1. Introduction
Prostate cancer (PCa) is a complex multifactorial disease. A twin study suggested that genetic factors may explain 42% of the etiological risk of PCa [
1]. Genome-wide association studies (GWAS) identified over 70 PCa susceptibility variants, providing evidence of genetic susceptibility in the development of PCa. However, these polymorphic loci were common variants, mostly with low penetrance [
2]. The odds ratios (ORs) of these PCa-associated single nucleotide polymorphisms (SNPs) were modest (1.02–1.66) and no one locus contributed highly to the risk of PCa [
3].
Gene-gene interactions play a role in potential mechanisms of the missing heritability in human genetics, and research has identified the phenomenon in PCa. For example, Tao et al. identified 1325 pairs of SNP–SNP interactions with a P cutoff of 1.0 × 10
-8 in 1176 PCa cases and 1101 control subjects from the National Cancer Institute Cancer Genetic Markers of Susceptibility study, although no SNP-SNP interaction reached a genome-wide significance level of 4.4 × 10
-13 [
4]. A study by Ciampa et al. showed that two biologically interesting interactions, one between rs748120 of NR2C2 and subregions of 8q24 and that between rs4810671 of SULF2 and both JAZF1 and HNF1B, were associated with PCa [
5]. In cases of Italian heredo-familial PCa, VDR1 T/T genotypes coupled with the VDR2 T/T genotype exhibited a five-fold higher probability of PCa [
6]. Additionally, some studies showed that the cumulative effect of such interactions could increase the ORs of PCa. Specifically, the risk of PCa in men with six or more risk alleles was higher than in men with two or fewer risk alleles (OR = 6.22) [
7]. In a Swedish population, the OR for PCa was 9.46 in men who had five or more SNPs associated with PCa, compared with men without any of these SNPs [
8].
Environmental factors play a significant role and perhaps can modify the genetic risk of PCa [
9]. Body mass index (BMI), smoking, and alcohol consumption are PCa-related environmental factors that affect the risk of this disease. A meta-analysis of prospective studies indicated that obesity may have a dual effect on the risk of PCa: a decreased risk for localized PCa and an increased risk for advanced PCa [
10]. Smoking cessation can reduce the risk of developing PCa [
11]. Alcohol consumption is related to an increased risk of PCa in a Chinese population [
12].
Despite identifying dozens of PCa risk variants using GWAS, as well as the emergence of evidence of environmental risk factors that contribute to PCa, the effect of gene-gene and gene-environment interactions on the risk of PCa is largely unknown. In the current study, 36 SNPs were selected from a GWAS and used to estimate their association with PCa in 286 cases and 288 control subjects. We determined the gene-gene interactions and cumulative effects between the confirmed PCa risk SNPs. Finally, the gene-environment interactions in 134 PCa patients were analyzed to identify the combination of factors yielding the greatest risk of PCa in Chinese men.
2. Materials and Methods
2.1. Study Population
This was a case-control study including 286 PCa patients and 288 healthy geographically matched controls. All subjects were unrelated Northern Han Chinese men, and were permanent residents of Beijing or Tianjin (Jingjin area). Detailed inclusion criteria of cases and controls were described previously [
13]. The age at diagnosis, the Gleason score, tumour stage, serum PSA levels of PCa patients were obtained and aggressive PCa was defined as tumours with a PSA level > 20 ng/mL, and/or a Gleason score ≥8 or higher, and/or a pathological stage ≥III [
14]. This study was approved by the ethics committee of the two participating hospitals (ethical approval number: 2013BJYYEC-047-01), and informed consent was obtained from all study participants.
Height and body weight were measured in 134 Beijing PCa cases, and information about smoking and alcohol consumption were collected. BMI was calculated using the individual’s height and weight (kilograms per square meter), and categorized according to standard cut-off points for the Chinese population (underweight <18.5, normal weight 18.5–24.0, overweight 24.0–28.0, and obese ≥28.0) [
15]. In our study, all patients were classified into normal, overweight, or obese groups.
Questionnaire data collected about smoking included addiction (current, past, or never smoked), age of smoking initiation, years of smoking, number of cigarettes smoked per day, and years since quitting smoking. Individuals smoking every day, or some days, were classified as current smokers. Those who responded “not at all” were classified as never smokers, and individuals with a smoking history but who had quit were classified as past smokers [
16]. Because of the small sample size in our study, the number of smoking classifications was reduced. Past smokers who had smoked more than 100 cigarettes in their lifetime were classified as current smokers and those who had smoked fewer than 100 cigarettes in their lifetime were classified as never smokers.
Individuals that consumed alcohol were grouped into four categories: non-drinking, drinking on special occasions only, often drinking, and drinking every day [
17]. Non-drinking and drinking occasionally individuals were classified as “seldom”. Drinking every day and drinking often were classified as “often”.
2.2. Selection of SNPs for Genotyping
Thirty-six PCa-associated SNPs, identified in a GWAS from 2007 to 2010 and a Multiethnic Cohort study of a functional locus, were selected for genotyping [
18,
19,
20,
21,
22,
23,
24,
25,
26,
27]. Blood genomic DNA was extracted and the selected loci were genotyped as described previously [
13]. rs11986220 was genotyped by sequencing while the other 36 SNPs were determined by using polymerase chain reaction-high resolution melting curves (PCR-HRM) of small amplicons and sequencing methods. Briefly, a final reaction volume of 10 µL included 1 µL of 1× LC-green PLUS fluorescent dye and 0.05 µL of each pair of low and high temperature calibrators (10 pmol/µL). After the PCR reaction, products were transferred into matching 96-well plates to be genotyped automatically and verified manually using a Lightscanner TMHR-I 96 (Idaho Technology, Inc., Salt Lake City, UT, USA). To validate the accuracy of genotyping, about 10% of the samples were selected randomly for duplicate analysis. In addition, five samples were selected randomly from the three different verified genotypes of each risk variant to be sequenced (Beijing Genomics Institute, Beijing, China) to confirm the genotyping results. All primers used for both PCR-HRM and PCR sequencing were designed using Oligo (version 6.0; Molecular Biology Insights, Inc., Cascade, CO, USA).
Tables S1 and S2 list the information about the primers that were used.
2.3. Gene-Gene Interaction Analyses
The Generalized Multifactor Dimensionality Reduction (GMDR) (GMDR Software, Beta version 0.7; Department of Biostatistics, University of Alabama, Birmingham, AL, USA) method, with age adjustment, was used to determine gene-gene interactions between PCa-associated SNPs [
28,
29]. Ten-fold cross-validation (CV) was set when running the software. The age-adjusted model with the smallest prediction error (the greatest testing balanced accuracy), the highest CV consistency, and
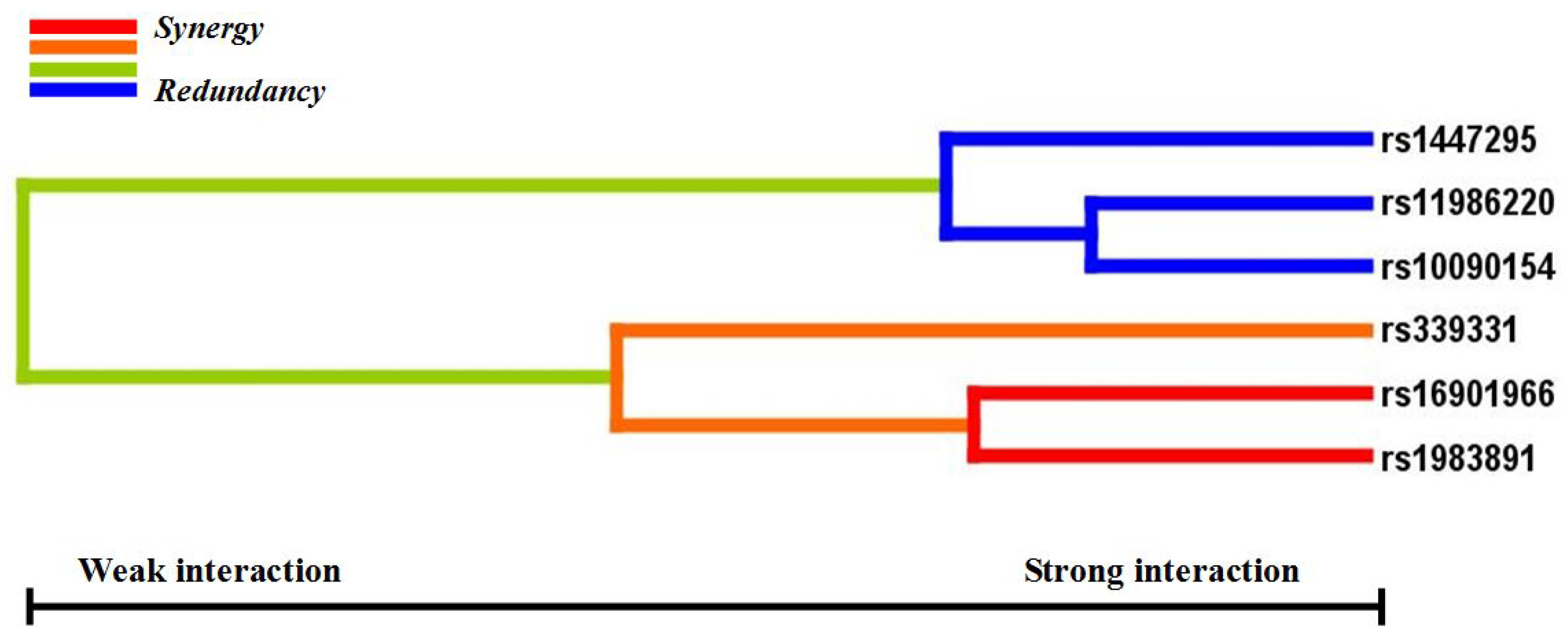
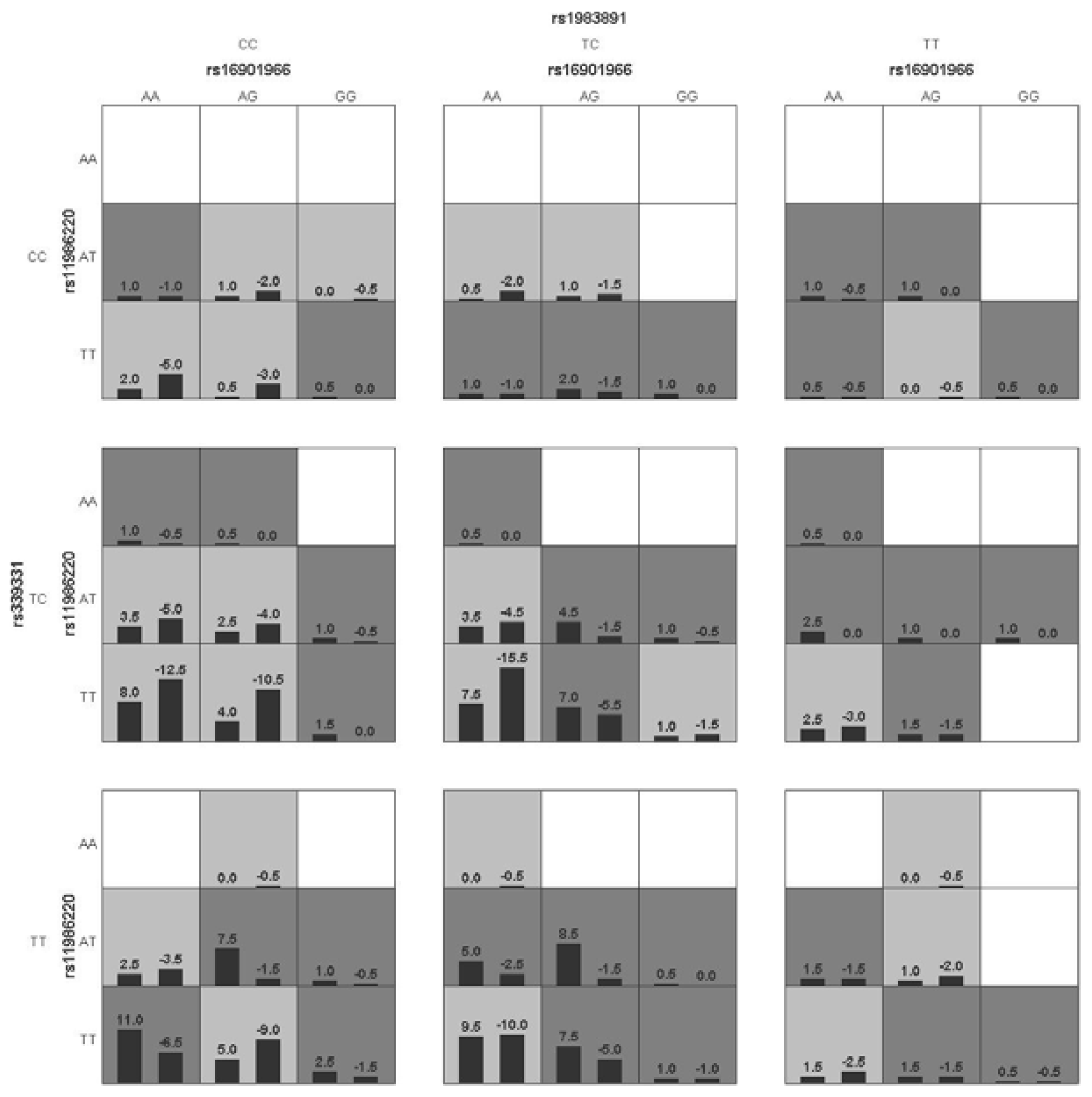
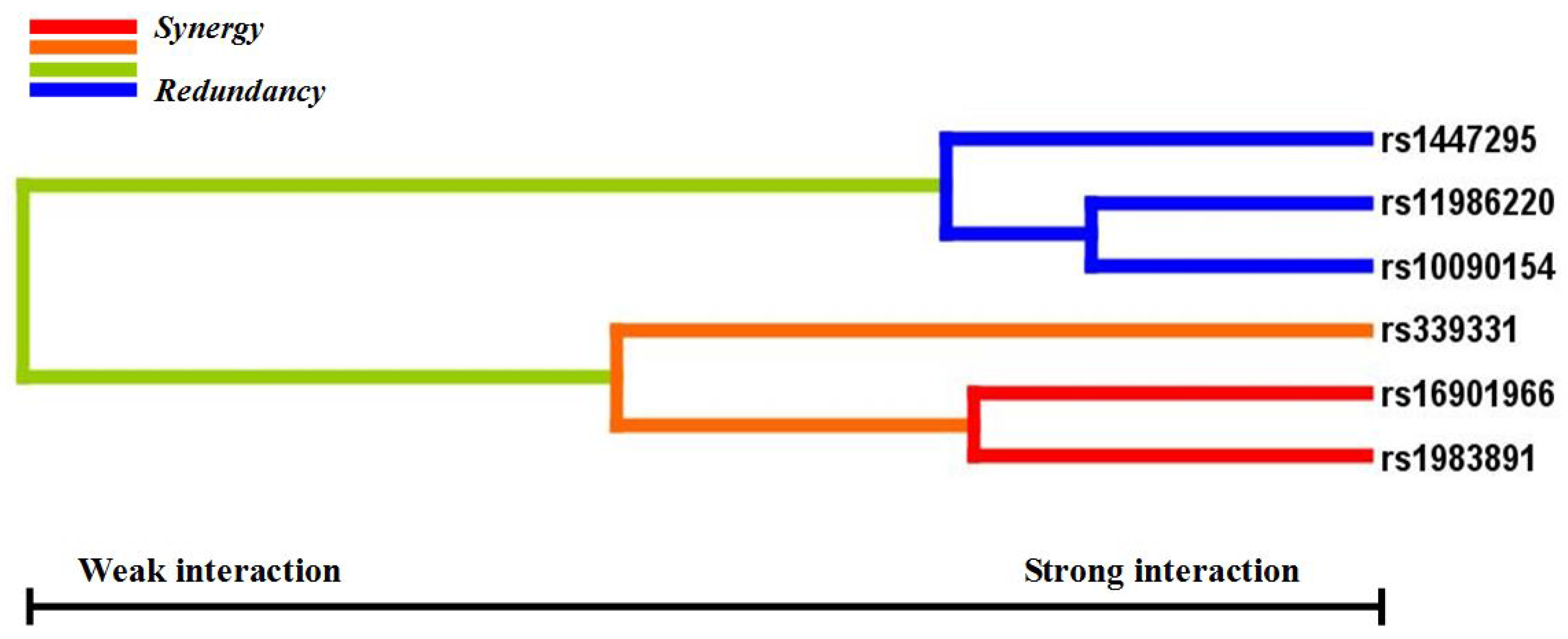
p < 0.05 in the sign test was selected as the best n-loci model. In this study, we analyzed the interaction among six SNPs associated with a risk for PCa (rs16901966, rs11986220, rs1447295, and rs10090154 at 8q24; rs1983891 at FOXP4; rs339331 at GPRC6A) by using a GMDR model that adjusted for age. Entropy-based interaction dendrograms were built by using Multifactor Dimensionality Reduction (MDR) to better confirm and visualize the interaction models identified by GMDR [
30].
2.4. Cumulative Effect Analysis
Based on the results of the association analyses between 36 SNPs and PCa, the cumulative effect was determined among five confirmed PCa risk loci, excluding rs11986220, which is linked with rs10090154. Combined with genotyping data, samples that carried none, one, two, or more than two of the risk genotypes were labeled 0, 1, 2, and ≥3, respectively. Risk genotypes were defined from the genetic model analyses of rs1983891 TT/TC, rs339331 TT, rs16901966 GG, rs11986220 AA/AT, rs1447295 AA/AC, and rs10090154 TC/TT. ORs and 95% confidence intervals (CIs) were calculated to compare the frequency of risk genotypes between PCa cases and controls. The logistic regression method was used to calculate the ORs and 95% CIs (or the age-adjusted ORs and 95% CIs).
2.5. Gene-Environment Interaction Analyses
A logistic regression test was used for the gene-environment interaction analyses. For gene-BMI interactions, BMI < 24.0, BMI = 24.0–28.0, and BMI ≥ 28.0 were defined as 1, 2, and 3, respectively. For gene-smoking interactions, never smokers and current smokers were defined as 1 and 2, respectively. For gene-drinking interactions, seldom and often were defined as 1 and 2, respectively. During multinomial logistic regression calculations, independent genotypes of the 36 SNPs were categorized as “dependent” and environmental factors were categorized as “factor”, while age was selected as “covariate” when adjusting the regression. In general, the non-risk genotypes and those in an unexposed environment (BMI < 24.0, never smoking, and seldom drinking) were used as the control group, and compared with the risk genotype and those in an exposed environment (BMI = 24.0–28.0, BMI ≥ 28.0, current smoking, and often drinking).
2.6. Statistical Analyses
Pearson’s χ2 was used to test the Hardy-Weinberg equilibrium (HWE) for each SNP separately among the control subjects. Logistic regression was used to estimate the unadjusted and age-adjusted ORs and 95% CIs for each risk allele (designated as “1”) versus each non-risk allele (designated as “2”). Genotypes 11, 12, and 22 represented risk homozygotes, risk heterozygotes, and non-risk homozygotes, respectively. When ORs and 95% CIs were calculated by logistic regression in the different models, genotypes 11 + 12 were designated as “1” and genotype 22 was designated as “2” in a dominant mode, and genotype 11 was designated as “1” and genotypes 12 + 22 were designated as “2” in a recessive mode. Logistic regression analyses were also used to estimate ORs and 95% CIs, and age adjusted ORs and 95% CIs, of cumulative effects. Statistical analyses were performed using the Statistical Package for the Social Sciences software package (version 16.0; SPSS, Inc., Chicago, IL, USA), and p < 0.05 was considered significant.
4. Discussion
GWAS have identified over 70 loci associated with PCa. However, it is largely unknown which genes and environmental risk factors jointly affect the risk of this disease. The present study investigated the relationships between 36 GWAS identified PCa risk variants and the risk of PCa in a northern Chinese population. Based on allelic and genotypic association analyses, we explored the gene-gene interactions among six confirmed PCa risk variants and their cumulative effect on the risk of PCa in a case-control study. We also examined the gene-environment interaction between 36 SNPs and the environmental factors BMI, smoking, and alcohol consumption. The best gene-gene interaction model involved rs16901966 and rs11986220 at 8q24, rs1983891 at FOXP4, and rs339331 at RFX6. The cumulative effect of these six loci increased the risk of PCa about 3.2-fold. We observed that cases with BMI ≥ 28 had five to seven times higher risk of PCa with homozygous rs16901966 GG and rs6983561 CC than cases with BMI < 24.0. Cases that carried the risk allele C of rs7679673 (CC, CA) and homozygous rs7679673 CC, and were current smokers and often drinkers, had a 1.77 and 3.37-fold higher risk of PCa, respectively, compared to cases with identical genotypes but who never smoked and seldom drank. Current smokers with the rs12653946 TT genotype had 3.11 times the risk of PCa than never smokers.
After age adjustment, the confirmed PCa risk loci were rs16901966, rs11986220, rs1447295, and rs10090154 at 8q24, rs1983891 at FOXP4, and rs339331 at RFX6. In our previous study, rs16901966, rs1447295, rs11986220, and rs10090154 at 8q24 (Region 1, Region 2) were associated with PCa and PCa-related clinical covariates [
13]. rs1983891 at FOXP4 and rs339331 at RFX6 were first identified as PCa risk loci in Japanese patients, and were further confirmed in subsequent studies [
18,
31,
32]. Our result that only 6 of 36 SNPs were associated with PCa suggests that this discrepancy could result from gene-gene and gene-environment interactions that would alter the effect of some disease-associated genes or loci.
Gene-gene interaction analyses showed that there were significant joint interactions among rs16901966, and rs11986220 at 8q24, rs1983891 at FOXP4, and rs339331 at RFX6. rs16901966 (region 2), rs11986220 (region 1) at 8q24 on chromosome 8 were verified to have consistent associations in various populations [
18,
27,
33,
34,
35]. rs1983891 is located in FOXP4, which belongs to subfamily P of the forkhead box (FOX) transcription factor family, and is associated with kidney tumors, larynx carcinoma, and breast tumors [
36,
37,
38]. At present, the potential role for FOXP4 in prostate tumorigenesis has not been determined. rs339331 was located at RFX6, a member of the regulatory factor X (RFX) family of transcription factors. A recent study reported that rs339331 can affect the risk of PCa by altering the expression of RFX6 [
39]. A family-based study reported an interaction between rs4242382 at 8q24 and rs10486567 at JAZF zinc finger1 gene (JAZF1), which encodes a transcriptional repressor, for non-aggressive PCa [
40]. These two transcription factors, FOXP4 and RFX6, may also interact with 8q24 by some regulated loci.
GMDR can effectively eliminate noise from the covariate, can increase prediction accuracy, and is applicable to both dichotomous and continuous phenotypes in various population-based study designs [
29]. The results of the GMDR analysis in our study established a biostatistical foundation for the study of functional epistasis between FOXP4, RFX6, and 8q24. To understand the cumulative effects of the confirmed six variants on the risk of PCa, we used a combined score of the total number of risk genotypes from the five SNPs that were independently associated with PCa (rs11986220 was excluded because of the linkage with rs10090154). Our data indicated that men who carried at least three risk genotypes had a three-fold increased risk of PCa compared with men who carried no risk genotypes. Because 33.1% of cases and 16.6% of control subjects carried three or more risk genotypes, the cumulative effects of these SNPs on PCa incidence in China are substantial. Furthermore, it has been verified that some variants at 8q24 are PCa-susceptible loci in different populations. Thus, confirming this cumulative effect in other populations is necessary.
PCa is a multifactorial process involving both genetic and environmental components. Studies on gene-environment interrelations can, therefore, provide a potentially powerful approach for identifying the causes of this disease [
41]. Although the current work is a case-only study, analyses based only on cases are valid, and offer better precision for estimating gene-environment interactions than those based on full data [
42]. Obesity, smoking, and alcohol consumption are associated with a moderate increase in the risk of PCa or aggressive PCa [
43,
44,
45]. Gene-environment interactions could either decrease or increase the risk of the disease. However, which genes and factors jointly affect the risk of PCa is largely unknown. In our case-only study, we analyzed the interactions between 36 SNPs and BMI, smoking, and alcohol consumption. Our results showed that rs6983561 and rs16901966, that exhibit a high degree of linkage disequilibrium at 8q24, interacted with BMI ≥ 28 (obesity) and contributed to a higher risk of PCa (ORs = 7.66, 5.33, respectively). The homozygous and risk allele carriers of rs7679673 at TET2, that encode a protein involved in myelopoiesis, interact with smoking and drinking to increase the risk of PCa (ORs = 2.77, 4.37, respectively). Homozygous rs12653946 at 5p15 in current smokers can also increase the risk of PCa (OR = 3.11).
Although some significant risk factors were identified in our study, there are several limitations to the results. The largest limitation is the small sample size that lowers the statistical power of the study. Although the sample quality is higher, replicating these results in larger populations is desirable. Second, the small sample size and the number of SNP variants studied limited us to exploring the gene-gene interactions among only six confirmed PCa risk SNPs, thus ignoring some potential interactions among other SNPs. Third, the case-only study involved only 134 Beijing PCa cases, limiting the gene-environment-phenotype analysis (e.g., PSA level, Gleason score, tumor stage, and aggressive PCa). Furthermore, some valuable factors those associated with development PCa such as presence of diabetes or use of diabetic medication should be designed in the future study. The lack of family history or treatment methods (surgical operation or medications used) for adjustment is another major limitation of the analysis. Therefore, populations with detailed PCa-related environmental exposures should be examined in multiple centers of China to confirm the association between these SNPs-environment interactions and PCa. This would establish a PCa databank about gene-gene-phenotype and gene-environment-phenotype, and provide a more effective approach to evaluate the risk of PCa, and to provide recommendations for preventing this disease.


 ,
, 
{kind=link}
{kind=link}