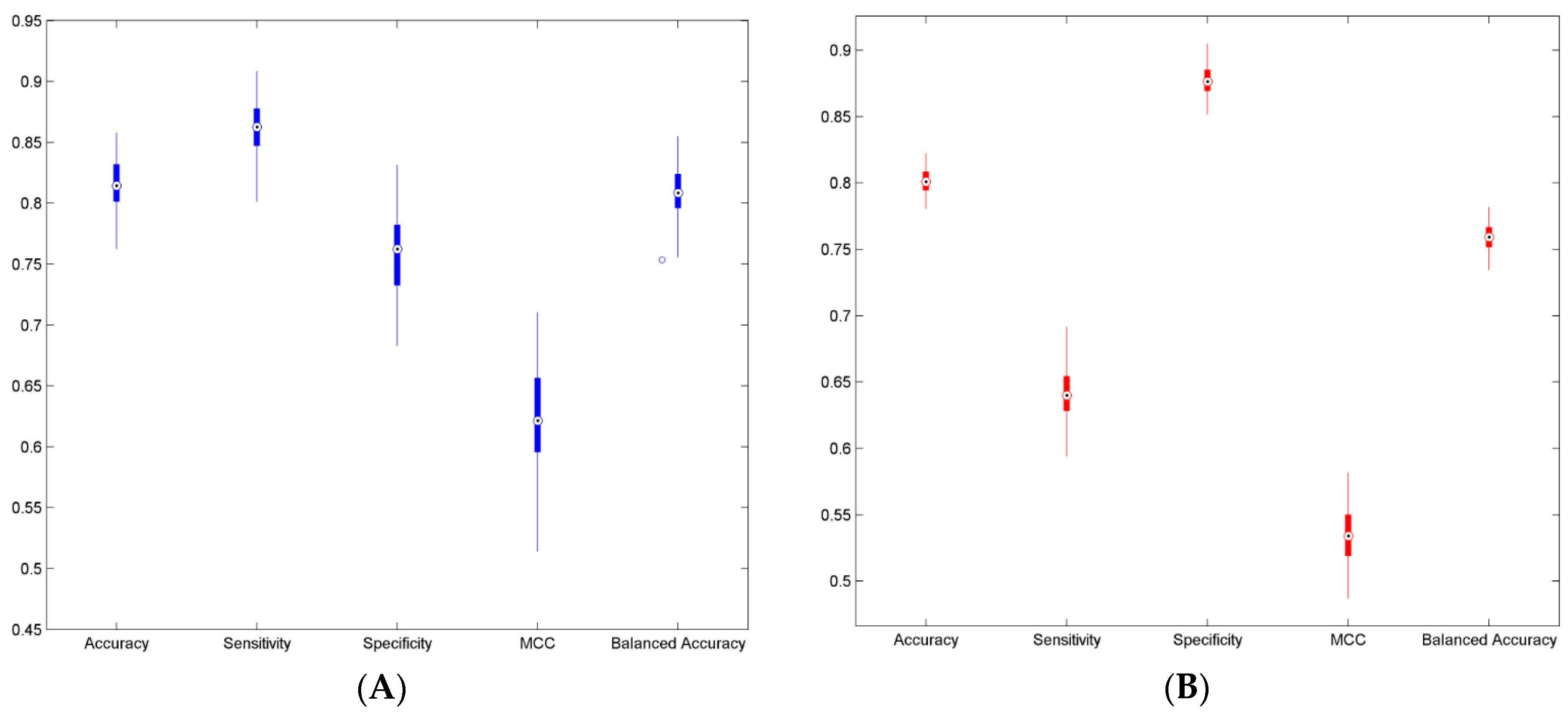
3.2. Cross Validations
3.3. External Validations
3.4. Qualitative Predictions
3.5. Quantitative Predictions
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pilato, L. Phenolic Resins: A Century of Progress; Springer: New York, NY, USA, 2010; p. 545. [Google Scholar]
- Bisphenol-A—A Global Market Overview. Available online: http://industry-experts.com/verticals/chemicals-and-materials/bisphenol-a-a-global-market-overview (accessed on 1 July 2016).
- Burridge, E. Bisphenol A: Product Profile; European Chemical News; IPC Industrial Press: London, UK, 2003; pp. 14–20. [Google Scholar]
- Kubwabo, C.; Kosarac, I.; Stewart, B.; Gauthier, B.R.; Lalonde, K.; Lalonde, P.J. Migration of Bisphenol A from plastic baby bottles, baby bottle liners and reusable polycarbonate drinking bottles. Food Addit. Contam. A 2009, 26, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Loganathan, S.N.; Kannan, K. Occurrence of Bisphenol A in indoor dust from two locations in the Eastern United States and implications for human exposures. Arch. Environ. Contam. Toxicol. 2011, 61, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Vom Saal, F.S.; Akingbemi, B.T.; Belcher, S.M.; Birnbaum, L.S.; Crain, D.A.; Eriksen, M.; Farabollini, F.; Guillette, L.J.; Hauser, R.; Heindel, J.J.; et al. Chapel hill Bisphenol A expert panel consensus statement: Integration of mechanisms, effects in animals and potential to impact human health at current levels of exposure. Reprod. Toxicol. 2007, 24, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Calafat, A.M.; Ye, X.Y.; Wong, L.Y.; Reidy, J.A.; Needham, L.L. Exposure of the U.S. population to Bisphenol A and 4-tertiary-octylphenol: 2003–2004. Environ. Health Perspect. 2008, 116, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Wetherill, Y.B.; Akingbemi, B.T.; Kanno, J.; McLachlan, J.A.; Nadal, A.; Sonnenscheing, C.; Watson, C.S.; Zoeller, R.T.; Belcher, S.M. In vitro molecular mechanisms of Bisphenol A action. Reprod. Toxicol. 2007, 24, 178–198. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, G.G.J.M.; Lemmen, J.G.; Carlsson, B.; Corton, J.C.; Safe, S.H.; van der Saag, P.T.; van der Burg, P.; Gustafsson, J.A. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 1998, 139, 4252–4263. [Google Scholar] [PubMed]
- Ng, H.W.; Perkins, R.; Tong, W.S.; Hong, H.X. Versatility or promiscuity: The estrogen receptors, control of ligand selectivity and an update on subtype selective ligands. Int. J. Environ. Res. Public Health 2014, 11, 8709–8742. [Google Scholar] [CrossRef] [PubMed]
- Durando, M.; Kass, L.; Piva, J.; Sonnenschein, C.; Soto, A.M.; Luque, E.H.; Munoz-de-Toro, M. Prenatal Bisphenol A exposure induces preneoplastic lesions in the mammary gland in wistar rats. Environ. Health Perspect. 2007, 115, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Timms, B.G.; Howdeshell, K.L.; Barton, L.; Bradley, S.; Richter, C.A.; Vom Saal, F.S. Estrogenic chemicals in plastic and oral contraceptives disrupt development of the fetal mouse prostate and urethra. Proc. Natl. Acad. Sci. USA 2005, 102, 7014–7019. [Google Scholar] [CrossRef] [PubMed]
- Goodman, J.E.; McConnell, E.E.; Sipes, I.G.; Witorsch, R.J.; Slayton, T.M.; Yu, C.J.; Lewis, A.S.; Rhomberg, L.R. An updated weight of the evidence evaluation of reproductive and developmental effects of low doses of Bisphenol A. Crit. Rev. Toxicol. 2006, 36, 387–457. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Magdalena, P.; Laribi, O.; Ropero, A.B.; Fuentes, E.; Ripoll, C.; Soria, B.; Nadal, A. Low doses of Bisphenol A and diethylstilbestrol impair Ca2+ signals in pancreatic alpha-cells through a nonclassical membrane estrogen receptor within intact islets of langerhans. Environ. Health Perspect. 2005, 113, 969–977. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Bisphenol A (BPA): Use in Food Contact Application; U.S. Food and Drug Administration: Silver Springs, MD, USA, 2015.
- Commission Directive 2011/8/EU Amending Directive 2002/72/EC as Regards the Restriction of Use of Bisphenol A in Plastic Infant Feeding Bottles. Official Journal of the European Union. Available online: http://faolex.fao.org/docs/pdf/eur100741.pdf (accessed on 22 February 2011).
- Order Amending Schedule I to the Hazardous Products Act (Bisphenol A). Canada. Available online: http://www.gazette.gc.ca/rp-pr/p2/2010/2010–03–31/html/sor-dors53-eng.html (accessed on 31 March 2010).
- Sommer, K. Correlation between primary chemical structure and property phenomena in polycondensates. Adv. Mater. 1991, 3, 590–599. [Google Scholar] [CrossRef]
- Neagu, L. Synthesis of Bisphenol A with Heterogeneous Catalysts; Queens University: Kingston, ON, Canada, 1998. [Google Scholar]
- Kamm, B.; Gruber, P.R.; Kamm, M. Biorefineries—Industrial Processes and Products; Wiley-VCH: Wienheim, Germany, 2010. [Google Scholar]
- Noordover, B.A.J.; van Staalduinen, V.G.; Duchateau, R.; Koning, C.E.; van Benthem, R.; Mak, M.; Heise, A.; Frissen, A.E.; van Haveren, J. Co- and terpolyesters based on isosorbide and succinic acid for coating applications: Synthesis and characterization. Biomacromolecules 2006, 7, 3406–3416. [Google Scholar] [CrossRef] [PubMed]
- Sablong, R.; Duchateau, R.; Koning, C.E.; de Wit, G.; van Es, D.; Koelewijn, R.; van Haveren, J. Incorporation of isosorbide into poly(butylene terephthalate) via solid-state polymerization. Biomacromolecules 2008, 9, 3090–3097. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, E.; Bassett, A.; Sadler, J.M.; La Scala, J.J.; Stanzione, J. Synthesis and characterization of bio-based epoxy resins derived from vanillyl alcohol. ACS Sustain. Chem. Eng. 2016, in press. [Google Scholar] [CrossRef]
- Harvey, B.G.; Guenthner, A.J.; Lai, W.W.; Meylemans, H.A.; Davis, M.C.; Cambrea, L.R.; Reams, J.T.; Lamison, K.R. Effects of o-methoxy groups on the properties and thermal stability of renewable high-temperature cyanate ester resins. Macromolecules 2015, 48, 3173–3179. [Google Scholar] [CrossRef]
- Meylemans, H.A.; Groshens, T.J.; Harvey, B.G. Synthesis of renewable bisphenols from creosol. ChemSusChem 2012, 5, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Meylemans, H.A.; Harvey, B.G.; Reams, J.T.; Guenthner, A.J.; Cambrea, L.R.; Groshens, T.J.; Baldwin, L.C.; Garrison, M.D.; Mabry, J.M. Synthesis, characterization, and cure chemistry of renewable bis(cyanate) esters derived from 2-methoxy-4-methylphenol. Biomacromolecules 2013, 14, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Harvey, B.G.; Guenthner, A.J.; Meylemans, H.A.; Haines, S.R.L.; Lamison, K.R.; Groshens, T.J.; Cambrea, L.R.; Davis, M.C.; Lai, W.W. Renewable thermosetting resins and thermoplastics from vanillin. Green Chem. 2015, 17, 1249–1258. [Google Scholar] [CrossRef]
- Harvey, B.G.; Guenthner, A.J.; Yandek, G.R.; Cambrea, L.R.; Meylemans, H.A.; Baldwin, L.C.; Reams, J.T. Synthesis and characterization of a renewable cyanate ester/polycarbonate network derived from eugenol. Polymer 2014, 55, 5073–5079. [Google Scholar] [CrossRef]
- Harvey, B.G.; Sahagun, C.M.; Guenthner, A.J.; Groshens, T.J.; Cambrea, L.R.; Reams, J.T.; Mabry, J.M. A high-performance renewable thermosetting resin derived from eugenol. ChemSusChem 2014, 7, 1964–1969. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.C.; Guenthner, A.J.; Groshens, T.J.; Reams, J.T.; Mabry, J.M. Polycyanurate networks from anethole dimers: Synthesis and characterization. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 4127–4136. [Google Scholar] [CrossRef]
- Cash, J.J.; Davis, M.C.; Ford, M.D.; Groshens, T.J.; Guenthner, A.J.; Harvey, B.G.; Lamison, K.R.; Mabry, J.M.; Meylemans, H.A.; Reams, J.T.; et al. High t-g thermosetting resins from resveratrol. Polym. Chem. 2013, 4, 3859–3865. [Google Scholar] [CrossRef]
- Laskoski, M.; Clarke, J.S.; Neal, A.; Harvey, B.G.; Ricks-Laskoski, H.L.; Hervey, W.J.; Keller, T.M. Sustainable high-temperature phthalonitrile resins derived from resveratrol and dihydroresveratrol. Polymer 2016, 55, 5073–5079. [Google Scholar]
- Harvey, B.G.; Guenthner, A.J.; Koontz, T.A.; Storch, P.J.; Reams, J.T.; Groshens, T.J. Sustainable hydrophobic thermosetting resins and polycarbonates from turpentine. Green Chem. 2016, 18, 2416–2423. [Google Scholar] [CrossRef]
- Garrison, M.D.; Harvey, B.G. Bio-based hydrophobic epoxy-amine networks derived from renewable terpenoids. J. Appl. Polym. Sci. 2016. [Google Scholar] [CrossRef]
- Maiorana, A.; Reano, A.F.; Centore, R.; Grimaldi, M.; Balaguer, P.; Allais, F.; Gross, R.A. Stucture property relationships of N-alkyl bisferulate epoxy resins. Green Chem. 2016. [Google Scholar] [CrossRef]
- Pion, F.; Reano, A.F.; Ducrot, P.H.; Allais, F. Chemo-enzymatic preparation of new bio-based bis and trisphenols: New versatile building blocks for chemistry. RSC Adv. 2013, 3, 8988–8997. [Google Scholar] [CrossRef]
- Shen, J.; Xu, L.; Fang, H.; Richard, A.M.; Bray, J.D.; Judson, R.S.; Zhou, G.X.; Colatsky, T.J.; Aungst, J.L.; Teng, C.; et al. EADB: An estrogenic activity database for assessing potential endocrine activity. Toxicol. Sci. 2013, 135, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Xu, L.; Fang, H.; Hong, H.X.; Perkins, R.; Harris, S.; Bearden, E.D.; Shi, L.M.; Tong, W.D. The EDKB: An established knowledge base for endocrine disrupting chemicals. BMC Bioinform. 2010, 11 (Suppl. S6), S5. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.W.; Shu, M.; Luo, H.; Ye, H.; Ge, W.G.; Perkins, R.; Tong, W.D.; Hong, H.X. Estrogenic activity data extraction and in silico prediction show the endocrine disruption potential of bisphenol A replacement compounds. Chem. Res. Toxicol. 2015, 28, 1784–1795. [Google Scholar] [CrossRef] [PubMed]
- Blair, R.M.; Fang, H.; Branham, W.S.; Hass, B.S.; Dial, S.L.; Moland, C.L.; Tong, W.D.; Shi, L.M.; Perkins, R.; Sheehan, D.M. The estrogen receptor relative binding affinities of 188 natural and xenochemicals: Structural diversity of ligands. Toxicol. Sci. 2000, 54, 138–153. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.X.; Tong, W.D.; Fang, H.; Shi, L.M.; Xie, Q.; Wu, J.; Perkins, R.; Walker, J.D.; Branham, W.; Sheehan, D.M. Prediction of estrogen receptor binding for 58,000 chemicals using an integrated system of a tree-based model with structural alerts. Environ. Health Perspect. 2002, 110, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Tong, W.; Fang, H.; Xie, Q.; Hong, H.; Perkins, R.; Wu, J.; Tu, M.; Blair, R.M.; Branham, W.S.; et al. An integrated “4-phase” approach for setting endocrine disruption screening priorities—Phase I and II predictions of estrogen receptor binding affinity. SAR QSAR Environ. Res. 2002, 13, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.D.; Fang, H.; Hong, H.X.; Xie, Q.; Perkins, R.; Anson, J.; Sheehan, D.M. Regulatory application of sar/qsar for priority setting of endocrine disruptors: A perspective. Pure Appl. Chem. 2003, 75, 2375–2388. [Google Scholar] [CrossRef]
- Tong, W.D.; Xie, W.; Hong, H.X.; Shi, L.M.; Fang, H.; Perkins, R. Assessment of prediction confidence and domain extrapolation of two structure-activity relationship models for predicting estrogen receptor binding activity. Environ. Health Perspect. 2004, 112, 1249–1254. [Google Scholar] [CrossRef] [PubMed]
- Sadler, J.M.; Nguyen, A.P.T.; Toulan, F.R.; Szabo, J.P.; Palmese, G.R.; Scheck, C.; Lutgen, S.; La Scala, J.J. Isosorbide-methacrylate as a bio-based low viscosity resin for high performance thermosetting applications. J. Mater. Chem. A 2013, 1, 12579–12586. [Google Scholar] [CrossRef]
- Hu, F.S.; La Scala, J.J.; Sadler, J.M.; Palmese, G.R. Synthesis and characterization of thermosetting furan-based epoxy systems. Macromolecules 2014, 47, 3332–3342. [Google Scholar] [CrossRef]
- Hu, F.S.; Yadav, S.K.; La Scala, J.J.; Sadler, J.M.; Palmese, G.R. Preparation and characterization of fully furan-based renewable thermosetting epoxy-amine systems. Macromol. Chem. Phys. 2015, 216, 1441–1446. [Google Scholar] [CrossRef]
- Stanzione, J.F.; Sadler, J.M.; La Scala, J.J.; Wool, R.P. Lignin model compounds as bio-based reactive diluents for liquid molding resins. Chem. Sus. Chem. 2012, 5, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Stanzione, J.F.; Giangiulio, P.A.; Sadler, J.M.; La Scala, J.J.; Wool, R.P. Lignin-based bio-oil mimic as biobased resin for composite applications. ACS Sustain. Chem. Eng. 2013, 1, 419–426. [Google Scholar] [CrossRef]
- Hong, H. Mold2, U.S. Food and Drug Administration. Available online: http://www.fda.gov/ScienceResearch/BioinformaticsTools/Mold2/default.htm (accessed on 26 March 2016).
- Hong, H.X.; Xie, Q.; Ge, W.G.; Qian, F.; Fang, H.; Shi, L.M.; Su, Z.Q.; Perkins, R.; Tong, W.D. Mold(2), molecular descriptors from 2d structures for chemoinformatics and toxicoinformatics. J. Chem. Inf. Model. 2008, 48, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- McPhail, B.; Tie, Y.F.; Hong, H.X.; Pearce, B.A.; Schnackenberg, L.K.; Ge, W.G.; Valerio, L.G.; Fuscoe, J.C.; Tong, W.D.; Buzatu, D.A.; et al. Modeling chemical interaction profiles: I. Spectral data-activity relationship and structure-activity relationship models for inhibitors and non-inhibitors of cytochrome p450 cyp3a4 and cyp2d6 isozymes. Molecules 2012, 17, 3383–3406. [Google Scholar] [CrossRef] [PubMed]
- Tie, Y.F.; McPhail, B.; Hong, H.X.; Pearce, B.A.; Schnackenberg, L.K.; Ge, W.G.; Buzatu, D.A.; Wilkes, J.G.; Fuscoe, J.C.; Tong, W.D.; et al. Modeling chemical interaction profiles: II. Molecular docking, spectral data-activity relationship, and structure-activity relationship models for potent and weak inhibitors of cytochrome p450 cyp3a4 isozyme. Molecules 2012, 17, 3407–3460. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.; Doughty, S.W.; Luo, H.; Ye, H.; Ge, W.G.; Tong, W.D.; Hong, H.X. Development and validation of decision forest model for estrogen receptor binding prediction of chemicals using large data sets. Chem. Res. Toxicol. 2015, 28, 2343–2351. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Shen, J.; Ng, H.W.; Sakkiah, S.; Ye, H.; Ge, W.; Gong, P.; Xiao, W.; Tong, W. Rat alpha-fetoprotein binding activity prediction model to facilitate assessment of endocrine disruption potential of environmental chemicals. Int. J. Environ. Res. Public Health 2016, 13, 372. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.X.; Xin, X.Q. Essesa, an expert system for structure elucidation from spectral-analysis 2. Novel algorithm of perception of the linear independent smallest set of smallest rings. Anal. Chim. Acta 1992, 262, 179–191. [Google Scholar] [CrossRef]
- Hong, H.X.; Xin, X.Q. Essesa—An expert system for structure elucidation from spectra. 3. Lnscs for chemical knowledge representation. J. Chem. Inf. Comp. Sci. 1992, 32, 116–120. [Google Scholar] [CrossRef]
- Hong, H.X.; Xin, X.Q. Essesa—An expert-system for structure elucidation from spectra. 4. Canonical representation of structures. J. Chem. Inf. Comp Sci. 1994, 34, 730–734. [Google Scholar] [CrossRef]
- Hong, H.X.; Xin, X.Q. Essesa—An expert-system for structure elucidation from spectra. 5. Substructure constraints from analysis of first-order H1-NMR spectra. J. Chem. Inf. Comp Sci. 1994, 34, 1259–1266. [Google Scholar] [CrossRef]
- Hong, H.X.; Han, Y.L.; Xin, X.Q.; Shi, Y.F. Essesa—An expert-system for structure elucidation from spectra. 6. Substructure constraints from analysis of C13-NMR spectra. J. Chem. Inf. Comp Sci. 1995, 35, 979–1000. [Google Scholar]
- Masui, H.; Hong, H.X. Spec2d: A structure elucidation system based on H1 NMR and H-HCOSY spectra in organic chemistry. J. Chem. Inf. Model. 2006, 46, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.X.; Xin, X.Q. Essesa—An expert system for elucidation of structures from spectra. 1. Knowledge base of infrared-spectra and analysis and interpretation programs. J. Chem. Inf. Comp Sci. 1990, 30, 203–210. [Google Scholar] [CrossRef]
- Tong, W.D.; Hong, H.X.; Fang, H.; Xie, Q.; Perkins, R. Decision forest: Combining the predictions of multiple independent decision tree models. J. Chem. Inf. Comp Sci. 2003, 43, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Tong, W.; Xie, Q.; Fang, H.; Perkins, R. An in silico ensemble method for lead discovery: Decision forest. SAR QSAR Environ. Res. 2005, 16, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.X.; Tong, W.D.; Perkins, R.; Fang, H.; Xie, Q.; Shi, L.M. Multiclass decision forest—A novel pattern recognition method for multiclass classification in microarray data analysis. DNA Cell Biol. 2004, 23, 685–694. [Google Scholar] [CrossRef] [PubMed]
- The Maestro Environment. Available online: http://www.schrodinger.com/Maestro (accessed on 6 April 2016).
- Gakh, A.A.; Anisimova, N.Y.; Kiselevsky, M.V.; Sadovnikov, S.V.; Stankov, I.N.; Yudin, M.V.; Rufanov, K.A.; Krasavin, M.Y.; Sosnov, A.V. Dihydro-resveratrol-A potent dietary polyphenol. Bioorg. Med. Chem. Lett. 2010, 20, 6149–6151. [Google Scholar] [CrossRef] [PubMed]
- De Medina, P.; Casper, R.; Savouret, J.F.; Poirot, M. Synthesis and biological properties of new stilbene derivatives of resveratrol as new selective aryl hydrocarbon modulators. J. Med. Chem. 2005, 48, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.T.; Han, G.Z.; Shim, J.Y.; Wen, Y.; Jiang, X.R. Quantitative structure-activity relationship of various endogenous estrogen metabolites for human estrogen receptor alpha and beta subtypes: Insights into the structural determinants favoring a differential subtype binding. Endocrinology 2006, 147, 4132–4150. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Compound | Qualitative Prediction * | Quantitative Prediction ** | ||||
|---|---|---|---|---|---|---|
| # | Name | TS-1 | TS-2 | Predict | Confidence | |
| 1 | Bisphenol-A (BPA) | −1.532 (±0.664, n = 21) | ||||
| 2 | Bisphenol-F (BPF) | −2.544 (±1.232, n = 3) | ||||
| 3 | Resveratrol | −2.489 (±0.016, n = 2) | ||||
| 4 | ||||||
| 5 | MDA | |||||
| 6 | 0.984 | 1.000 | + | 0.984 | −1.004 | |
| 7 | 0.984 | 0.800 | + | 0.784 | −0.831 | |
| 8 | 0.788 | 0.600 | + | 0.388 | 0.786 | |
| 9 | 0.984 | 1.000 | + | 0.984 | −0.704 | |
| 10 | 0.984 | 1.000 | + | 0.984 | −1.903 | |
| 11 | 0.984 | 1.000 | + | 0.984 | −1.064 | |
| 12 | 0.984 | 0.943 | + | 0.927 | −0.380 | |
| 13 | 0.784 | 0.543 | + | 0.327 | −2.338 | |
| 14 | 0.784 | 0.743 | + | 0.527 | −0.214 | |
| 15 | 0.984 | 0.972 | + | 0.962 | −2.222 | |
| 16 | Triguaiacol | 0.984 | 0.600 | + | 0.584 | NA |
| 17 | Bisguaiacol E | 0.834 | 0.600 | + | 0.434 | −1.117 |
| 18 | BGF-Catechol | 0.984 | 0.943 | + | 0.927 | −1.862 |
| 19 | Bisguaiacol-F (BGF) | 0.984 | 0.600 | + | 0.584 | −1.760 |
| 20 | MDA-13 | 0.003 | 0.004 | − | 0.993 | |
| 21 | Me-DFDA | 0.003 | 0.404 | − | 0.592 | |
| 22 | DFDA | 0.203 | 0.404 | − | 0.392 | |
| 23 | MDA-30 | 0.123 | 0.401 | − | 0.475 | |
| 24 | MDA-13 | 0.317 | 0.444 | − | 0.238 | |
| 25 | p-Cymene | 0.453 | 0.333 | − | 0.213 | |
| 26 | 0.216 | 0.400 | − | 0.384 | ||
| 27 | 0.616 | 0.300 | − | 0.084 | ||
| 28 | 0.316 | 0.350 | − | 0.334 | ||
| 29 | 0.566 | 0.300 | − | 0.134 | ||
| 30 | 0.566 | 0.300 | − | 0.134 | ||
| 31 | BHMF | 0.033 | 0.363 | − | 0.604 | |
| 32 | Isosorbide | 0.203 | 0.363 | − | 0.434 | |
| 33 | Bisguaiacol A | 0.516 | 0.250 | − | 0.234 | |
| 34 | BGF-Syringol | 0.366 | 0.400 | − | 0.234 | |
| Parameter | Result-1 (Mean ± Std) | Result-2 (Mean ± Std) |
|---|---|---|
| Accuracy | 0.812 (±0.019) | 0.801 (±0.009) |
| Sensitivity | 0.861 (±0.020) | 0.641 (±0.018) |
| Specificity | 0.758 (±0.033) | 0.877 (±0.011) |
| MCC | 0.624 (±0.039) | 0.534 (±0.021) |
| Balanced Accuracy | 0.809 (±0.020) | 0.759 (±0.010) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, H.; Harvey, B.G.; Palmese, G.R.; Stanzione, J.F.; Ng, H.W.; Sakkiah, S.; Tong, W.; Sadler, J.M. Experimental Data Extraction and in Silico Prediction of the Estrogenic Activity of Renewable Replacements for Bisphenol A. Int. J. Environ. Res. Public Health 2016, 13, 705. https://doi.org/10.3390/ijerph13070705
Hong H, Harvey BG, Palmese GR, Stanzione JF, Ng HW, Sakkiah S, Tong W, Sadler JM. Experimental Data Extraction and in Silico Prediction of the Estrogenic Activity of Renewable Replacements for Bisphenol A. International Journal of Environmental Research and Public Health. 2016; 13(7):705. https://doi.org/10.3390/ijerph13070705
Chicago/Turabian StyleHong, Huixiao, Benjamin G. Harvey, Giuseppe R. Palmese, Joseph F. Stanzione, Hui Wen Ng, Sugunadevi Sakkiah, Weida Tong, and Joshua M. Sadler. 2016. "Experimental Data Extraction and in Silico Prediction of the Estrogenic Activity of Renewable Replacements for Bisphenol A" International Journal of Environmental Research and Public Health 13, no. 7: 705. https://doi.org/10.3390/ijerph13070705