N-Acetylglucosamine: Production and Applications

Abstract
:1. Introduction
2. Production of N-Acetyl-d-glucosamine
2.1. Chemical methods to produce GlcNAc
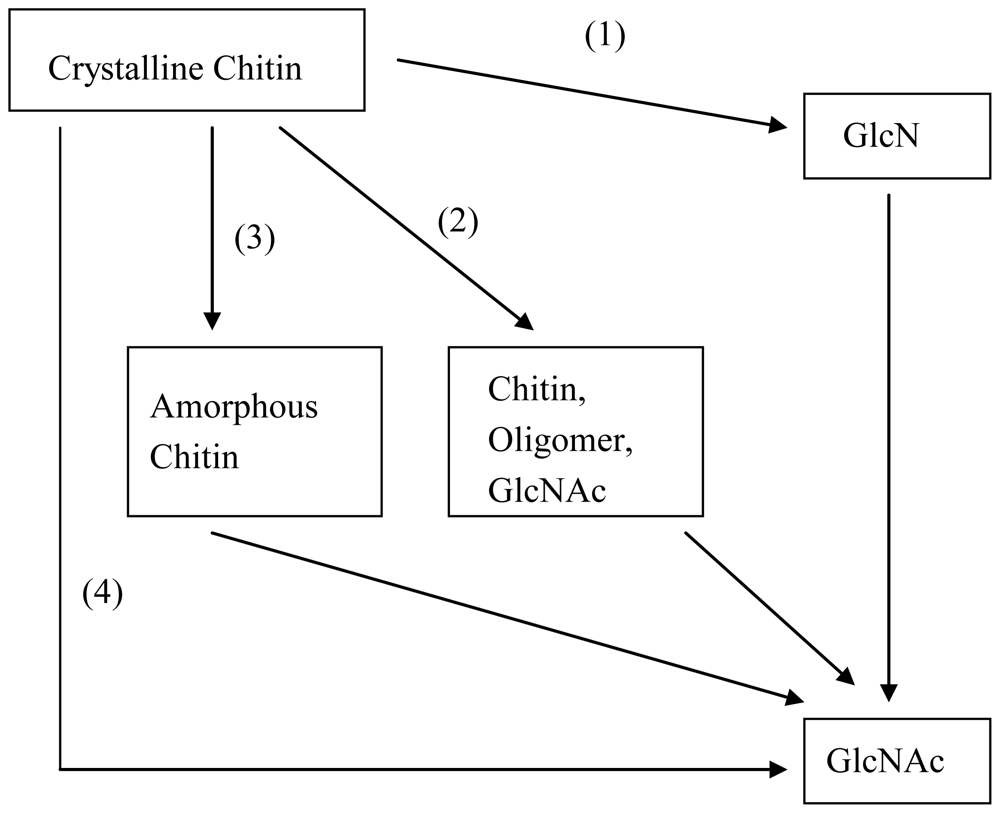
2.2. Enzymatic methods to produce GlcNAc
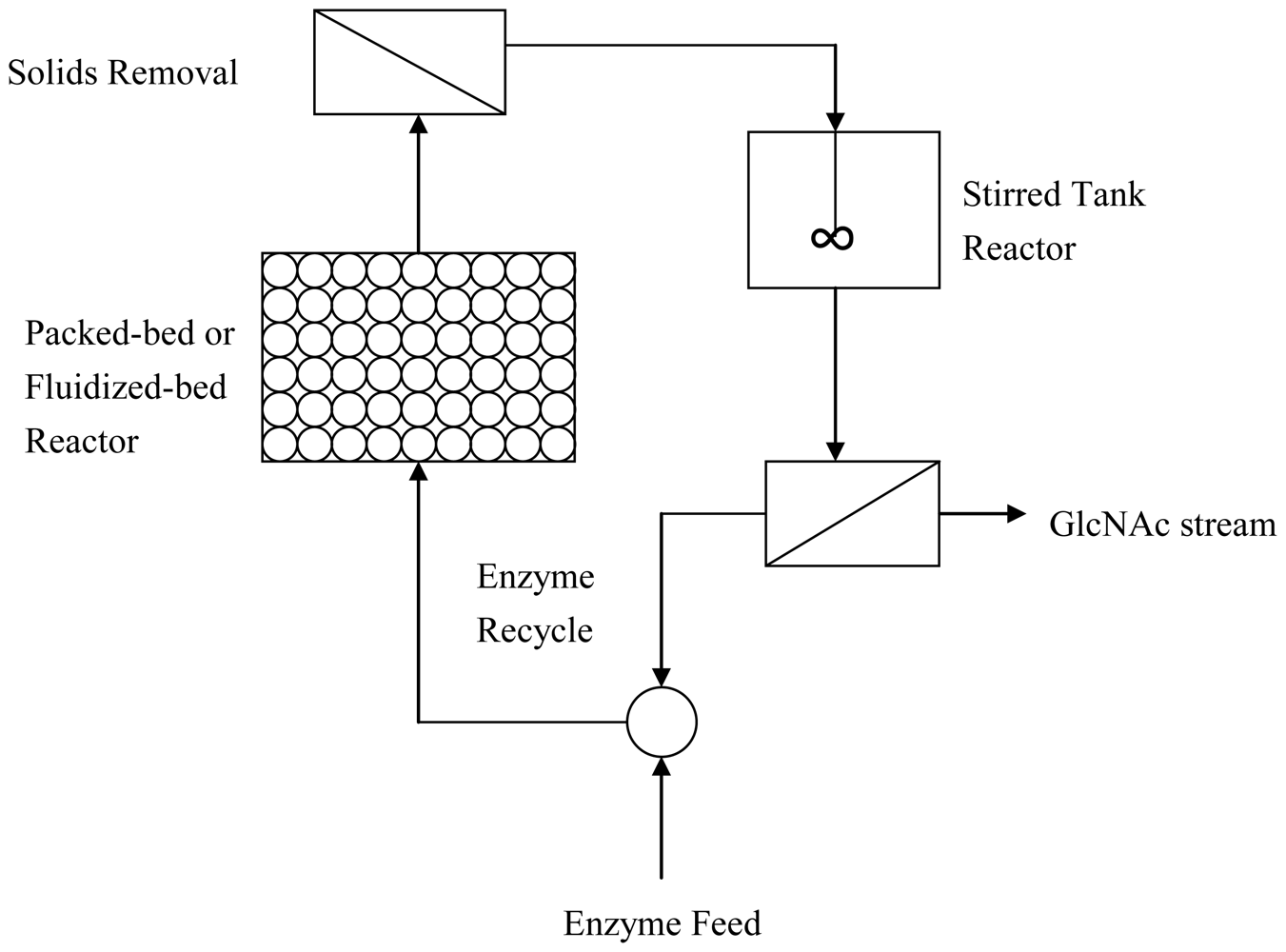
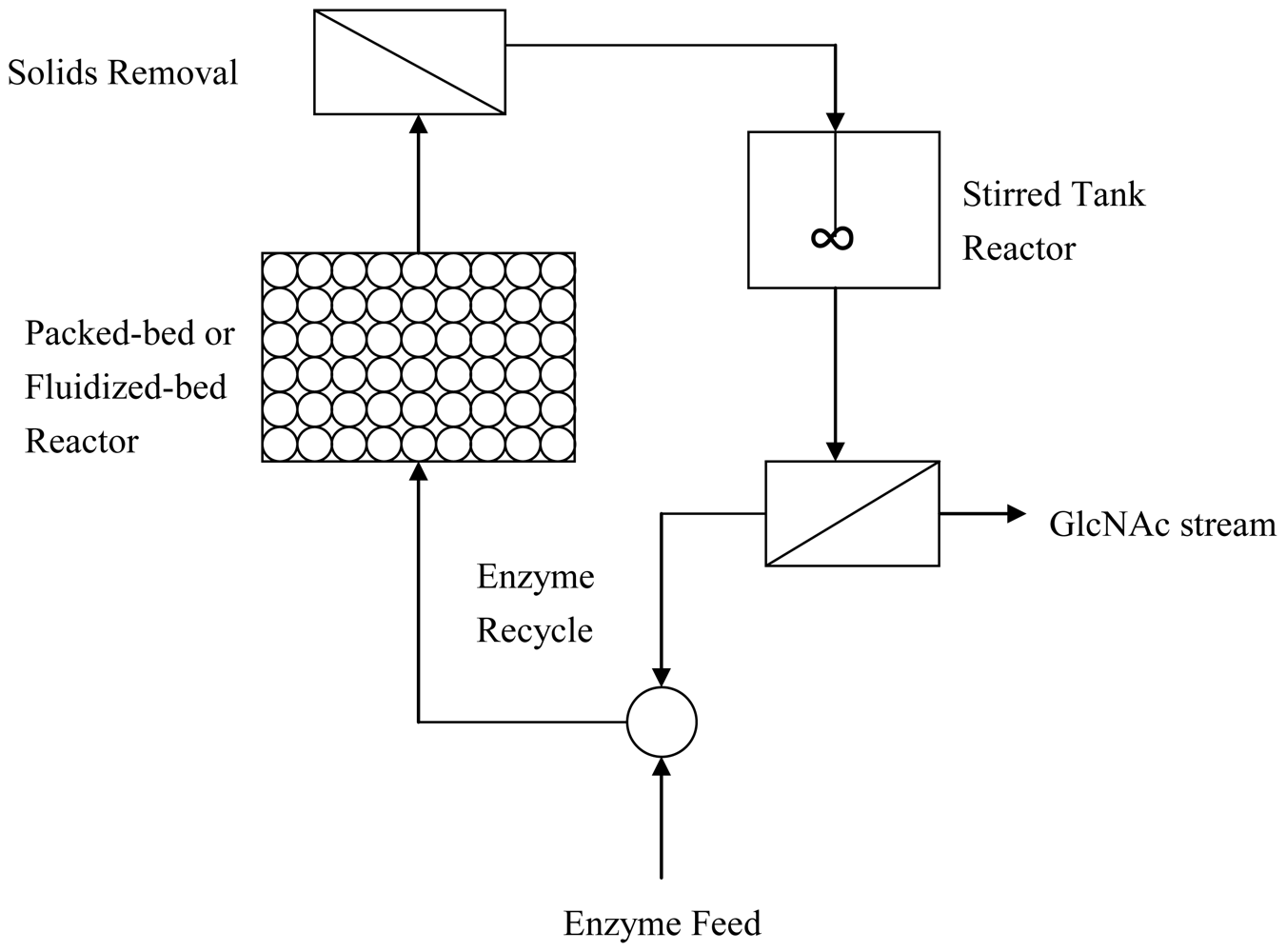
2.3. Improved enzymatic methods to produce GlcNAc
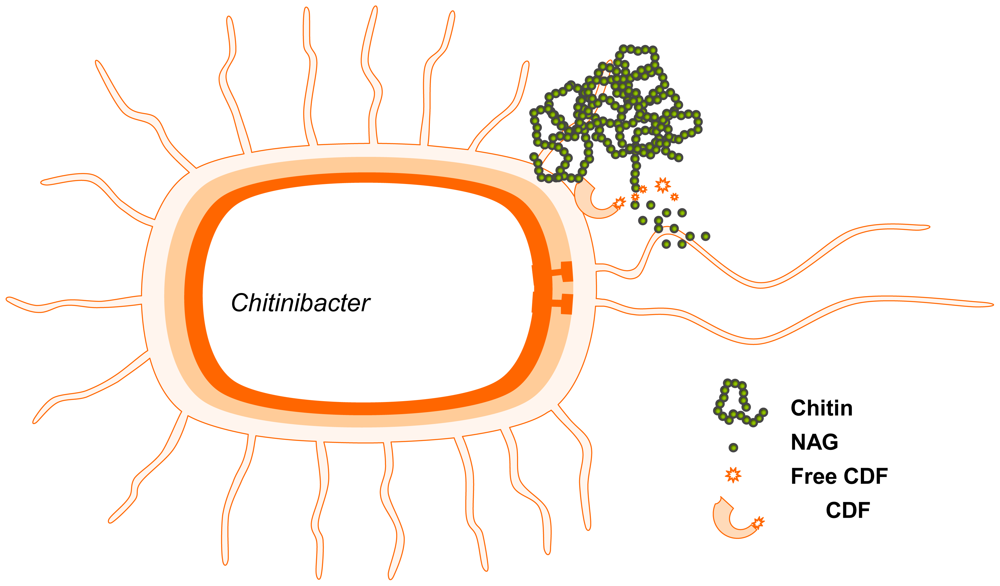
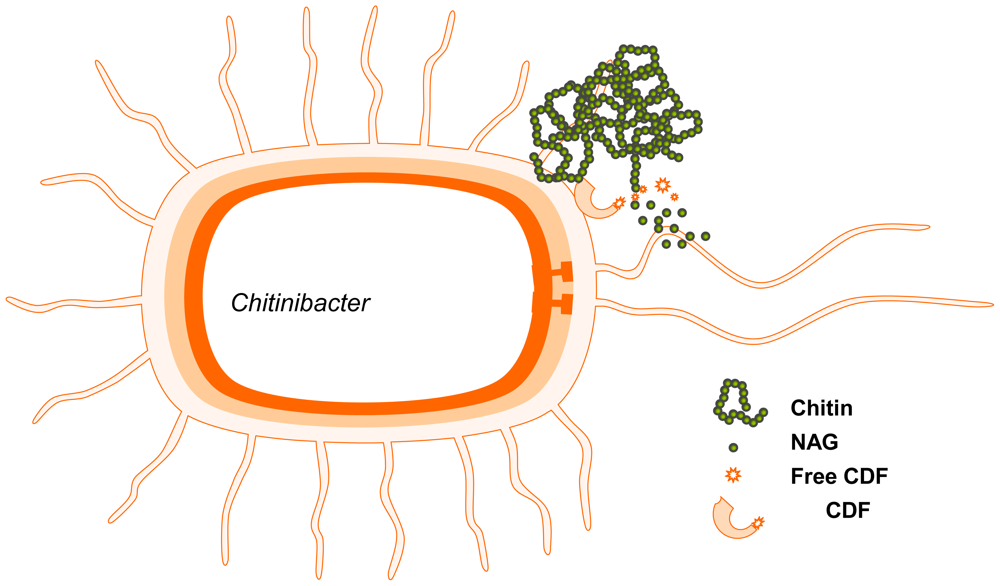
2.4. Production of GlcNAc by biotransformation
3. Applications of GlcNAc
3.1. Safety of GlcNAc
3.2. GlcNAc is used to treat joint damage
3.3. GlcNAc is a potential candidate to treat inflammatory bowel disease (IBD)
3.4. Application of GlcNAc in cosmetics
3.5. GlcNAc is used as a substrate in sialic acid production
3.6. Other applications of GlcNAc
4. Conclusions
Acknowledgements
References
- Álvarez-Añorve, LI; Calcagno, ML; Plumbridge, J. Why Does Escherichia coli Grow More Slowly on Glucosamine Than on N-Acetylglucosamine? Effects of Enzyme Levels and Allosteric Activation of GlcN6P Deaminase (NagB) on Growth Rates. J. Bacteriol 2005, 187, 2974–2982. [Google Scholar]
- Chiu, PCN; Tsang, HY; Koistinen, H; Seppala, M; Lee, KF; Yeung, WSB. The Contribution of d-Mannose, l-Fucose, N-Acetylglucosamine, and Selectin Residues on the Binding of Glycodenlin Isoforms to Human Spermatozoa. Biol. Reprod 2004, 70, 1710–1719. [Google Scholar]
- Promé, JC; Denarié, J; Truchet, G. Acylated Chitooligomers Are Molecular Signals That Mediate the Symbiotic Interactions Between Nitrogen-fixing Bacteria and Their Host Plants. Pure Appl. Chem 1998, 70, 55–60. [Google Scholar]
- Oldroyd, GED; Mitra, RM; Wais, RJ; Long, SR. Evidence for Structurally Specific Negative Feedback in the Nod Factor Signal Transduction Pathway. Plant J 2001, 28, 191–199. [Google Scholar]
- Watkins, WM. Biochemistry and Genetics of the ABO, H, Lewis and P Blood Group Systems. Adv. Hum. Genet 1980, 10, 1–136. [Google Scholar]
- Izawa, M; Kumamoto, K; Mitsuoka, C; Kanamori, A; Ohmori, K; Ishida, H; Nakamura, S; Kurata-Miura, K; Sasaki, K; Nishi, T; Kannagi, R. Expression of Sialyl 6-Sulfo Lewis X Is Inversely Correlated with Conventional Sialyl Lewis X Expression in Human Colorectal Cancer. Cancer Res 2000, 60, 1410–1416. [Google Scholar]
- Kennedy, JF; White, CA. Bioactive Carbohydrates in Chemistry, Biochemistry and Biology; Ellis Horwood: Chichester, UK, 1983. [Google Scholar]
- D’Ambrosio, E; Casa, B; Bompani, R; Scali, G; Scali, M. Glucosamine Sulfate: a Controlled Clinical Investigation in Arthrosis. Pharmatherpeutica 1981, 2, 504–508. [Google Scholar]
- Laverty, S; Sandy, JD; Celeste, C; Vanchon, P; Marier, JF. Synovial Fluid Levels and Serum Pharmacokinetics in a Large Animal Model Following Treatment with Oral Glucosamine at Clinically Relevant Doses. Arthritis Rheum 2005, 52, 181–191. [Google Scholar]
- Gooday, GW. The Ecology of Chitin Degradation. Adv. Microb. Ecol 1990, 11, 387–430. [Google Scholar]
- Hackman, RH; Goldberg, M. Light-scattering and Infrared-spectrophotometric Studies of Chitin and Chitin Derivatives. Carbohydr. Res 1974, 38, 35–45. [Google Scholar]
- Jollès, P; Muzzarelli, RAA (Eds.) Chitin and Chitinases; Birkhauser Verlag: Basel, Switzerland, 1999.
- Tanaka, T; Fujiwara, S; Nishikori, S; Fukui, T; Takagi, M; Imanaka, TA. Unique Chitinase with Dual Active Sites and Triple Substrate Binding Sites from the Hyperthermophilic Archaeon Pyrococcus kodakaraensis KOD1. Appl. Environ. Microbiol 1999, 65, 5338–5344. [Google Scholar]
- Ashry, ESHE; Aly, MRE. Synthesis and Biological Relevance of N-Acetylglucosamine-containing Oligosaccharides. Pure Appl. Chem 2007, 12, 2229–2242. [Google Scholar]
- DeAngelis, PL. Molecular Directionality of Polysaccharide Polymerization by the Pasteurella multocida Hyaluronan Synthase. J. Biol. Chem 1999, 274, 26557–26562. [Google Scholar]
- Turely, EA; Bowman, P; Kytryk, MA. Effects of Hyaluronate and Hyaluronate Binding Proteins on Cell Motile and Contact Behaviour. J. Cell Sci 1985, 78, 133–145. [Google Scholar]
- Alho, AM; Underhill, CB. The Hyaluronate Receptor Is Preferentially Expressed on Proliferating Epithelial Cells. J. Cell Biol 1989, 108, 1557–1565. [Google Scholar]
- Toole, BP; Jackson, G; Gross, J. Hyaluronate in Morphogenesis: Inhibition of Chondrogenesis in Vitro. Proc. Natl. Acad. Sci. USA 1972, 69, 1384–1386. [Google Scholar]
- Brecht, M; Mayer, U; Schlosser, E; Prehm, P. Increased Hyaluronate Synthesis Is Required for Fibroblast Detachment and Mitosis. Biochem. J 1986, 239, 445–450. [Google Scholar]
- Dube, B; Luke, HJ; Aumailley, M; Prehm, P. Hyaluronan Reduces Migration and Proliferation in CHO Cells. Biochim. Biophys. Acta 2001, 1538, 283–289. [Google Scholar]
- Takaku, H; Ishida, HK; Fujita, M; Inazu, T; Ishida, H; Kiso, M. A Chemical Synthesis of GlcNAc[beta](1–4)GlcUA-UDP to Elucidate to Catalytic Mechanism of Hyaluronic Acid Synthesis (HAS). Synlett 2007, 5, 818–820. [Google Scholar]
- Aronson, NN; Kuranda, MJ. Lysosomal Degradation of Asn-Linked Glycoproteins. FASEB J 1989, 3, 2615–2622. [Google Scholar]
- Chien, LJ; Lee, CK. Hyaluronic Acid Production by Recombinant Lactococcus lactis. Appl. Microbiol. Biotechnol 2007, 77, 339–346. [Google Scholar]
- Chien, LJ; Lee, CK. Enhanced Hyaluronic Acid Production in Bacillus subtilis by Coexpressing Bacterial Hemoglobin. Biotechnol. Prog 2007, 23, 1017–1022. [Google Scholar]
- Danishefsky, I; Steiner, H; Bella, A; Friedlander, A. Investigations on the Chemistry of Heparin: VI. Position of the Sulfate Ester Groups. J. Biol. Chem 1969, 244, 1741–1745. [Google Scholar]
- Bhavanandan, VP; Meyer, K. Mucopolysaccharides: N-Acetylglucosamine- and Galactose-6- sulfates from Keratosulfate. Science 1966, 151, 1404–1405. [Google Scholar]
- Scott, LJ; Clarke, NW; George, NJR; Shanks, JH; Testa, NG; Lang, SH. Interactions of Human Prostatic Epithelial Cells with Bone Marrow Endothelium: Binding and Invasion. Br. J. Cancer 2001, 84, 1417–1423. [Google Scholar]
- Ugorski, M; Laskowska, A. Sialyl Lewis (a): a Tumor-Associated Carbohydrate Antigen Involved in Adhesion and Metastatic Potential of Cancer Cells. Acta Biochim. Pol 2002, 49, 303–311. [Google Scholar]
- Patel, KD; Cuvelier, SL; Wiehler, S. Selectins: Critical Mediators of Leukocyte Recruitment. Semin. Immunol 2002, 14, 73–81. [Google Scholar]
- Parasadarao, NV; Wass, CA; Kim, KS. Endothelial Cell GlcNAc Beta 1–4GlcNAc Epitopes for Outer Membrane Protein A Enhance Traversal of Escherichia coli across the Blood-brain Barrier. Infect. Immun 1996, 64, 154–160. [Google Scholar]
- Aly, MRE; Rochaix, P; Amessou, M; Johannes, L; Florent, JC. Synthesis of Globo- and Isoglobotriosides Bearing a Cinnamoylphenyl Tag as Novel Electrophilic Thiol-Specific Carbohydrate Reagents. Carbohydr. Res 2006, 341, 2026–2036. [Google Scholar]
- Hartweck, LM; Scott, CL; Olszewski, NE. Two O-Linked N-Acetylglucosamine Transferase Genes of Arabidopsis thaliana L. Heynh. Have Overlapping Functions Necessary for Gamete and Seed Development. Genetics 2002, 161, 1279–1291. [Google Scholar]
- Holt, GD; Snow, CM; Senior, A; Haltiwanger, RS; Gerace, L; Hart, GW. Nuclear Pore Complex Glycoproteins Contain Cytoplasmically Disposed O-Linked N-Acetylglucosamine. J. Cell Biol 1987, 104, 1157–1164. [Google Scholar]
- Schlesinger, PH; Rodman, JS; Doebber, TW; Stahl, PD; Lee, YC; Stowell, CP; Kuhlenschmidt, TB. The Role of Extra-hepatic Tissues in the Receptor-mediated Plasma Clearance of Glycoproteins Terminated by Mannose or N-Acetylglucosamine. Biochem. J 1980, 192, 597–606. [Google Scholar]
- Lord, JM. Precursors of Ricin and Ricinus communis Agglutinin. Eur. J. Biochem 1985, 146, 411–416. [Google Scholar]
- Liu, CL; Tsai, CC; Lin, SC; Wang, LI; Hsu, CI; Hwang, MJ; Lin, JY. Primary Structure and Function Analysis of the Abrus precatorius Agglutinin A Chain by Site-directed Mutagenesis. J. Biol. Chem 2000, 257, 1897–1901. [Google Scholar]
- Wu, AM; Wu, JH; Liu, JH; Chen, YY; Singha, B; Chow, LP; Lin, JY. Roles of Mammalian Structural Units, Ligand Cluster and Polyvalency in the Abrus precatorius Agglutinin and Glycoprotein Recognition Process. Mol. Immunol 2009, 16, 3427–3437. [Google Scholar]
- Spellman, MW; Basa, LJ; Leonard, CK; Chakel, JA; O'Connor, JV; Wilson, S; van Halbeek, H. Carbohydrate Structures of Human Tissue Plasminogen Activator Expressed in Chinese Hamster Ovary Cells. J. Biol. Chem 1989, 264, 14100–14111. [Google Scholar]
- Houpt, JB; McMillan, R; Wein, C; Paget-Dellio, SD. Effect of Glucosamine Hydrochloride in the Treatment of Pain of Osteoarthritis of the Knee. J. Rheumatol 1999, 26, 2423–243. [Google Scholar]
- Breidenbach, MA; Gallagher, JE; King, DS; Smart, BP; Wu, P; Bertozzi, CR. Targeted Metabolic Labeling of Yeast N-Glycans with Unnatural Sugars. Proc. Natl. Acad. Sci. USA 2010, 107, 3988–3993. [Google Scholar]
- Shen, CR; Juang, JH; Tsai, ZT; Wu, ST; Tsai, FY; Wang, JJ; Liu, CL; Yen, TC. Preparation, Characterization and Application of Superparamagnetic Iron Oxide Encapsulated with N-[(2-Hydroxy-3-trimethylammonium) propyl] Chitosan Chloride. Carbohydr. Polym 2010. [Google Scholar] [CrossRef]
- Levin, RM; Krieger, NN; Winzler, RJ. Glucosamine and Acetylglucosamine Tolerance in Man. J. Lab. Clin. Med 1961, 58, 927–932. [Google Scholar]
- Liu, Y; Li, Z; Liu, G; Jia, J; Li, S; Yu, C. Liquid Chromatography–Tandem Mass Spectrometry Method for Determination of N-Acetylglucosamine Concentration in Human Plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci 2008, 862, 150–154. [Google Scholar]
- Kobata, A; Ginsburg, V. Oligosaccharides of Human Milk: I. Isolation and Characterization. Arch. Biochem. Biophys 1969, 130, 509–513. [Google Scholar]
- Miller, JB; Bull, S; Miller, J; McVeagh, P. The Oligosaccharide Composition of Human Milk: Temporal and Individual Variations in Monosaccharide Components. J. Pediatri. Gastroenterol. Nutr 1994, 19, 371–376. [Google Scholar]
- Bakkers, J. An Important Developmental Role for Oligosaccharides during Early Embryogenesis of Cyprinid Fish. Proc. Natl. Acad. Sci. USA 1997, 94, 7982–7986. [Google Scholar]
- Tharanathan, RN; Kittur, FS. Chitin—the Undisputed Biomolecule of Great Potential. Crit. Rev. Food Sci. Nutr 2003, 43, 61–87. [Google Scholar]
- Howard, MB; Ekborg, NA; Weiner, RM; Hutcheson, SW. Detection and Characterization of Chitinases and Other Chitin-modifying Enzymes. J. Ind. Microbiol. Biotechnol 2003, 30, 627–635. [Google Scholar]
- Kurita, K. Chitin and Chitosan: Functional Biopolymers from Marine Crustaceans. Mar. Biotechnol 2006, 8, 203–226. [Google Scholar]
- Bohlman, JA; Schisler, DO; Hwang, KO; Hennling, JP; Trinkle, JR; Anderson, TB; Steinke, JD; Vanderhoff, A. N-Acetyl-d-glucosamine and Process for Producing N-Acetyl-d-glucosamine. US Patent 6693188B2, 2004. [Google Scholar]
- Sashiwa, H; Fujishima, S; Yamano, N; Kawasaki, N; Nakayama, A; Muraki, E; Aiba, S. Production of N-Acetyl-d-glucosamine from β-Chitin by Enzymatic Hydrolysis. Chem. Lett 2001, 31, 308–309. [Google Scholar]
- Roseman, S; Ludoweig, J. N-Acetylation of Hexosamines. J. Am. Chem. Soc 1954, 76, 301–302. [Google Scholar]
- Kohn, P; Winzler, RJ; Hoffmann, RC. Metabolism of d-Glucosamine and N-Acetyl-d-glucosamine in the Intact Rat. J. Biol. Chem 1962, 237, 304–308. [Google Scholar]
- Zhan, WS. Process for Preparing Refined N-Acetyl-d-aminoglucose. CN Patent 1907993, 2007. [Google Scholar]
- Ryosuke, K; Yoshiharu, M; Kazuaki, K; Kazuo, S. Production of Natural-type N-Acetyl-d-glucosamine. JP Patent 200281696, 2002. [Google Scholar]
- Seo, S; King, JM; Prinyawiwatkul, W. Simultaneous Depolymerization and Decolorization of Chitosan by Ozone Treatment. J. Food Sci 2007, 72, C522–C526. [Google Scholar]
- Gooday, BW. Biosynthesis of the Fungal Wall—Mechanisms and Implications. The First Fleming Lecture. J. Gen. Microbiol 1977, 99, 1–11. [Google Scholar]
- Fuchs, RL; McPherson, SA; Drahos, DJ. Cloning of a Serratia marcescens Gene Encoding Chitinase. Appl. Environ. Microbiol 1986, 51, 504–509. [Google Scholar]
- Abeles, FB; Bosshart, RP; Forrence, LE; Habig, WH. Preparation and Purification of Glucanase and Chitinase from Bean Leaves. Plant Physiol 1971, 47, 129–134. [Google Scholar]
- Cohen-Kupiec, R; Chet, I. The Molecular Biology of Chitin Digestion. Curr. Opin. Biotechnol 1998, 9, 270–277. [Google Scholar]
- Zhu, Z; Zheng, T; Homer, RJ; Kim, YK; Chen, NY; Cohn, L; Hamid, Q; Elias, JA. Acidic Mammalian Chitinase in Asthmatic Th2 Inflammation and IL-13 Pathway Activation. Science 2004, 304, 1678–1682. [Google Scholar]
- Yang, CJ; Liu, YK; Liu, CL; Shen, CN; Kuo, ML; Su, CC; Tseng, CP; Yen, TC; Shen, CR. Inhibition of Acidic Mammalian Chitinase by RNA Interference Suppresses OVA-Sensitized Allergic Asthma. Hum. Gene Ther 2009, 20, 1597–1606. [Google Scholar]
- Cohen-Kupiec, R; Chet, I. The Molecular Biology of Chitin Digestion. Curr. Opin. Biotechnol 1998, 9, 270–277. [Google Scholar]
- Lee, EA; Pan, CH; Son, JM; Kim, SI. Isolation and Characterization of Basic Exochitinase from Leaf Extract of Rehmannia glutinosa. Biosci. Biotechnol. Biochem 1999, 63, 1781–1783. [Google Scholar]
- Mori, T; Ichikawa, W; Kita, Y; Tetsuka, Y. Method for Fermentative Production of N-Acetyl- d-glucosamine by Microorganism. US Patent 20100055746, 2010. [Google Scholar]
- Roberts, RL; Cabib, E. Serratia marcescens Chitinase: One-step Purification and Use for the Determination of Chitin. Anal. Biochem 1982, 127, 402–412. [Google Scholar]
- Oppenheim, AB; Chet, I. Cloned Chitinase in Fungal Plant—Pathogen Control Strategies. Trends Biotechnol 1992, 10, 392–394. [Google Scholar]
- Pan, CH; Rim, SL; Kim, SI. Expression of Two cDNAs Encoding Class I Chitinases of Rice in Escherichia coli. Biosci. Biotech. Biochem 1996, 60, 1346–1348. [Google Scholar]
- Yalpani, M; Pantaleone, D. An Examination of the Unusual Susceptibilities of Aminoglycans to Enzymatic Hydrolysis. Carbohydr. Res 1994, 256, 159–175. [Google Scholar]
- Muzarelli, RAA. Muzzarelli, RAA, Peter, MG, Eds.; Depolymerization of Chitins and Chitosans with Hemicellulase, Lysozyme, Papain and Lipase. In Chitin Handbook; Atec: Grottammare, Italy, 1997; pp. 153–163. [Google Scholar]
- Aiba, S; Muraki, E. Chen, RH, Chen, HC, Eds.; Preparation of Higher N-Acetylchitooligosaccharides in High Yields. In Advance in Chitin Science; Rita Advertising Co. Ltd: Taipei, Taiwan, 1999; Volume III, pp. 89–96. [Google Scholar]
- Sashiwa, H; Fujishima, S; Yamano, N; Kawasaki, N; Nakayama, A; Muraki, E; Hiraga, K; Oda, K; Aiba, S. Production of N-Acetyl-d-glucosamine from α-Chitin by Crude Enzymes from Aeromonas hydrophila H2330. Carbohydr. Res 2002, 337, 761–763. [Google Scholar]
- Pichyangkura, R; Kudan, S; Kuttiyawang, K; Sukwattanasinitt, M; Aiba, S. Quantitative Production of 2-Acetoamodo-2-d-glucose from Crystalline Chitin by Bacterial Chitinase. Carbohydr. Res 2002, 337, 557–559. [Google Scholar]
- Sashiwa, H; Fujishima, S; Yamano, N; Kawasaki, N; Nakayama, A; Muraki, E; Sukwattanasinitt, M; Pichyangkura, R; Aiba, S. Enzymatic Production of N-Acetyl-d-glucosamine from Chitin. Degradation Study of N-Acetylchitooligosaccharide and the Effect of Mixing of Crude Enzymes. Carbohydr. Polym 2003, 51, 391–395. [Google Scholar]
- Rinaudo, MA. Chitin and Chitosan: Properties and Applications. Prog. Polym. Sci 2006, 31, 603–632. [Google Scholar]
- Ilankovan, P; Hein, S; Ng, C; Trung, TS; Stevens, WF. Production of N-Acetyl Chitobiose from Various Chitin Substances Using Commercial Enzymes. Carbohydr. Polym 2006, 63, 245–250. [Google Scholar]
- Kurita, K; Kaji, Y; Mori, T; Nishiyama, Y. Enzymatic Degradation of β-Chitin: Susceptibility and the Influence of Deacetylation. Carbohydr. Polym 2003, 42, 19–21. [Google Scholar]
- Shen, CR; Chen, YS; Yang, CJ; Chen, JK; Liu, CL. Colloid Chitin Azure Is a Dispersible, Low-cost, Substrate for Chitinase Measurements in a Sensitive, Fast, Reproducible, Assay. J. Biomol. Screen 2010, 15, 213–217. [Google Scholar]
- Kawasaki, IK; Morita, T. Enzymatic Decomposition of Chitin Containing Materials. US Patent 5262310, 1993. [Google Scholar]
- Immanaka, T; Fukui, T; Fujiwara, S. Chitinase from Thermococcus kodakaraensis KOD1. Methods Enzymol 2001, 330, 319–329. [Google Scholar]
- Guo, SH; Chen, JK; Lee, WC. Purification and Characterization of Extracellular Chitinase from Aeromonas schubertii. Enzyme Microb. Technol 2004, 35, 550–556. [Google Scholar]
- Liu, CL; Shen, CR; Hsu, FF; Chen, JK; Wu, PT; Guo, SH; Lee, WC; Yu, FW; Mackey, ZB; Turk, J; Gross, ML. Isolation and Identification of Two Novel SDS-Resistant Secreted Chitinases from Aeromonas schubertii. Biotechnol. Prog 2009, 25, 124–134. [Google Scholar]
- Chen, MH; Chan, HY; Wu, CL; Chuang, SH; Hwang, IE; Chen, YL; Yuan, GF. Production of Chitin and Chitosan. US Patent 6255085 B1, 2001. [Google Scholar]
- Barnhill, JG; Fye, CL; Reda, DJ; Harris, CL; Clegg, DO. Is All Glucosamine Alike? Clarifying the Controversies for Product Selection and Clinical Research. J. Compl. Integr. Med 2009, 6, 17. [Google Scholar]
- Raetz, E; Leuba, JL; Di Giambattista, R; Federici, F; Fenice, M. Chitinolytic Enzymes Production by Penicillium janthinellum. EP Patent 0885954 A1, 1998. [Google Scholar]
- Louise, CA; Pedro, A; Charles, AH. Process for Producing N-Acetyl-d-glucosamine. US Patent 5998173, 1999. [Google Scholar]
- Li, YL; Wu, ST; Yu, ST; Too, JR. Screening of a Microbe to Degrade Chitin. Taiwanese J. Agric. Chem. Food Sci 2005, 43, 410–418, (Chinese). [Google Scholar]
- Chern, LL; Stackebrandt, E; Lee, SF; Lee, FL; Chen, JK; Fu, HM. Chitinibacter tainanensis Gen. nov. sp. nov., a Chitin-degrading Aerobe from Soil in Taiwan. Int. J. Syst. Evol. Microbiol 2004, 54, 1387–1391. [Google Scholar]
- Chen, JK; Shen, CR; Fang, BS; Huang, TL; Liu, CL. The N-Acetyl-glucosamine Obtained from Chitin with Chitinibacter tainanensis. Carbohydr. Polym 2010. submitted. [Google Scholar]
- Doi, RH; Kosugi, A; Murashima, K; Tamura, Y; Han, SO. Cellulosomes from Mesophilic Bacteria. J. Bacteriol 2003, 185, 5907–5914. [Google Scholar]
- Deng, MD; Angerer, JD; Cyron, D; Grund, AD; Jerrell, TA, Jr; Leanna, C; Mathre, O; Rossen, R; Running, J; Severson, D. Process and Materials for Producing of Glucosamine and N-Acetylglucosamine. KR Patent 20050053534, 2005. [Google Scholar]
- Yoon, SH; Rhee, JS. Lipid from Yeast Fermentation: Effects of Cultural Conditions on Lipid Production and Its Characteristics of Rhodotorula glutinis. J. Am. Oil Chem. Soc 1983, 60, 1281–1286. [Google Scholar]
- Hamme, JV; Singh, A; Ward, O. Physiological Aspects Part 1 in a Series of Papers Devoted to Surfactants in Microbiology and Biotechnology. Biotechnol. Adv 2006, 24, 604–620. [Google Scholar]
- Wendland, J; Schaub, Y; Walther, A. N-Acetylglucosamine Utilization by Saccharomyces cerevisiae Based on Expression of Candida albicans NAG Genes. Appl. Environ. Microbiol 2009, 75, 5840–5845. [Google Scholar]
- Vidal, Y; Plana, PR; Bizzari, D; Rovati, A. Articular Cartilage Pharmacology: I. In Vitro Studies on Glucosamine and Non-Steroidal Anti-inflammatory Drugs. Pharmacol. Res. Commun 1978, 10, 557–569. [Google Scholar]
- Heim, HK; Oestmann, A; Thiele, H; Sewing, KH. Incorporation of N-Acetyl-[14C]d-glucosamine and [3H]l-Leucine by Isolated Pig Gastric Mucosal Cells. Digestion 1989, 44, 26–35. [Google Scholar]
- Shoji, A; Iga, T; Inagaki, S; Kobayashi, K; Matahira, Y; Sakai, K. Metabolic Disposition of [14C] N-Acetylglucosamine in Rats. Chitin Chitosan Res 1999, 5, 34–42. [Google Scholar]
- Lee, KY; Shibutani, M; Takagi, H; Arimura, T; Takigami, S; Uneyama, C; Kato, N; Hirose, M. Subchronic Toxicity Study of Dietary N-Acetylglucosamine in F344 Rats. Food Chem. Toxicol 2004, 42, 687–695. [Google Scholar]
- Xu, Q; Liu, J. Use of N-Acetyl d-glucosamine in Treatment of Organ Lesion Related to Toxicosis of Drugs or Chemicals. US Patent 2006281707, 2006. [Google Scholar]
- Felson, DT. Epidemiology of Hip and Knee Osteoarthritis. Epidemiol. Rev 1988, 10, 1–28. [Google Scholar]
- Creamer, P. Osteoarthritis Pain and Its Treatment. Curr. Opin. Rheumatol 2000, 12, 450–455. [Google Scholar]
- Rovati, L; Casula, P; Mascherpa, S. N-Acetylglucosamine for Treating Degenerative Afflictions of the Joints. US Patent 3697652, 1972. [Google Scholar]
- Talent, JM; Gracy, RW. Pilot Study of Oral Polymeric N-Acetyl-d-glucosamine as a Potential Treatment for Patients with Osteoarthritis. Clin. Ther 1996, 18, 1184–1190. [Google Scholar]
- Chrisope, GL; Rose, R. Product and Method for Treating Joint Disorders in Vertebrates. US Patent 6344220, 1999. [Google Scholar]
- Tamai, Y; Miyatake, K; Okamoto, Y; Takamori, Y; Sakamoto, K; Minami, S. Enhanced Healing of Cartilaginous Injuries by N-Acetyl-d-glucosamine and Glucuronic Acid. Carbohydr. Polym 2003, 54, 251–262. [Google Scholar]
- Wilkie, D. Topical Composition for the Treatment of Joint Damage or Pain. GB Patent 2403405, 2005. [Google Scholar]
- Marcum, FD; Seanor, JW. Composition and Method for Treating Rheumatoid Arthritis. US Patent 2008003258, 2007. [Google Scholar]
- Kamel, M; Hanafi, N; Bassiouni, M. Inhibition of Elastase Enzyme Release from Human Polymorphonuclear Leukocytes by N-Acetylgalactosamine and N-Acetylglucosamine. Clin. Exp. Rheumatol 1991, 9, 17–21. [Google Scholar]
- Kamel, M; Alnahdi, M. Inhibition of Superoxide Anion Release from Human Polymorphonuclear Leukocytes by N-Acetylgalactosamine and N-Acetylglucosamine. Clin. Rheumatol 1992, 11, 254–260. [Google Scholar]
- Murch, SH; Braegger, CP; Walker-Smith, JA; McDonald, TT. Localization of Tumor Necrosis Factor α by Immunohistochemistry in Chronic Inflammatory Bowel Disease. Gut 1993, 34, 1705–1709. [Google Scholar]
- Kanazawa, M; Fukudo, S. Effects of Fasting Therapy on Irritable Bowel Syndrome. Int. J. Behav. Med 2006, 13, 214–220. [Google Scholar]
- Monnikes, H; Schmidtmann, M; van der Voort, IR. Drug Therapy for Irritable Bowel Syndrome. What Works, What Doesn't Work and for Whom? Internist (Berl) 2006, 47. [Google Scholar]
- Burton, AF; Gislason, S. Methods and Composition for Treatment of Food Allergy. US Patent 5192750, 1993. [Google Scholar]
- Burton, AF; Mclean, DI. Method and Composition for Treating Psoriasis. US Patent 5217962, 1993. [Google Scholar]
- Karzed, K; Domenjoz, R. Effects of Hexosamine Derivatives and Uronic Acid Derivatives on Glycosaminoglycan Metabolism of Fibroblast Cultures. Pharmocology 1971, 5, 337–345. [Google Scholar]
- Burtan, AF. N-Acetyl Glucosamine as a Cytoprotective Agent. KR Patent 0145715 B, 1998. [Google Scholar]
- Burtan, AF; Freeman, HJ. N-Acetyl Glucosamine as a Gastroprotective Agent. WO Patent 9323055, 1993. [Google Scholar]
- Salvatore, S; Heuschkel, R; Tomlin, S; Davies, SE; Edwards, S; Walker-Smith, JA; French, I; Murch, SH. A Pilot Study of N-Acetyl Glucosamine, a Nutritional Substrate for Glycosaminoglycan Synthesis, in Paediatric Chronic Inflammatory Bowel Disease. Aliment. Pharmacol. Ther 2000, 14, 1567–1579. [Google Scholar]
- Wertz, PW; van der Bergh, B. The Physical, Chemical and Functional Properties of Lipid in the Skin and Other Biological Barrier. Chem. Phys. Lipids 1998, 91, 85–96. [Google Scholar]
- Laurent, TC; Fraser, JR. Hyaluronan. FASEB J 1992, 6, 2397–2404. [Google Scholar]
- Ghersetich, I; Lotti, T; Campanile, G; Grappone, C; Dini, G. Hyaluronic Acid in Cutaneous Intrinsic Agent. Int. J. Dermatol 1994, 33, 119–122. [Google Scholar]
- Sayo, T; Sakai, S; Inoue, S. Synergestic Effect of N-Acetylglucosamine and Retinoids on Hyaluronan Production in Human Keratinocytes. Skin Pharmacol. Physiol 2004, 17, 77–83. [Google Scholar]
- Chen, RH; Hsu, CN; Chung, MY; Tsai, WL; Liu, CH. Effect of Different Concentrations of Collagen, Ceramides, N-Acetyl Glucosamine, or Their Mixture on Enhancing the Proliferation of Keratinocytes, Fibroblasts and the Secretion of Collagen and/or the Expression of mRNA of Type I Collagen. J. Food Drug Anal 2008, 16, 66–74. [Google Scholar]
- Bissett, D; Robinson, LR; Raleigh, PS; Miyamoto, K; Hakozaki, T; Li, J; Klem, GR. Reduction in the Appearance of Facial Hyperpigmentation by Topical N-Acetyl Glucosamine. J. Cosmet. Dermatol 2007, 6, 20–26. [Google Scholar]
- Bissett, D; Farmer, T; McPhail, S; Reichling, T; Tiesman, JP; Juhlim, KD; Hurley, GJ; Robinson, MK. Genomic Expression Changes Induced by Topical N-Acetylglucosamine in Skin Equivalent Cultures in Vitro. J. Cosmet. Dermatol 2007, 6, 232–238. [Google Scholar]
- Riordan, NH. Skin Treatment System. US Patent 5866142, 1999. [Google Scholar]
- Hwang, JI; Kim, KS. Cosmetics Composition Comprising Extract of Natural Materials for Improving Acne and Skin Wrinkles and Whitening Skin. KR Patent 20050004355 A, 2005. [Google Scholar]
- Minami, S; Okamoto, Y. Drug for Remedy or Treatment of Wound. EP Patent 1749532, 2007. [Google Scholar]
- Flessner, MF; Lofthouse, J; Williams, A. Chronic Alteration of Sub-Peritoneal Tissue and Peritoneal Transport. Adv. Perit. Dial 2002, 18, 12–14. [Google Scholar]
- Vimr, E; Lichtensteiger, C. To Sialylate, or not to Sialylate: That Is the Question. Trends Microbiol 2002, 10, 254–257. [Google Scholar]
- Tanner, ME. The Enzymes of Sialic Acid Biosynthesis. Bioorg. Chem 2005, 33, 216–228. [Google Scholar]
- Maru, I; Ohnishi, J; Ohta, Y; Tsukada, Y. Why Is Sialic Acid Attracting Interest Now? Complete Enzymatic Synthesis of Sialic Acid with N-Acetylglucosamine 2-Epimerase. J. Biosci. Bioeng 2002, 93, 258–265. [Google Scholar]
- Gubareva, LV; Kaiser, L; Hayden, FG. Influenza Virus Neuraminidase Inhibitors. Lancet 2000, 827–835. [Google Scholar]
- Tabata, K; Koizumi, S; Endo, T; Ozaki, A. Production of N-Acetyl-d-neuraminic Acid by Coupling Bacteria Expressing N-Acetyl-d-glucosamine 2-Epimerase and N-Acetyl-d-neuraminic Acid Synthetase. Enzyme Microb. Technol 2002, 30, 327–333. [Google Scholar]
- Maru, I; Ohnishi, J; Ohta, Y; Tsukada, T. Simple and Large-Scale Production of N-Acetyl-neuraminic Acid from N-Acetyl-d-glucosamine and Pyruvate Using N-Acetyl-d-glucosamine 2- Epimerase and N-Acetyl-neuraminic Acid Lyase. Carbohydr. Res 1998, 306, 575–578. [Google Scholar]
- Lee, YC; Chien, HR; Hsu, WH. Production of N-Acetyl-neuraminic Acid by Recombinant Whole Cells Expressing Anabaena sp. CH1 N-Acetyl-d-glucosamine 2-epimerase and Escherichia coli N-Acetyl-neuraminic Acid Lyase. J. Biotechnol 2007, 129, 453–460. [Google Scholar]
- Rauprich, O; Matsushita, M; Weijer, CJ; Siegert, F; Esipov, SE; Shapiro, JA. Periodic Phenomena in Proteus mirabilis Swarm Colony Development. J. Bacteriol 1996, 178, 6525–6538. [Google Scholar]
- Xu, Q; Liu, J; Yuan, Z. Use of N-Acetyl-d-glucosamine in Preparation of Drugs for the Treatment of Cancer and Metastasis. EP Patent 1666046 A1, 2006. [Google Scholar]
- Xu, Q; Liu, J; Yuan, Z. Use of N-Acetyl-d-glucosamine in Treatment of Local Lesions or Systematic Symptoms Related to Autoimmune Reactions. EP Patent 1611896 A1, 2006. [Google Scholar]
- Xu, Q; Liu, J; Yuan, Z. Use of N-Acetyl-d-glucosamine for Preparing Medicines for Urogenital Tract Infection’s Treatment and Prevention. US Patent 2006/0142243 A1, 2006. [Google Scholar]
- Xu, Q; Liu, J; Yuan, Z. Use of N-Acetyl-d-glucosamine in Treatment of Local Lesions and Systematic Symptoms Related to Infections of Virus and Bacteria. US Patent 2007/0042995 A1, 2007. [Google Scholar]
- Xu, Q; Liu, J; Yuan, Z. Use of N-Acetyl-d-glucosamine in the Manufacture of Pharmaceutical Useful for Preventing and Treating Sexual Disorder. US Patent 7015207 B2, 2006. [Google Scholar]
- Xu, Q; Liu, J; Yuan, Z. Use of N-Acetyl-d-glucosamine in Treatment of Non-Specific Infections Related to Physical or Chemical Factors. EP Patent 1611894, 2006. [Google Scholar]
- Xu, Q; Liu, J; Yuan, Z. Use of N-Acetyl-d-glucosamine in the Manufacture of Pharmaceutical Useful for Adjuvant Treatment of Perianal Disease. US Patent 2005119224, 2005. [Google Scholar]
- Xu, Q; Liu, J; Yuan, Z. Compound Antibacterial Drugs Comprising N-Acetyl-d-glucosamine. US Patent 20070191291, 2007. [Google Scholar]
- Xu, Q; Liu, J; Yuan, Z. Use of N-Acetyl-d-glucosamine in the Manufacture of Pharmaceutical Useful for Treating Cervical Erosion. US Patent 2004138174 A1, 2004. [Google Scholar]
- Xu, Q. Application of N-Aceto-d-aminoglucose in Medicinal Preparation for Curing Respiratory Tract Disease. CN Patent 1156026, 1997. [Google Scholar]
- Xu, Q; Liu, J. Use of N-Acetyl-d-glucosamine in Treatment of Organ Lesions Related to Toxicosis of Drugs or Chemicals. US Patent 7345030 B2, 2008. [Google Scholar]
- Xu, Q; Liu, J; Yuan, Z. Antibacterial Compositions of N-Acetyl-d-aminoglycosamine and Antibiotics. EP Patent 1669077, 2006. [Google Scholar]
- Xu, Q; Liu, J; Yuan, Z. Use of N-Acetyl-d-glucosamine in the Manufacture of Pharmaceutical Useful for Suppress Side Effect of Radiotherapy and Chemotherapy. US Patent 7037904 B2, 2006. [Google Scholar]
- Xu, Q; Liu, J; Yuan, Z. Use of N-Acetyl-d-glucosamine in the Manufacture of Pharmaceutical Useful for Treating Cardio-Cerebrovascular Anoxemia. US Patent 7074774 B2, 2006. [Google Scholar]
- Xu, Q; Liu, J; Yuan, Z. Use of N-Acetyl-d-glucosamine in the Manufacture of Pharmaceutical Useful for Treating Motion Sickness. US Patent 6946452 B2, 2005. [Google Scholar]
- Xu, Q; Yuan, Z; Liu, J. Use of N-Acetyl-d-glucosamine in Preparation of Drugs for Modulating Microorganisms on Mucus Membrane. CA Patent 2539286 A1, 2005. [Google Scholar]
- Xu, Q. Application of N-Acetyl-d-aminoglucose for Preparing Skin Sanitary Article Preparation. CN Patent 1156028, 1997. [Google Scholar]
- Xu, Q; Liu, J; Yuan, Z. Use of N-Acetyl-d-glucosamine in Preparation of Skin Membrane Mucosa Microecological Balance Regulator Medicines. CN Patent 1470244, 2004. [Google Scholar]
- Xu, Q; Liu, J; Yuan, Z. The Use of N-Acetyl Glucosamine as Additive in Milk Products. WO Patent 2004093556, 2004. [Google Scholar]
- Xu, Q; Liu, J; Yuan, Z. The Use of N-Acetyl Glucosamine as Additive in Beer and Beer Comprising N-Acetyl Glucosamine. WO Patent 2004085603, 2004. [Google Scholar]
- Xu, Q; Liu, J; Yuan, Z. The Use of N-Acetyl Glucosamine as Additive in Wine and Wine Comprising N-Acetyl Glucosamine. WO Patent 2004085604, 2004. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Function | Object | Trial | Reference |
|---|---|---|---|
| Disease Treatment | cancer and metastasis | animal, human | [138] |
| autoimmune reactions | cell, animal, human | [139] | |
| urogenital tract infection | cell, human | [140] | |
| viral or bacterial infection | cell, animal | [141] | |
| sexual disorder | human | [142] | |
| non-specific inflammation | animal, human | [143] | |
| perianal disease | cell, animal, human | [144] | |
| intestinal disease | human | [145] | |
| cervical erosion | bacteria, human | [146] | |
| respiratory tract disease | human | [147] | |
| Disease Prevention | toxicosis of drugs or chemicals | cell, animal | [148] |
| antibiotics | bacteria | [149] | |
| side effects of radiotherapy and chemotherapy | human | [150] | |
| cardio-cerebrovascular anoxemia | animal | [151] | |
| motion sickness | human | [152] | |
| Dermatology | modulation of microorganisms on mucus membrane | animal, human | [153] |
| skin sanitary article preparation | human | [154] | |
| microecological balance of mucosa | bacteria, human | [155] | |
| Food supplement | additive in milk | animal, human | [156] |
| additive in beer | animal, human | [157] | |
| additive in wine | animal, human | [158] | |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, J.-K.; Shen, C.-R.; Liu, C.-L. N-Acetylglucosamine: Production and Applications. Mar. Drugs 2010, 8, 2493-2516. https://doi.org/10.3390/md8092493
Chen J-K, Shen C-R, Liu C-L. N-Acetylglucosamine: Production and Applications. Marine Drugs. 2010; 8(9):2493-2516. https://doi.org/10.3390/md8092493
Chicago/Turabian StyleChen, Jeen-Kuan, Chia-Rui Shen, and Chao-Lin Liu. 2010. "N-Acetylglucosamine: Production and Applications" Marine Drugs 8, no. 9: 2493-2516. https://doi.org/10.3390/md8092493