New One-Pot Methodologies for the Modification or Synthesis of Alkaloid Scaffolds

Abstract
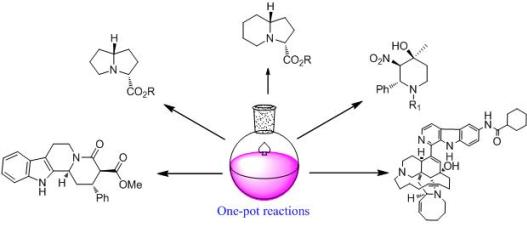
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
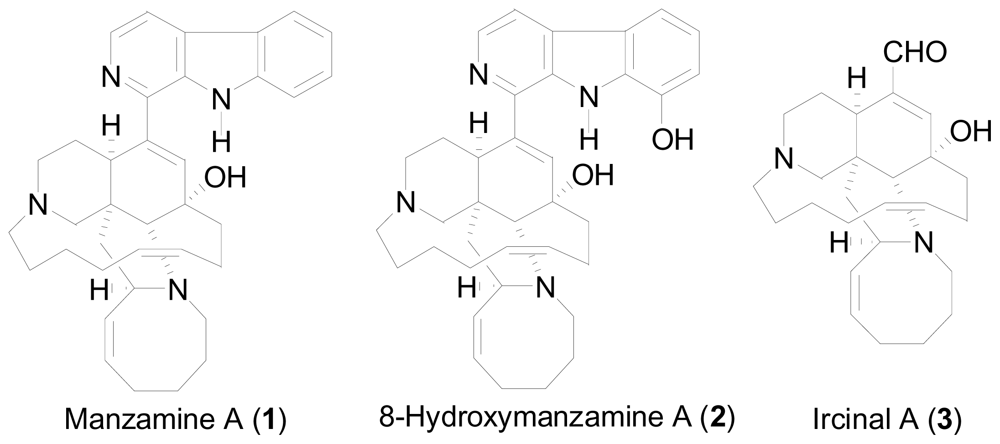
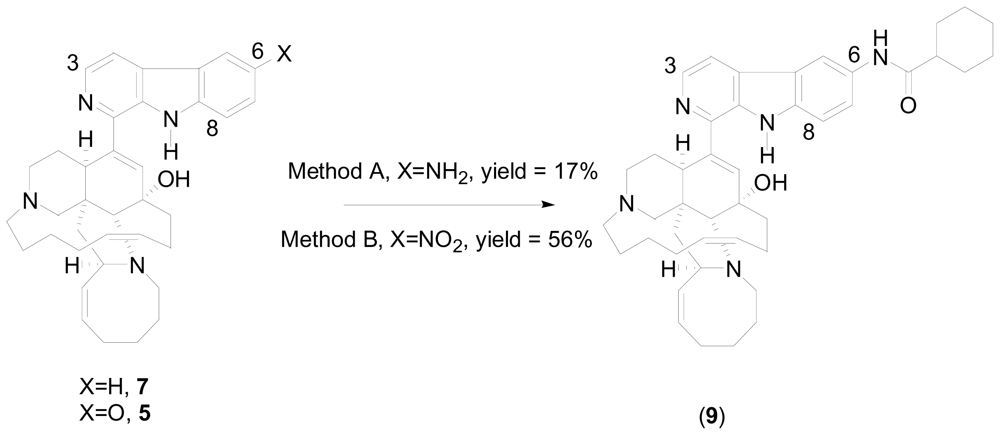
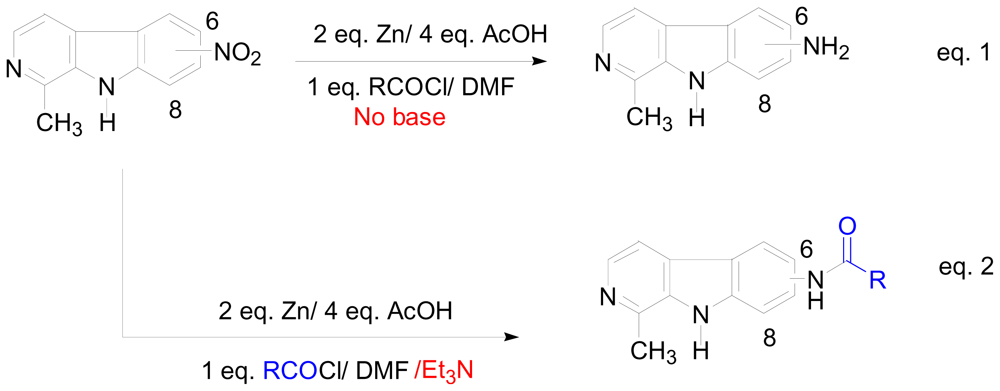
2. New One-Pot Methods Used in the Modification of Marine Alkaloids
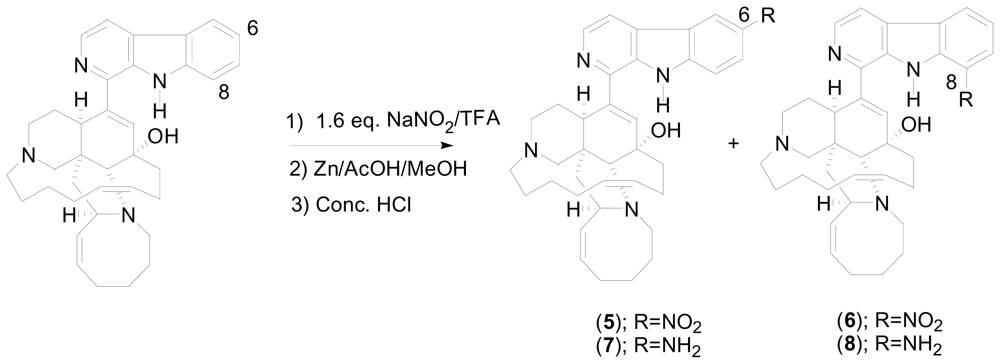
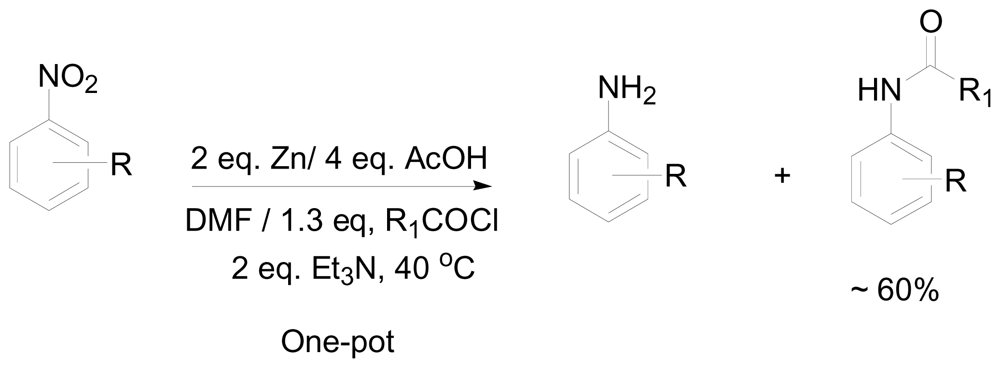
One-pot reductive amidation of nitroarenes
3. New One-Pot Reactions Used in the Asymmetric Construction of Some Important Alkaloid Moieties
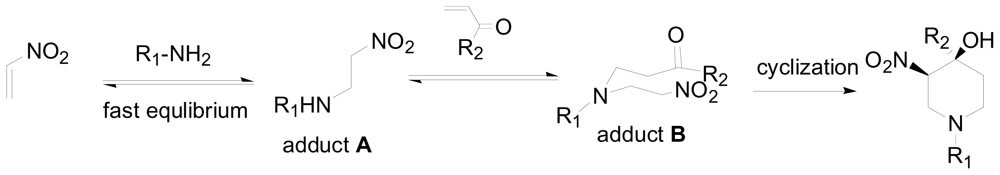
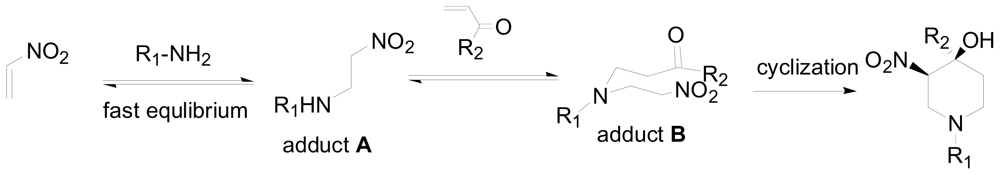
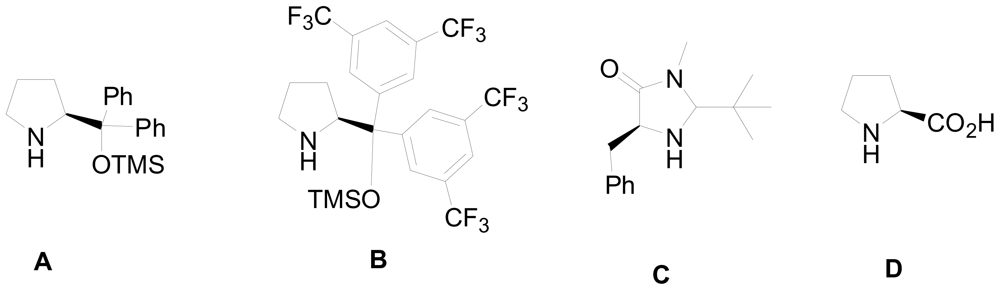
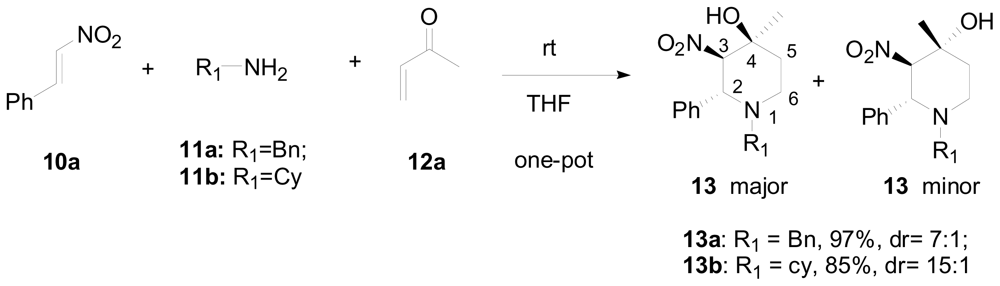
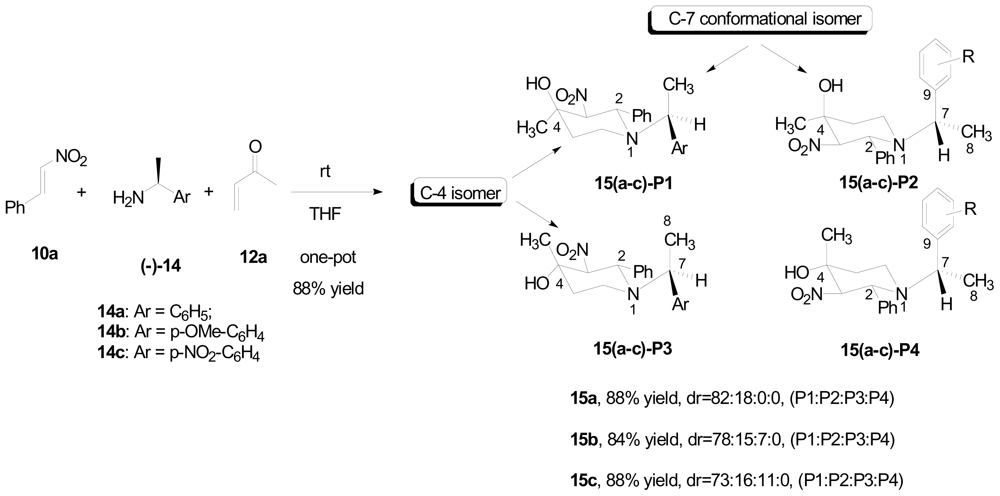
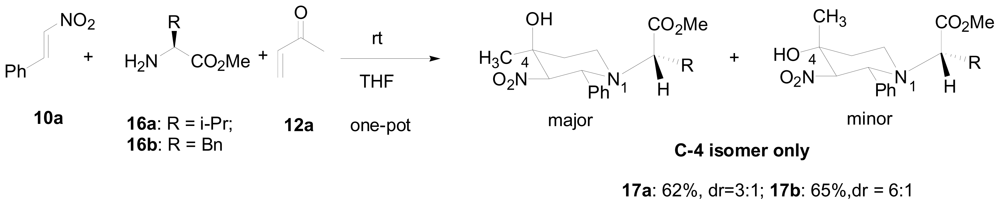
3.1. One-pot asymmetric synthesis of substituted piperidines
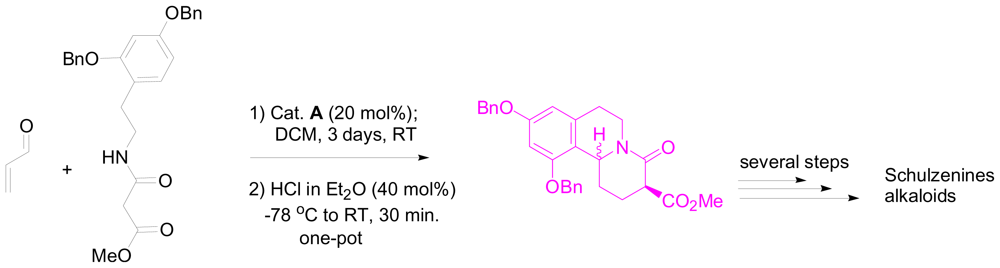
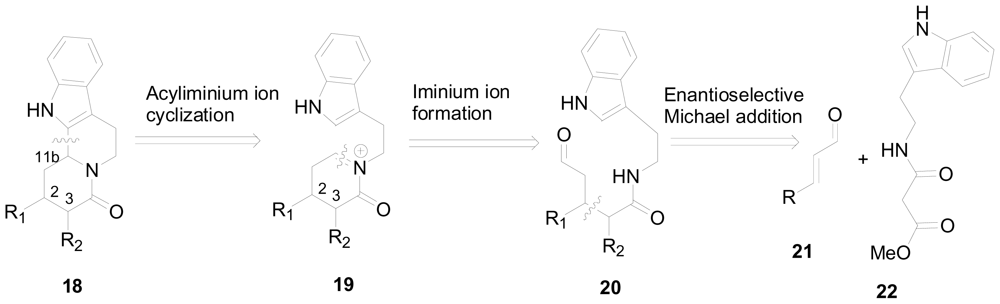
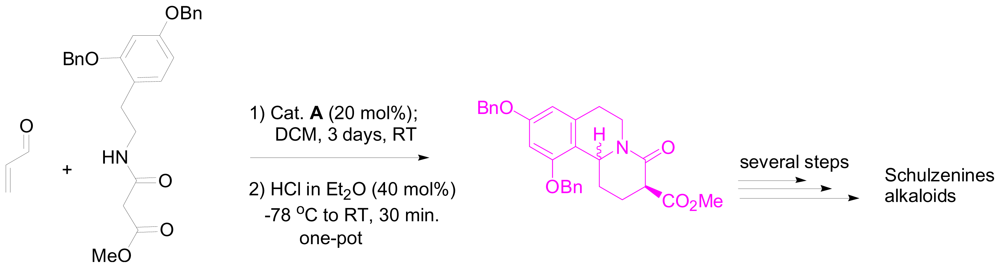
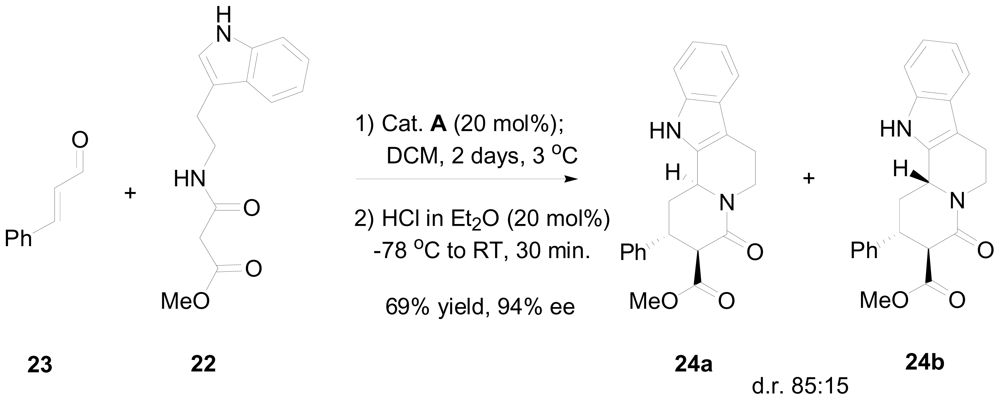
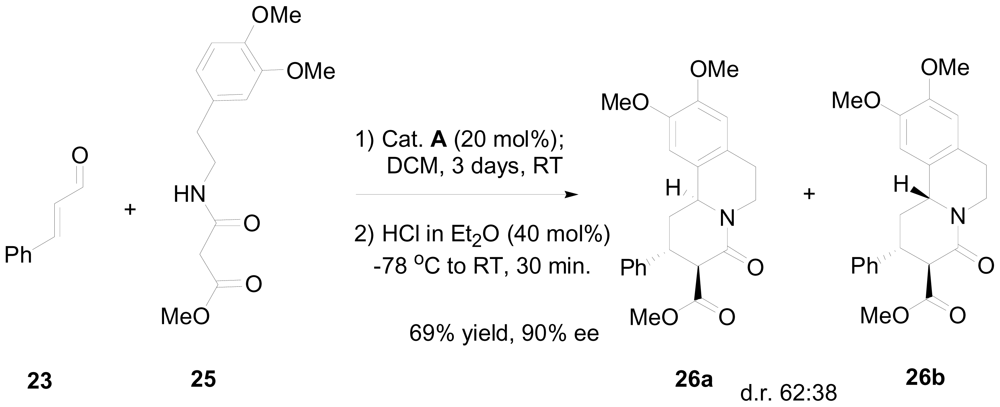
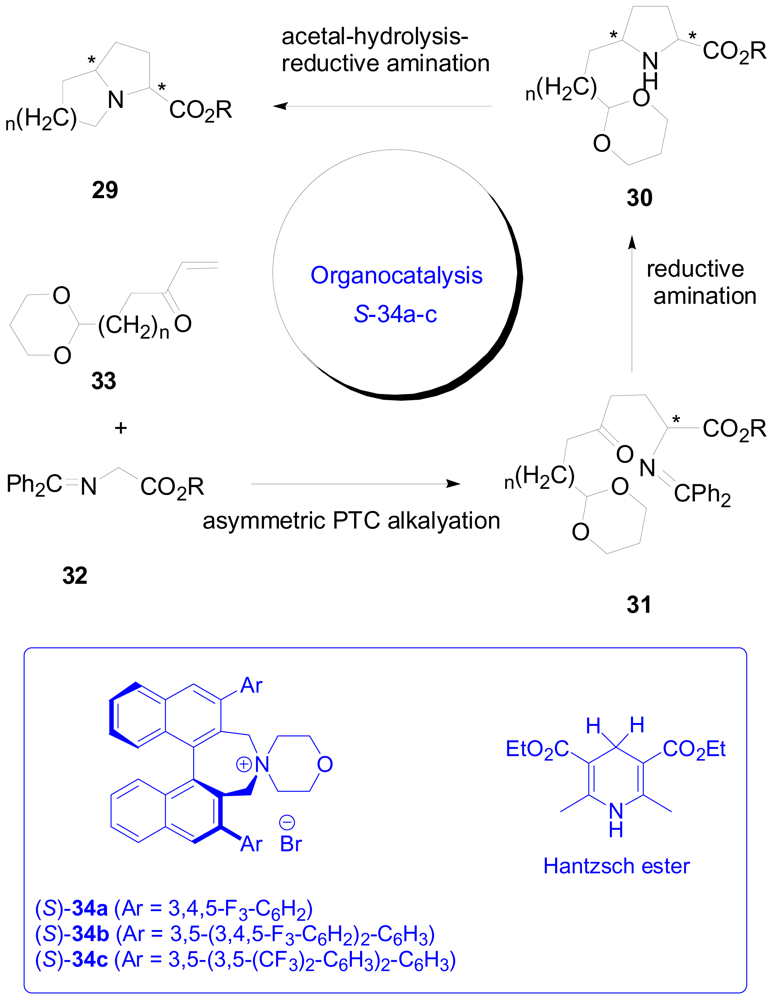
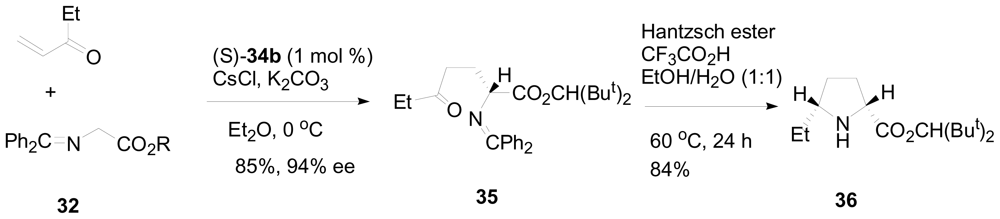
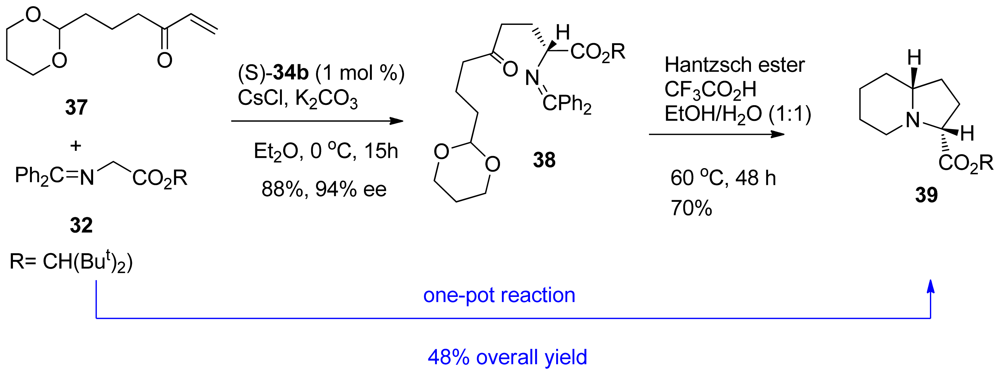
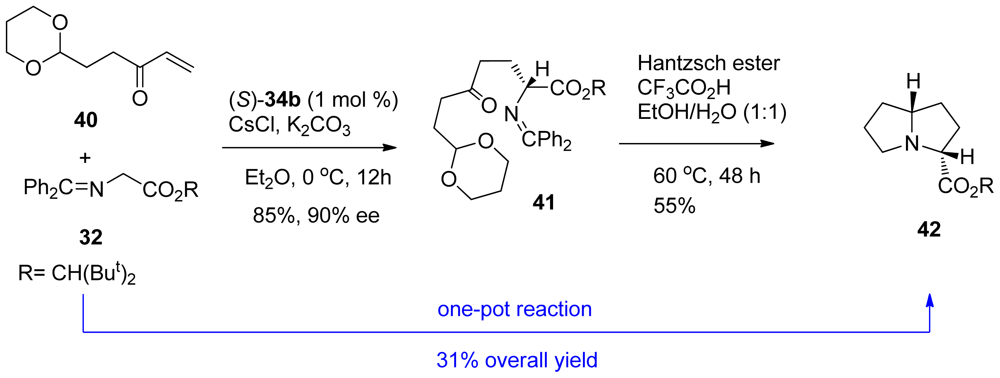
3.2. One-pot organo-catalytic synthesis of quinolizidine derivatives
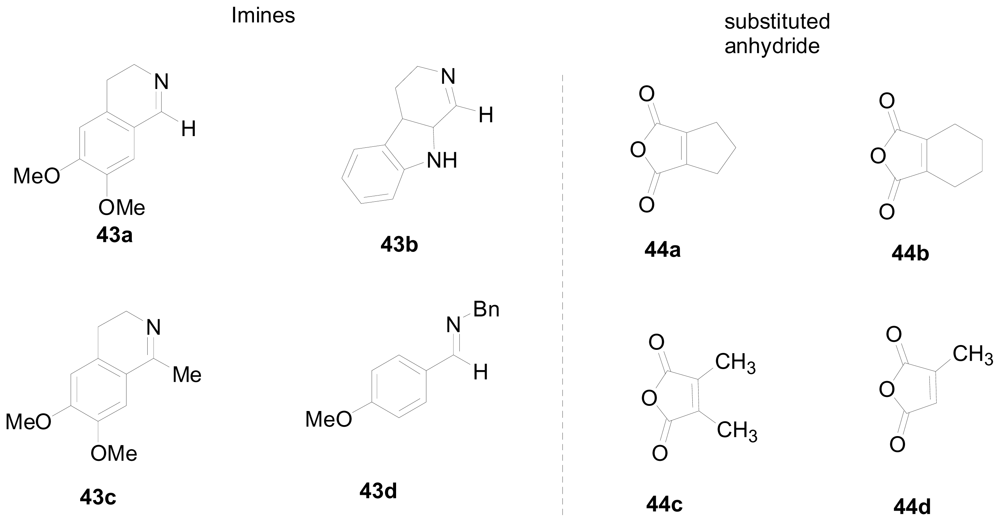
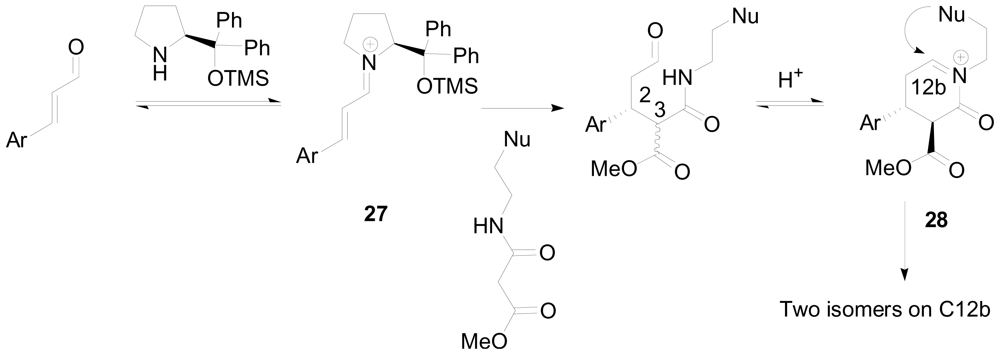
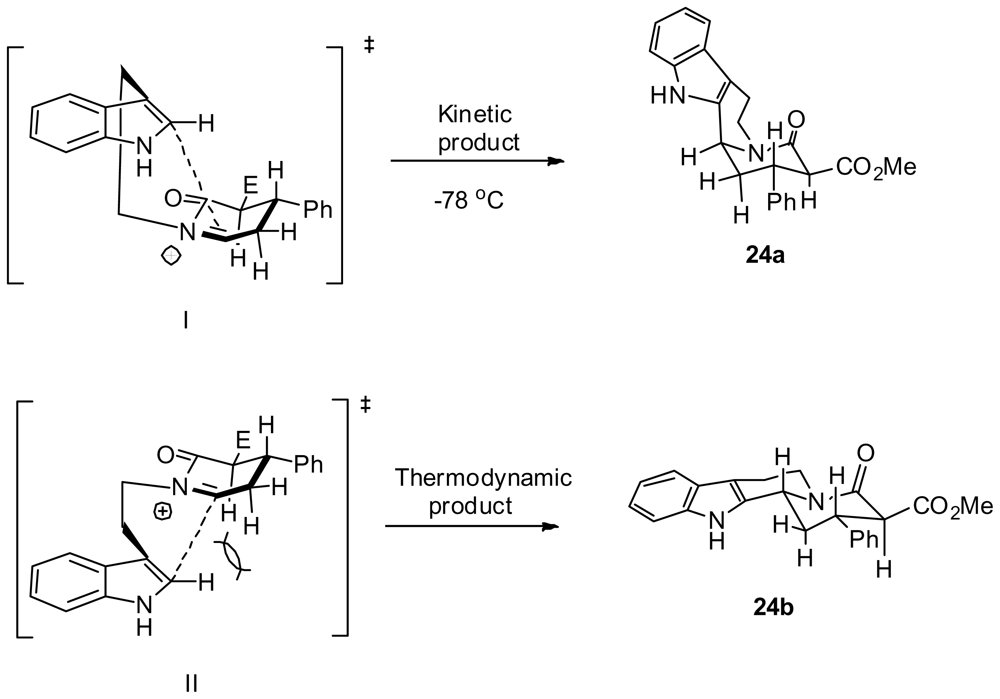
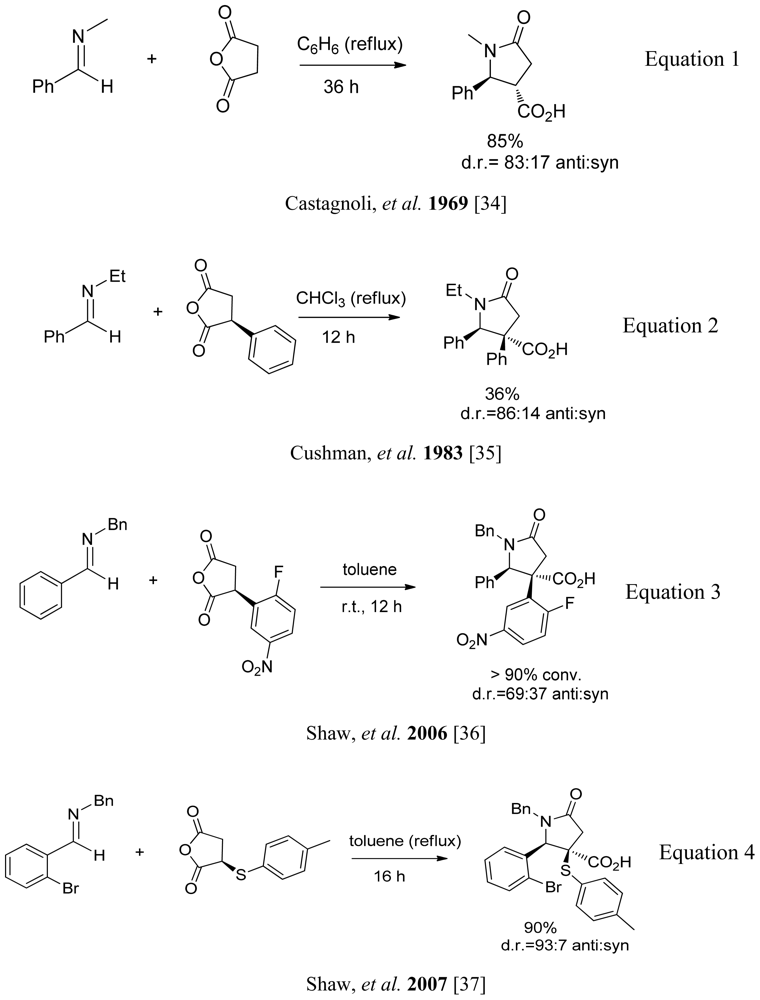
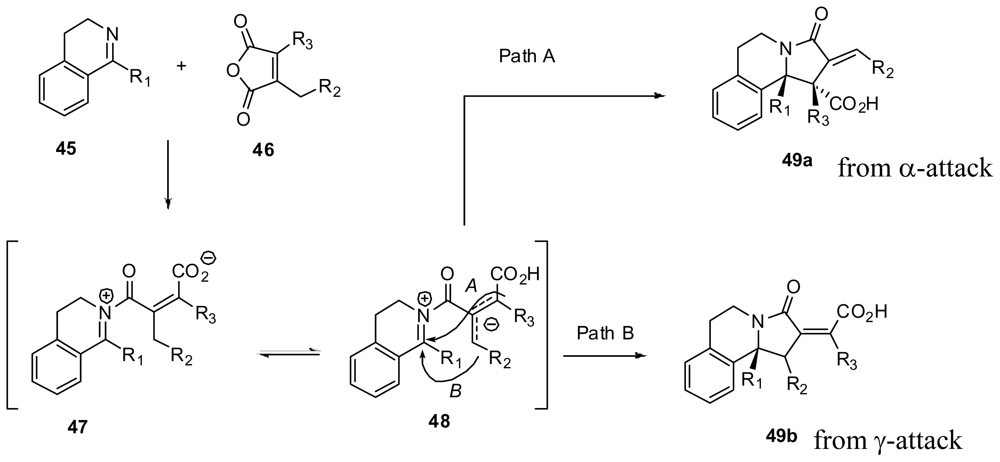
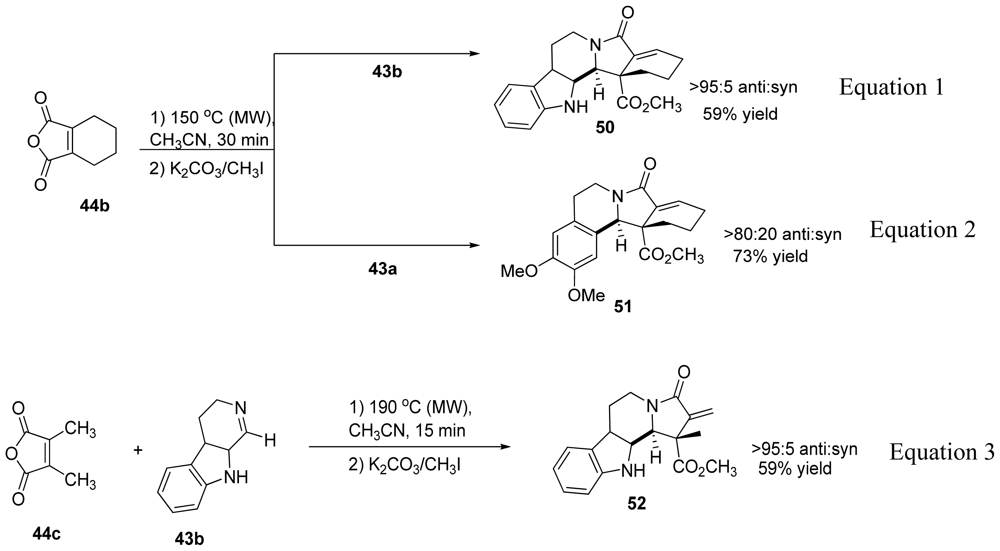
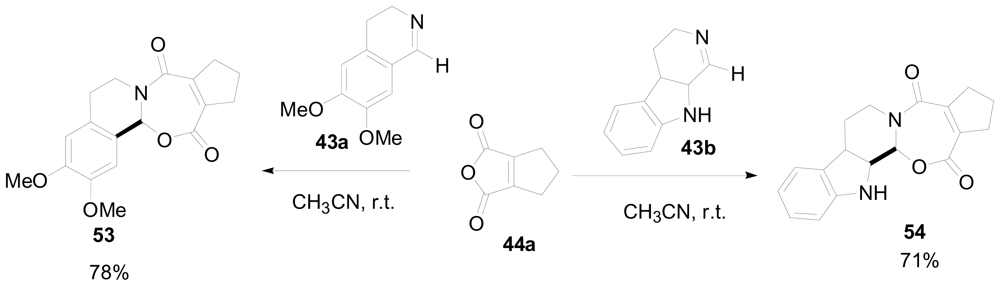
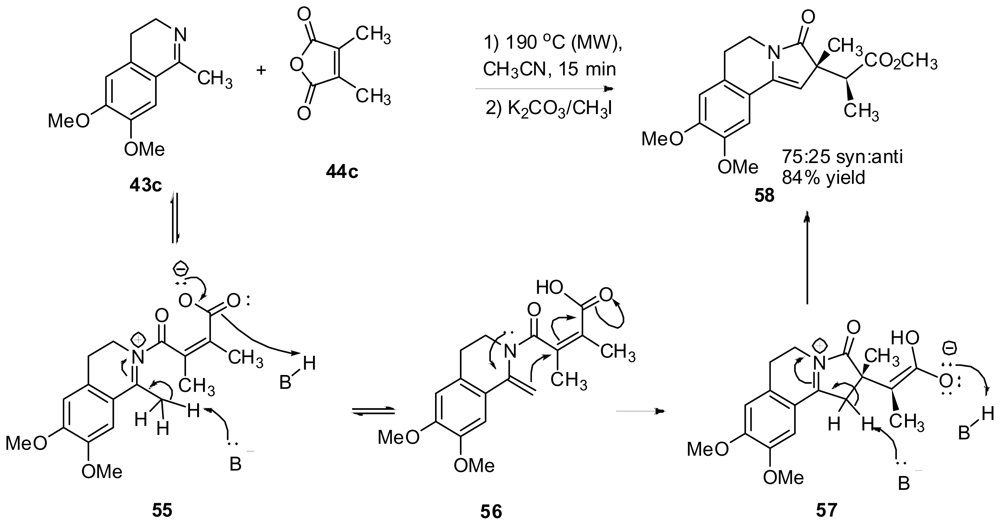
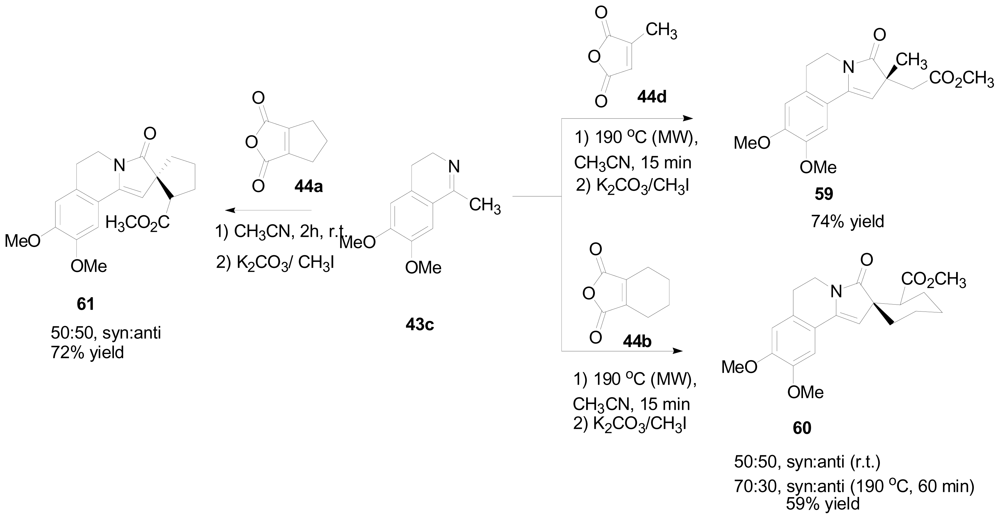
3.4. One-pot diastereoselective synthesis of γ-lactams
4. Conclusion
Acknowledgments
- Samples Availability: Available from the authors.
References
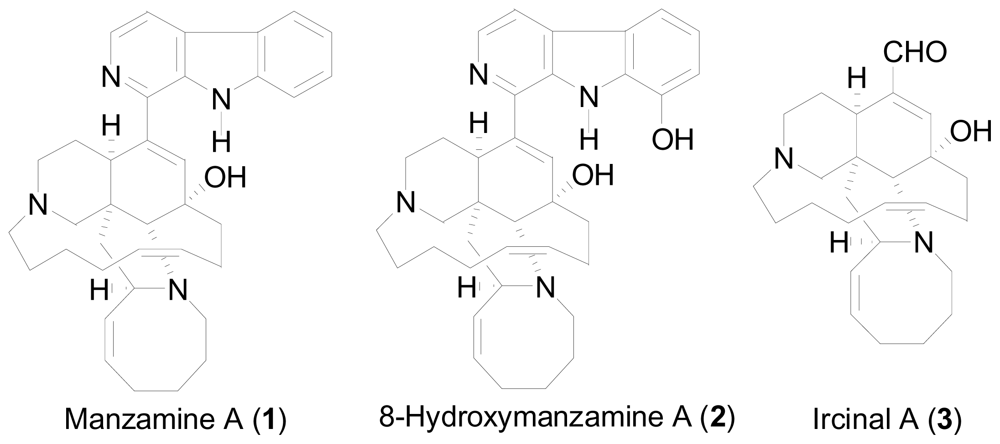
- Peng, J; Rao, KV; Choo, YM; Hamann, MT. Fattorusso, E, Taglialatela-Scafati, O, Eds.; Manzamine alkaloids. In Modern Alkaloids; Wiley-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2007; pp. 189–232. [Google Scholar]
- Tietze, LF. Domino reactions in organic synthesis. Chem. Rev 1996, 96, 115–136. [Google Scholar]
- Tietze, LF; Beifuss, U. Sequential transformations in organic chemistry: A synthetic strategy with a future. Angew. Chem. Int. Ed. Engl 1993, 32, 131–163. [Google Scholar]
- Nicolaou, KC; Montagnon, T; Snyder, SA. Tandem reactions, cascade sequences, and biomimetic strategies in total synthesis. Chem. Commun 2003, 5, 551–564. [Google Scholar]
- Nicolaou, KC; Edmonds, DJ; Bulger, PG. Cascade reactions in total synthesis. Angew. Chem. Int. Ed. Engl 2006, 45, 7134–7186. [Google Scholar]
- Nicolaou, KC; Chen, SJ. The art of total synthesis through cascade reactions. Chem. Soc. Rev 2009, 38, 2993–3009. [Google Scholar]
- Pellissier, H. Asymmetric domino reactions. Part A: reactions based on the use of chiral auxiliaries. Tetrahedron 2006, 62, 1619–1665. [Google Scholar]
- Pellissier, H. Asymmetric domino reactions. Part B: reactions based on the use of chiral catalysts and biocatalysts. Tetrahedron 2006, 62, 2143–2173. [Google Scholar]
- Padwa, A. Domino reactions of rhodium(ii) carbenoids for alkaloid synthesis. Chem. Soc. Rev 2009, 38, 3072–3081. [Google Scholar]
- Padwa, A; Bur, SK. The domino way to heterocycles. Tetrahedron 2007, 63, 5341–5378. [Google Scholar]
- Kibayashi, C. Development of new synthetic methods and its applications to total synthesis of nitrogen-containing bioactive natural products. Chem. Pharm. Bull 2005, 53, 1375–1386. [Google Scholar]
- Sakai, R; Higa, T; Jefford, CW; Bernardinelli, G. Manzamine A, a novel antitumor alkaloid from a sponge. J. Am. Chem. Soc 1986, 108, 6404–6405. [Google Scholar]
- Rao, KV; Kasanah, N; Wahyuono, S; Tekwani, BL; Schinazi, RF; Hamann, MT. Three new manzamine alkaloids from a common Indonesian sponge and their activity against infectious and tropical parasitic diseases. J. Nat. Prod 2004, 67, 1314–1318. [Google Scholar]
- Peng, J; Kudrimoti, S; Prasanna, S; Odde, S; Doerksen, RJ; Pennaka, HK; Choo, Y; Rao, KV; Tekwani, BL; Madgula, V; Khan, SI; Wang, B; Mayer, AMS; Jacob, MR; Tu, LC; Gertsch, J; Hamann, MT. Structure-activity relationship and mechanism of action studies of manzamine analogues for the control of neuroinflammation and cerebral infections. J. Med. Chem 2010, 53, 61–76. [Google Scholar]
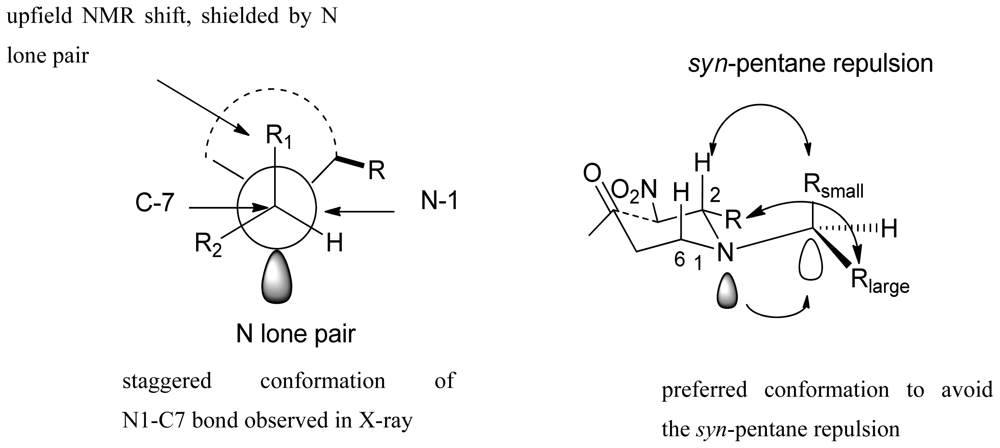
- Wahba, AE; Peng, J; Kudrimoti, S; Tekwani, BL; Hamann, MT. Structure activity relationship studies of manzamine A: amidation of positions 6 and 8 of the β-carboline moiety. Bioorg. Med. Chem 2009, 17, 7775–7782. [Google Scholar]
- Shilabin, AG; Kasanah, N; Tekwani, BL; Hamann, MT. Kinetic studies and bioactivity of potential manzamine prodrugs. J. Nat. Prod 2008, 71, 1218–1221. [Google Scholar]
- El Sayed, KA; Khalil, AA; Yousaf, M; Labadie, G; Kumar, GM; Franzblau, SG; Mayer, AMS; Avery, MA; Hamann, MT. Semisynthetic studies on the manzamine alkaloids. J. Nat. Prod 2008, 71, 300–308. [Google Scholar]
- Ibrahim, MA; Shilabin, AG; Prasanna, S; Jacob, M; Khan, SI; Doerksen, RJ; Hamann, MT. 2-Methyl modifications and SAR studies of manzamine A. Bioorg. Med. Chem 2008, 16, 6702–6706. [Google Scholar]
- Wahba, AE; Peng, J; Hamann, MT. Reductive amidation of nitroarenes: a practical approach for the amidation of natural products. Tetrahedron Lett 2009, 50, 3901–3904. [Google Scholar]
- De Oliveira, JHHL; Nascimento, AM; Kossuga, MH; Cavalcanti, BC; Pessoa, CO; Moraes, MO; Macedo, ML; Ferreira, AG; Hajdu, E; Pinheiro, US; Berlinck, RGS. Cytotoxic alkylpiperidine alkaloids from the Brazilian marine sponge pachychalina alcaloidifera. J. Nat. Prod 2007, 70, 538–543. [Google Scholar]
- Matsunaga, S; Miyata, Y; van Soest, RWM; Fusetani, N. Tetradehydrohalicyclamine A and 22-hydroxyhalicyclamine A, new cytotoxic bis-piperidine alkaloids from a marine sponge amphimedon sp. J. Nat. Prod 2004, 67, 1758–1760. [Google Scholar]
- Ishiguro, Y; Kubota, T; Ishiuchi, K; Fromont, J; Kobayashi, J. Plakoridine C, a novel piperidine alkaloid from an Okinawan marine sponge Plakortis sp. Tetrahedron Lett 2009, 50, 3202–3204. [Google Scholar]
- Escolano, C; Amat, M; Bosch, J. Chiral Oxazolopiperidone Lactams: Versatile Intermediates for the Enantioselective Synthesis of Piperidine-Containing Natural Products. Chem. Eur. J 2006, 12, 8198–8207. [Google Scholar]
- Felpin, F; Lebreton, J. Recent advances in the total synthesis of piperidine and pyrrolidine natural alkaloids with ring-closing metathesis as a key step. Eur. J. Org. Chem 2003, 2003, 3693–3712. [Google Scholar]
- Laschat, S; Dickner, T. Stereoselective synthesis of piperidines. Synthesis 2000, 2000, 1781–1813. [Google Scholar]
- Sun, X; Sengupta, S; Petersen, JL; Wang, H; Lewis, JP; Shi, X. Intermolecular cross-double-Michael addition between nitro and carbonyl activated olefins as a new approach in C-C bond formation. Org. Lett 2007, 9, 4495–4498. [Google Scholar]
- Chen, Y; Zhong, C; Petersen, JL; Akhmedov, NG; Shi, X. One-pot asymmetric synthesis of substituted piperidines by exocyclic chirality induction. Org. Lett 2009, 11, 2333–2336. [Google Scholar]
- Franzen, J; Fisher, A. Asymmetric alkaloid synthesis: a one-pot organocatalytic reaction to quinolizidine derivatives. Angew. Chem. Int. Ed. Engl 2009, 48, 787–791. [Google Scholar]
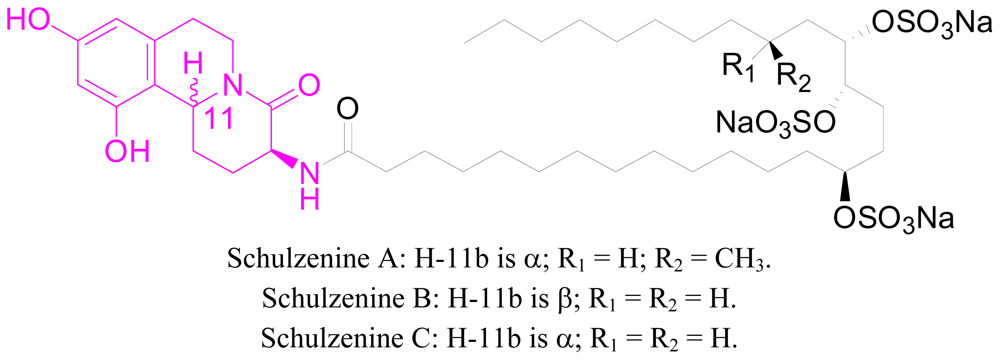
- Takada, K; Uehara, T; Nakao, Y; Matsunaga, S; van Soest, RW; Fusetani, N. Schulzeines A-C, new α-glucosidase inhibitors from the marine sponge penares schulzei. J. Am. Chem. Soc 2004, 126, 187–193. [Google Scholar]
- Enders, D; Grondal, C; Huttl, ARM. Asymmetric organocatalytic domino reactions. Angew. Chem. Int. Ed. Engl 2007, 46, 1570–1581. [Google Scholar]
- Wang, Y; Kumano, T; Kano, T; Maruoka, K. Organocatalytic approach to enantioselective one-pot synthesis of pyrrolidine, hexahydropyrrolizine, and octahydroindolizine core structures. Org. Lett 2009, 11, 2027–2029. [Google Scholar]
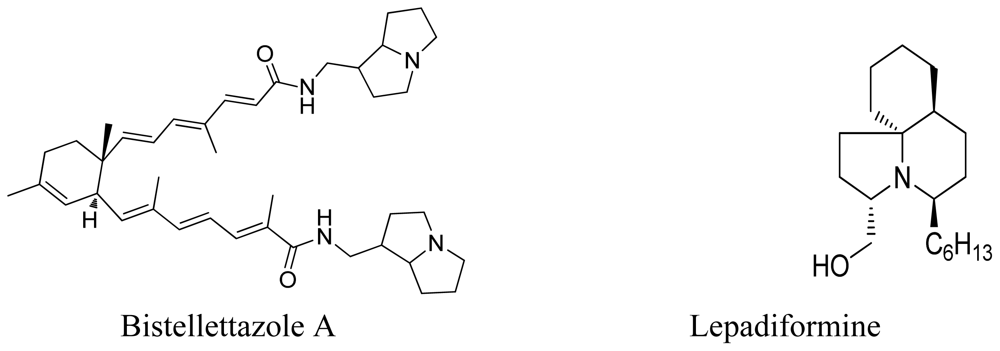
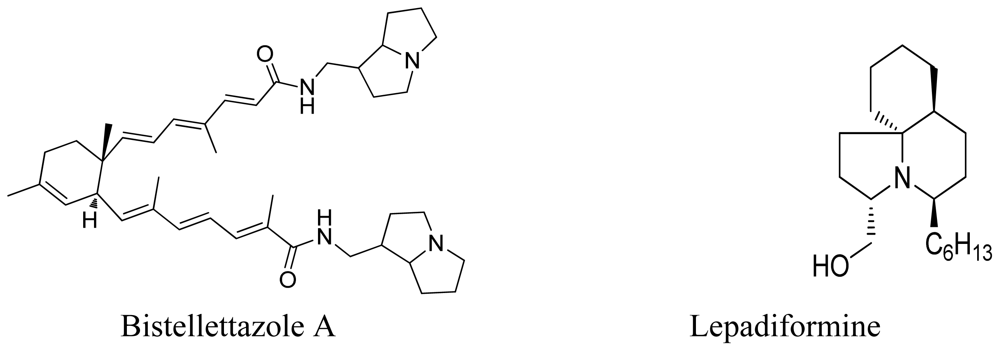
- El-Naggar, M; Piggott, AM; Capon, RJ. Bistellettazines A-C and Bistellettazole A: New Terpenyl-Pyrrolizidine and Terpenyl-Imidazole Alkaloids from a Southern Australian Marine Sponge, Stelletta sp. Org. Lett 2008, 10, 4247–4250. [Google Scholar]
- Biard, JF; Guyot, S; Roussakis, C; Verbist, JF; Vercauteren, J; Weber, JF; Boukef, K. Lepadiformine, a new marine cytotoxic alkaloid from Clavelina lepadiformis Mueller. Tetrahedron Lett 1994, 35, 2691–2694. [Google Scholar]
- Castagnoli, NJ. N-Acylenamines from oxazolines. New route to 2-acetamidogylcals. J. Org. Chem 1969, 34, 3187–3189. [Google Scholar]
- Cushman, M; Abbaspour, A; Gupta, YP. Total synthesis of (±)-14-epicorynoline, (±)-corynoline, and (±)-6-oxocorynoline. J. Am. Chem. Soc 1983, 105, 2873–2879. [Google Scholar]
- Masse, CE; Ng, PY; Fukase, Y; Sanchez-Rosello, M; Shaw, JT. Divergent Structural Complexity from a Linear Reaction Sequence: Synthesis of Fused and Spirobicyclic γ-Lactams from Common Synthetic Precursors. J. Comb. Chem 2006, 8, 293–296. [Google Scholar]
- Wei, J; Shaw, JT. Diastereoselective Synthesis of γ-Lactams by a One-Pot, Four-Component Reaction. Org. Lett 2007, 9, 4077–4080. [Google Scholar]
- Tang, Y; Fettinger, JC; Shaw, JT. One-Step Synthesis of Complex Nitrogen Heterocycles from Imines and Alkyl-Substituted Maleic Anhydrides. Org. Lett 2009, 11, 3802–3805. [Google Scholar]

© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wahba, A.E.; Hamann, M.T. New One-Pot Methodologies for the Modification or Synthesis of Alkaloid Scaffolds. Mar. Drugs 2010, 8, 2395-2416. https://doi.org/10.3390/md8082395
Wahba AE, Hamann MT. New One-Pot Methodologies for the Modification or Synthesis of Alkaloid Scaffolds. Marine Drugs. 2010; 8(8):2395-2416. https://doi.org/10.3390/md8082395
Chicago/Turabian StyleWahba, Amir E., and Mark T. Hamann. 2010. "New One-Pot Methodologies for the Modification or Synthesis of Alkaloid Scaffolds" Marine Drugs 8, no. 8: 2395-2416. https://doi.org/10.3390/md8082395