Synthesis of Marine Polycyclic Polyethers via Endo-Selective Epoxide-Opening Cascades

Abstract
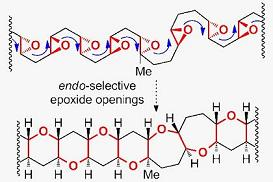
:
1. Introduction
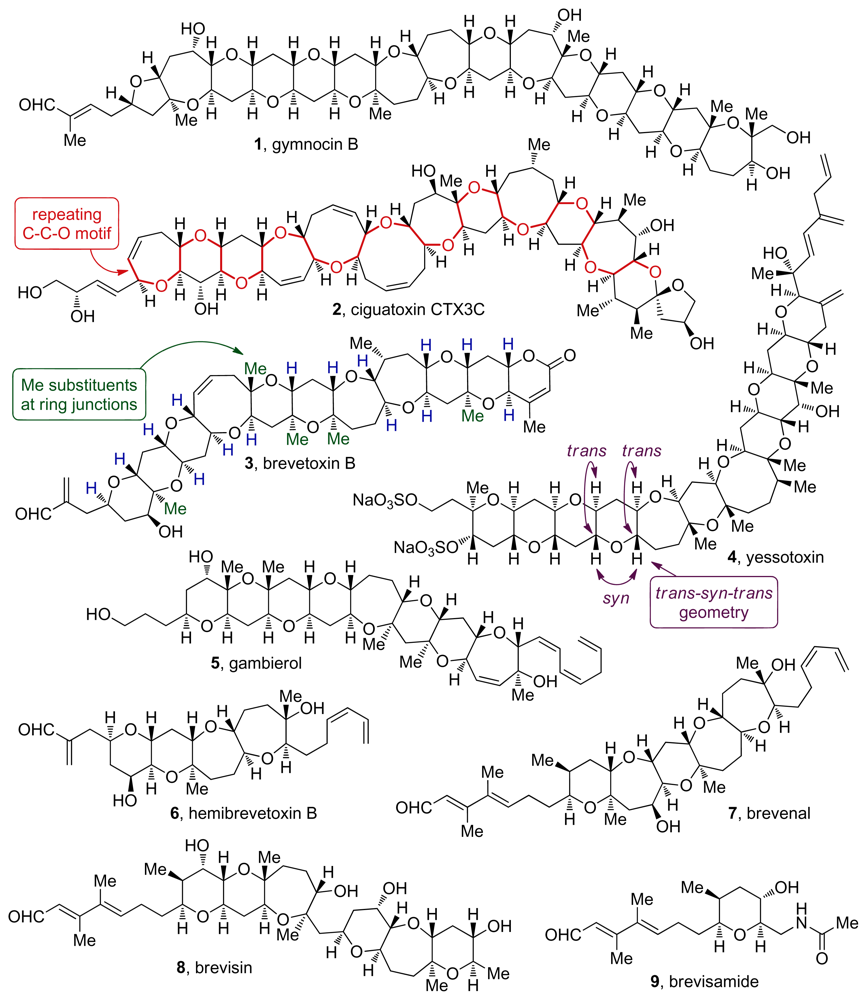
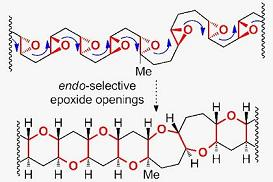
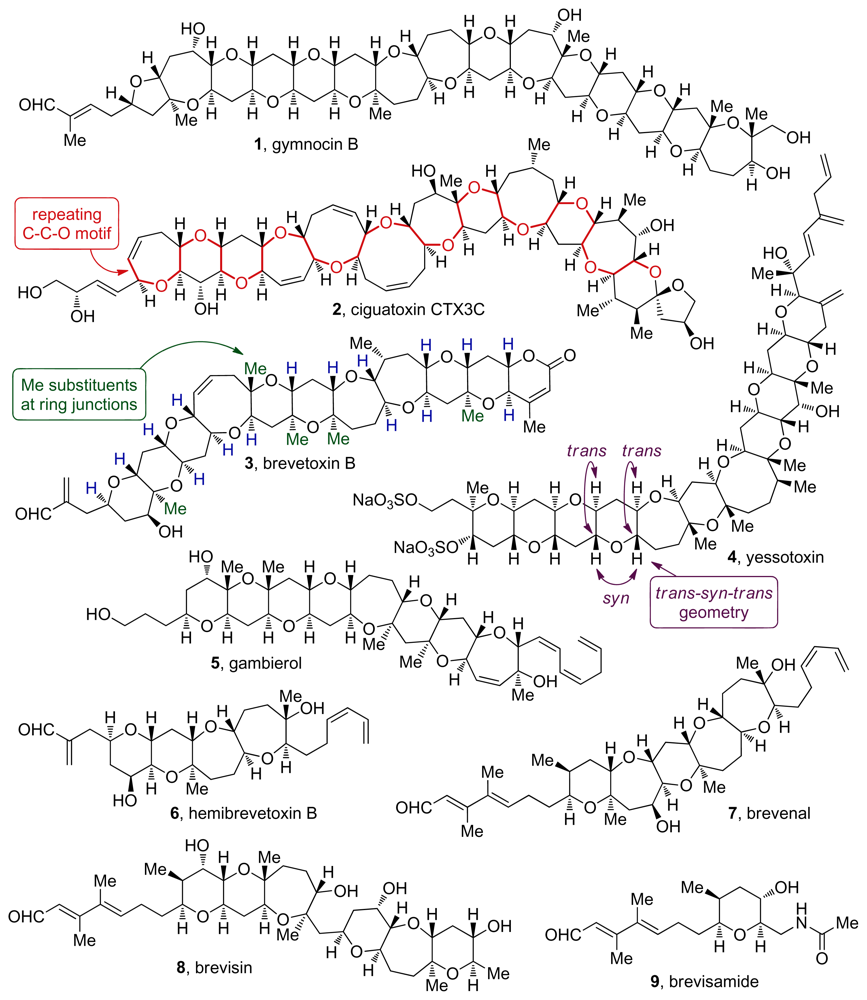
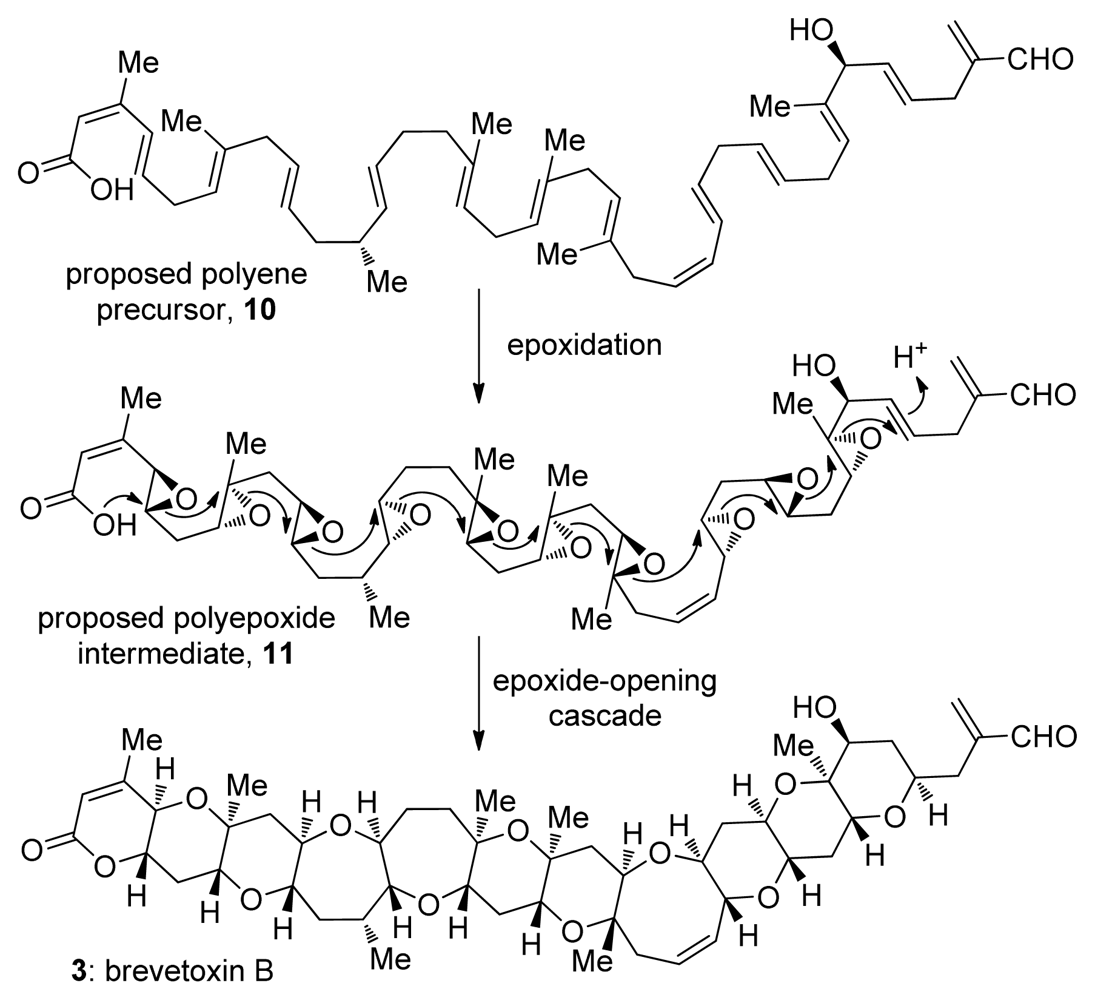
2. Ladder Polyethers
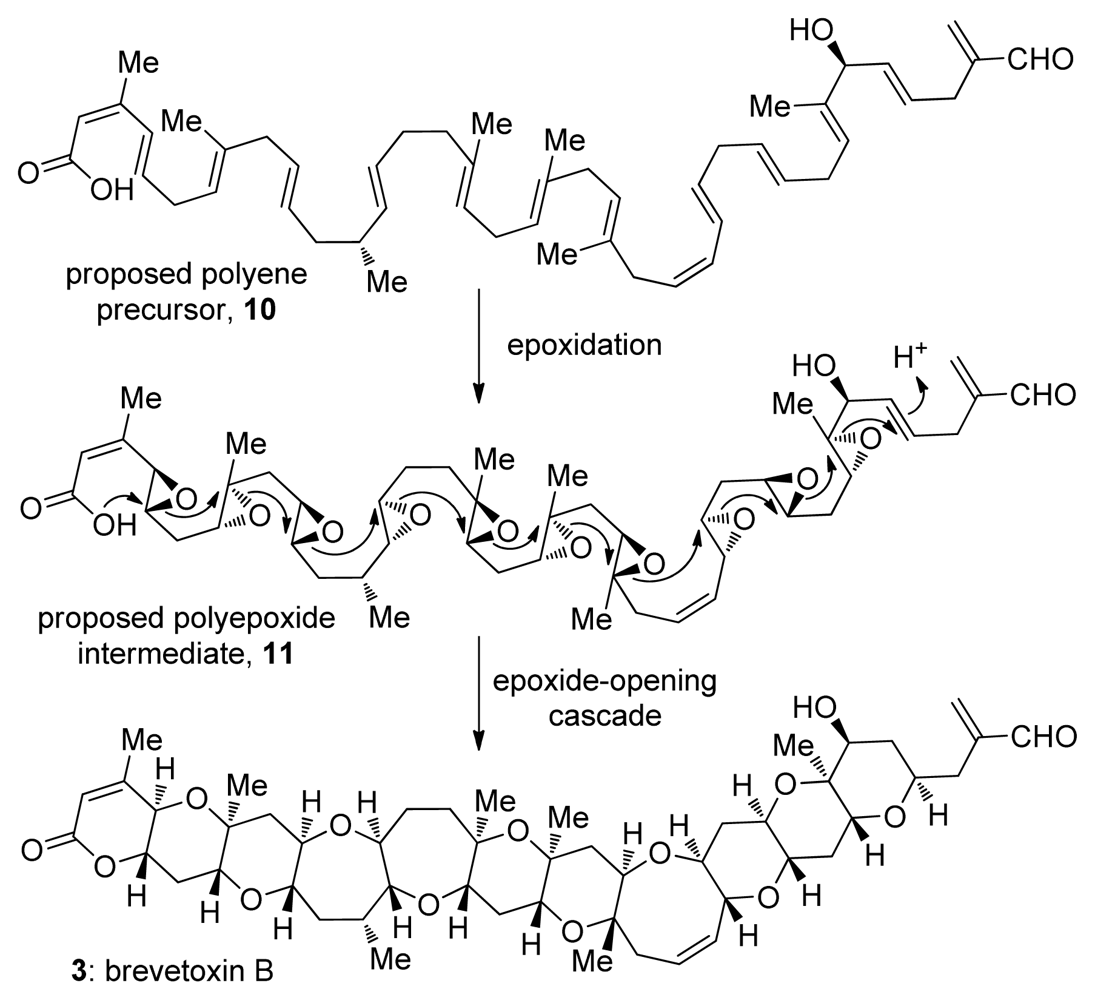
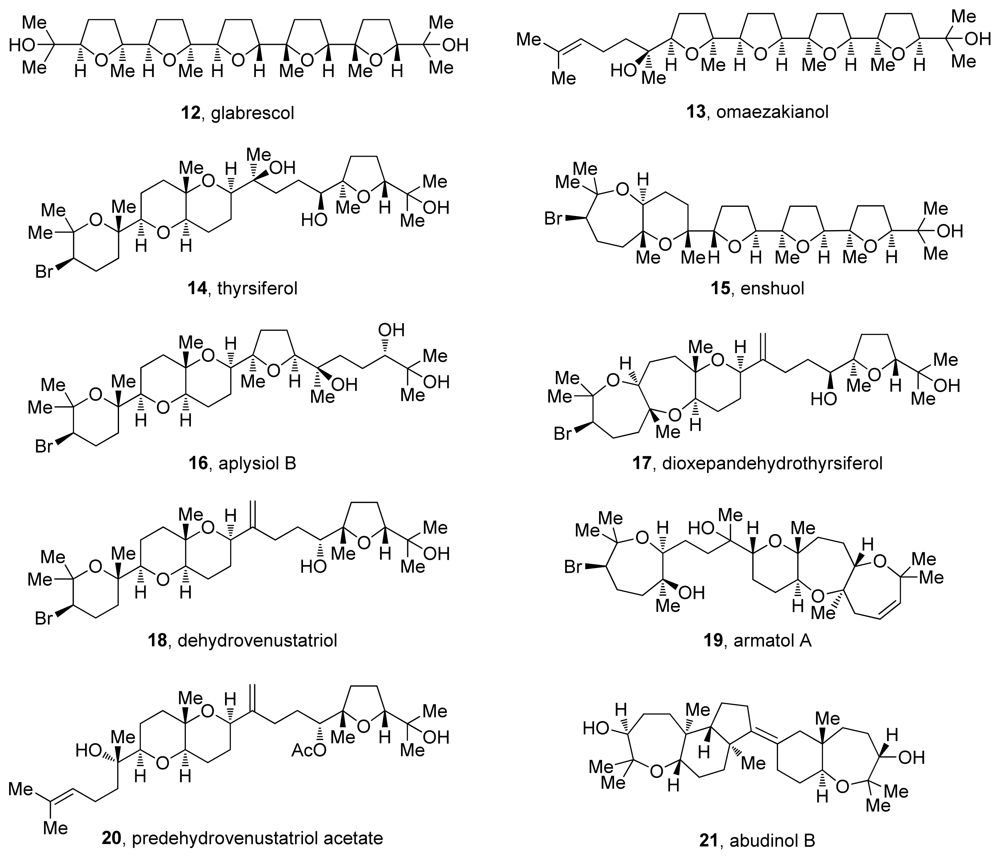
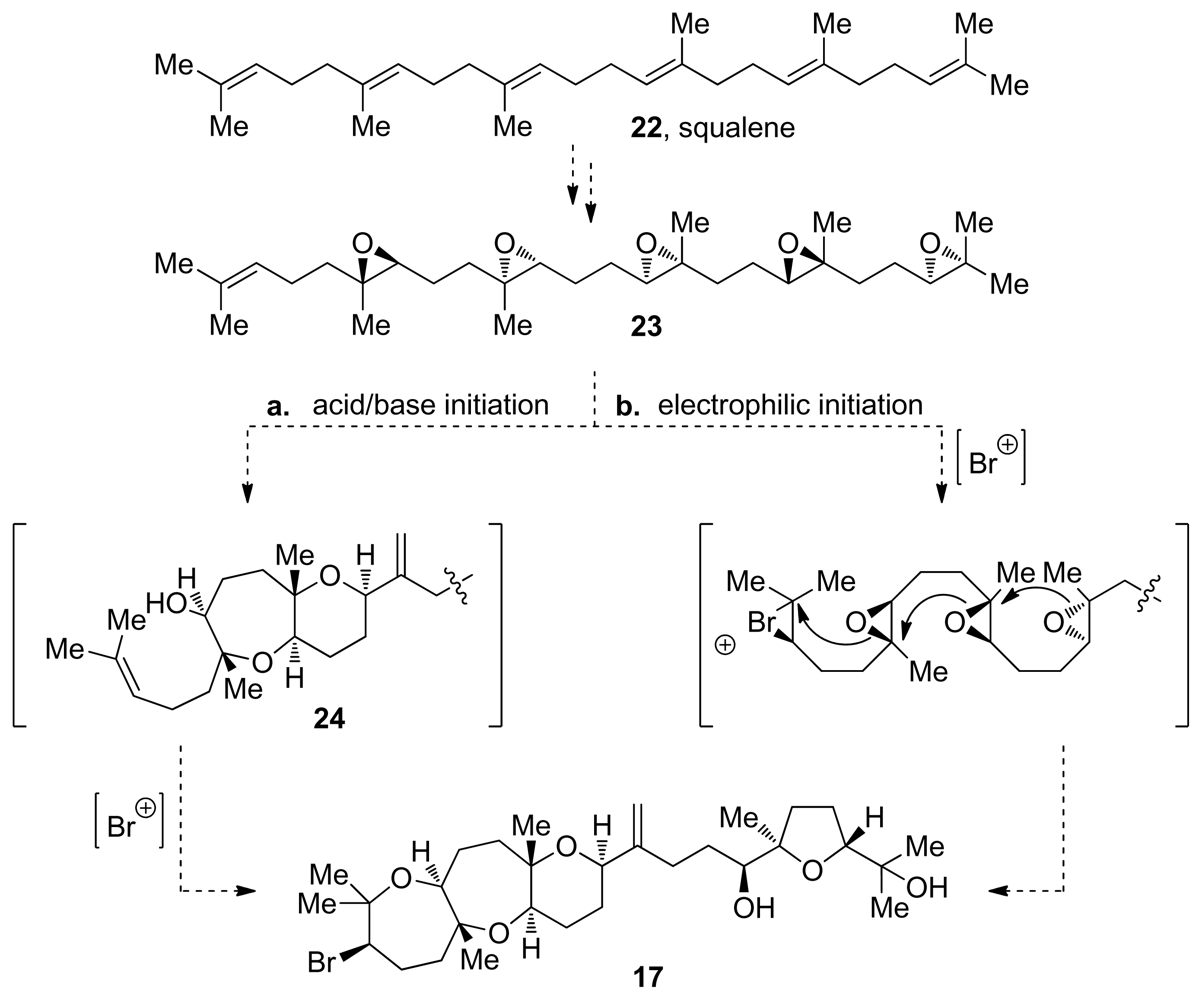
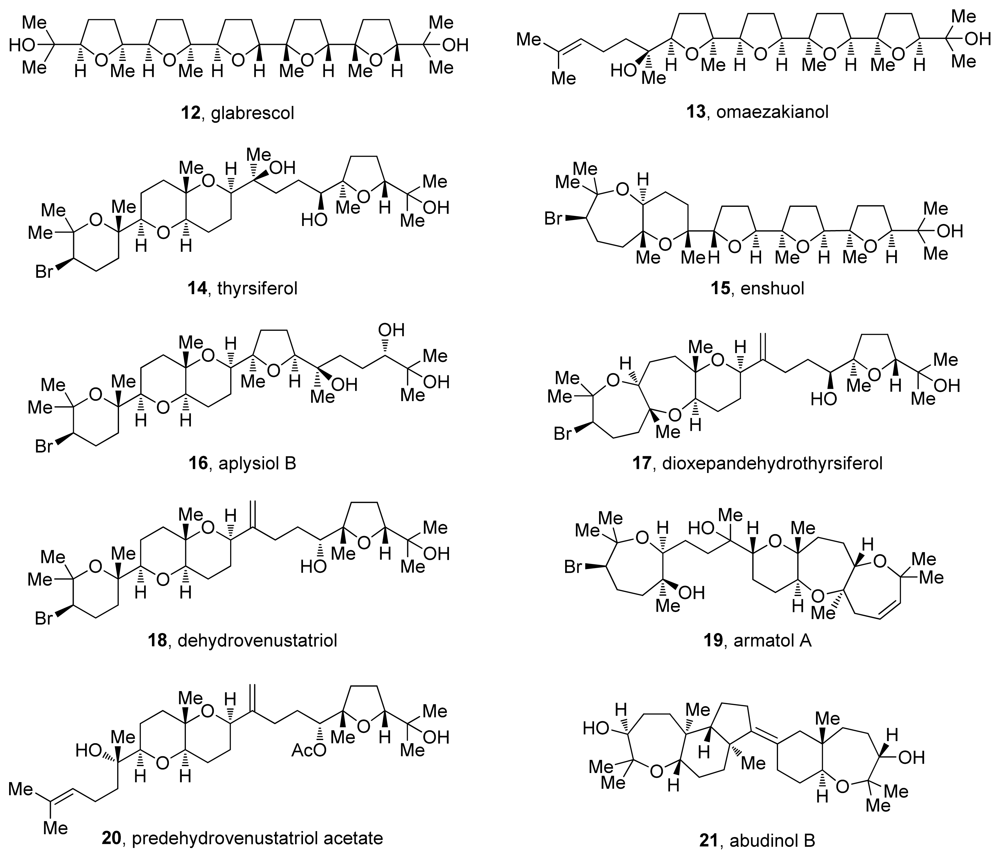
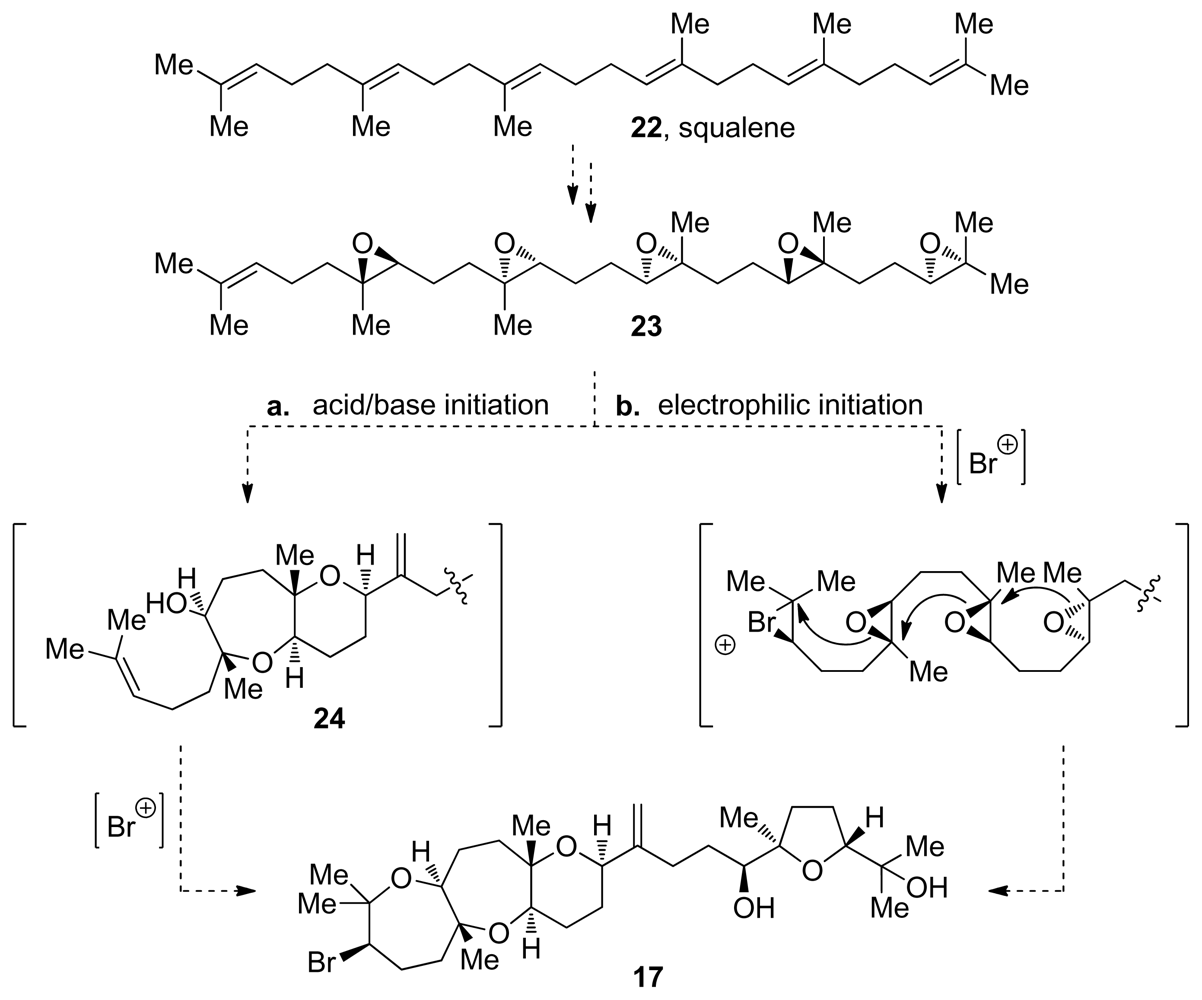
3. Oxasqualenoids, Polyethers Derived from Squalene
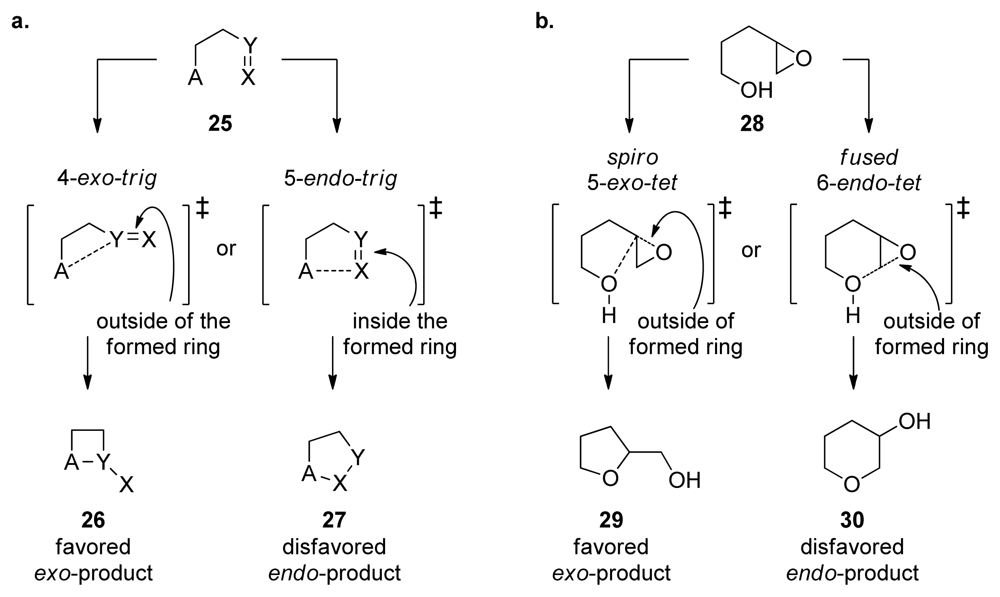
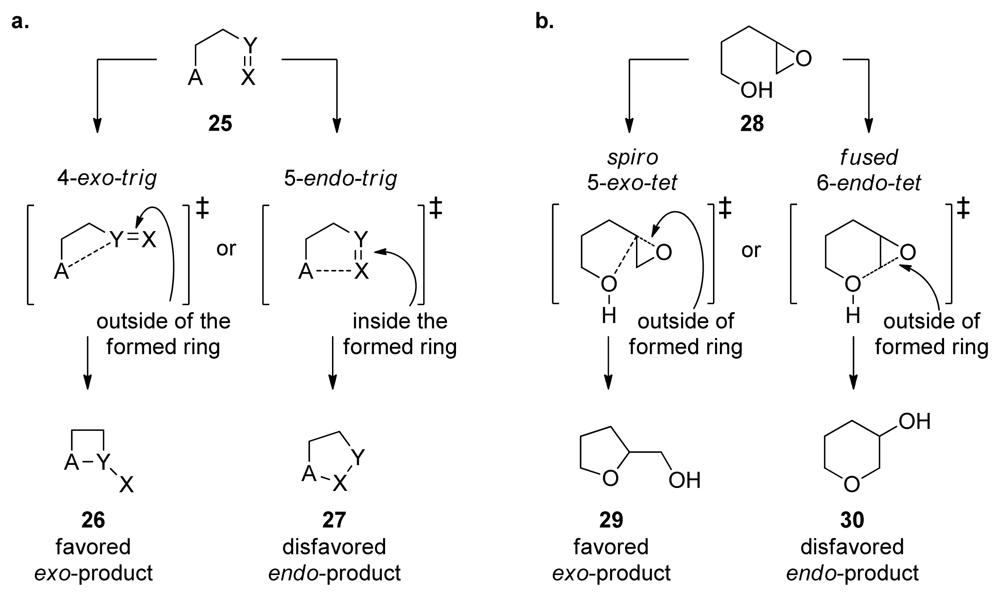
4. Baldwin’s Rules
5. Construction of Tetrahydropyrans via Endo-Selective Epoxide-Opening Reactions
5.1. Reactions of electronically biased epoxides
5.1.1. Alkenylepoxides
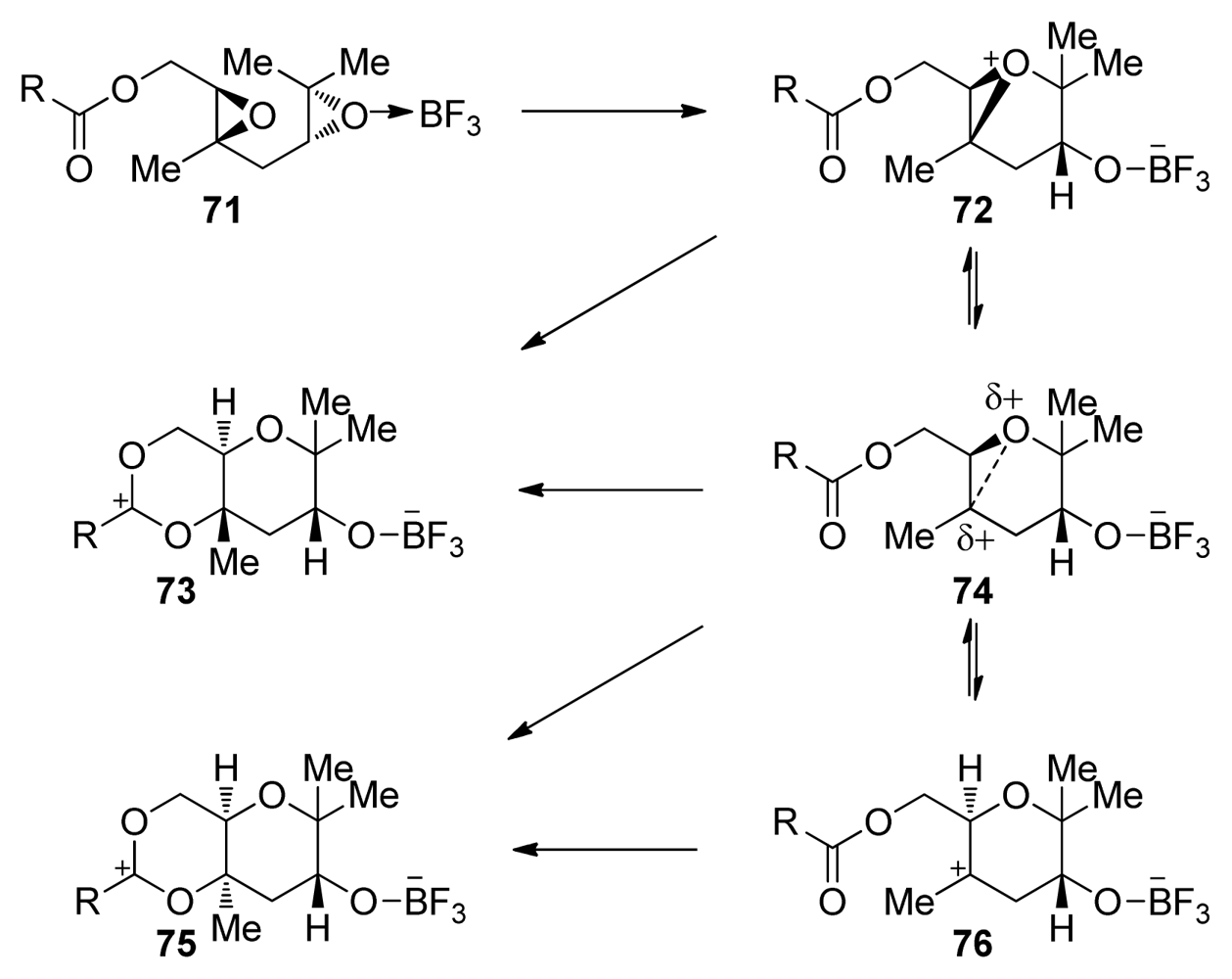
5.1.2. α,β-Epoxysulfones
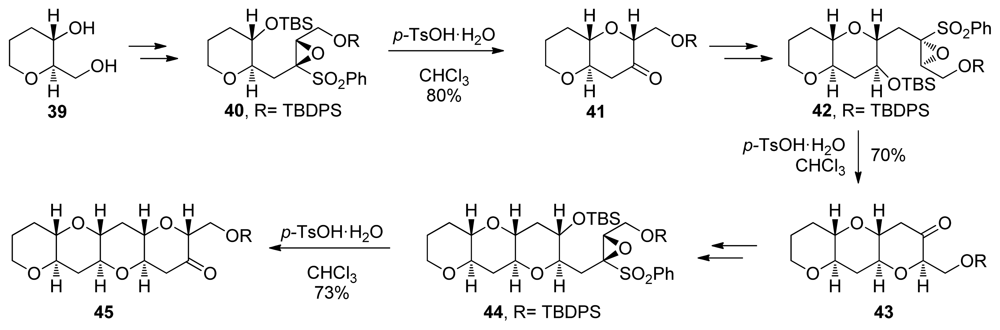
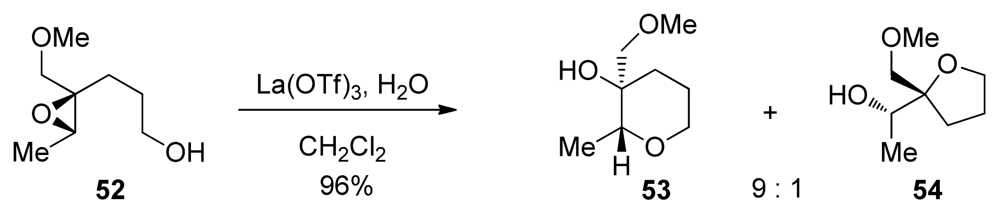
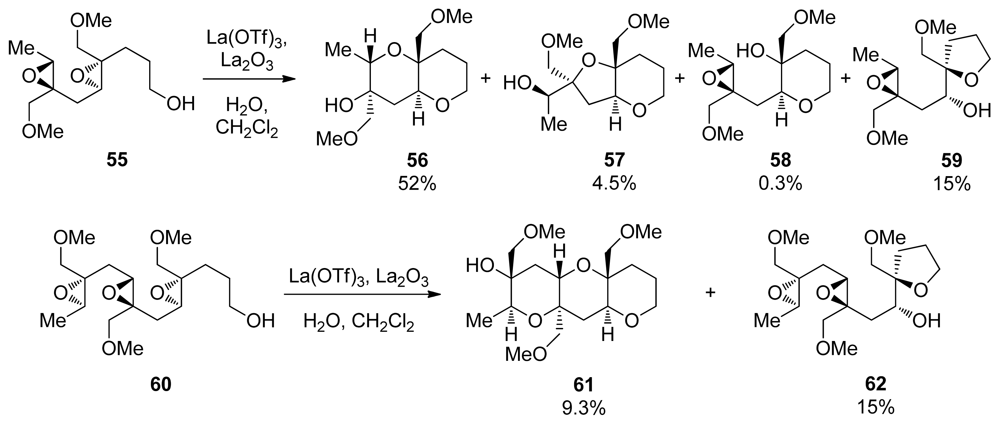
5.1.3. Methoxymethyl substituted epoxides
5.1.4. Trialkyl substituted epoxides
5.1.5. Epoxysilanes
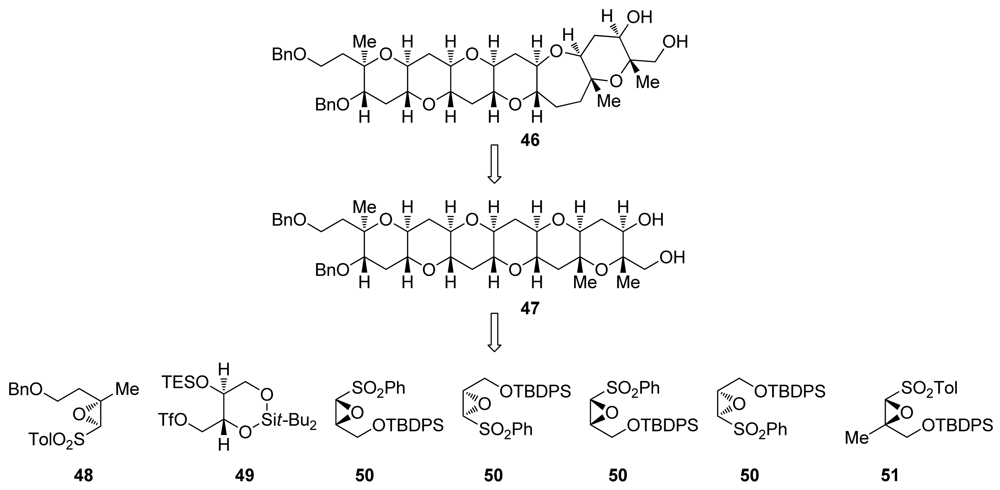
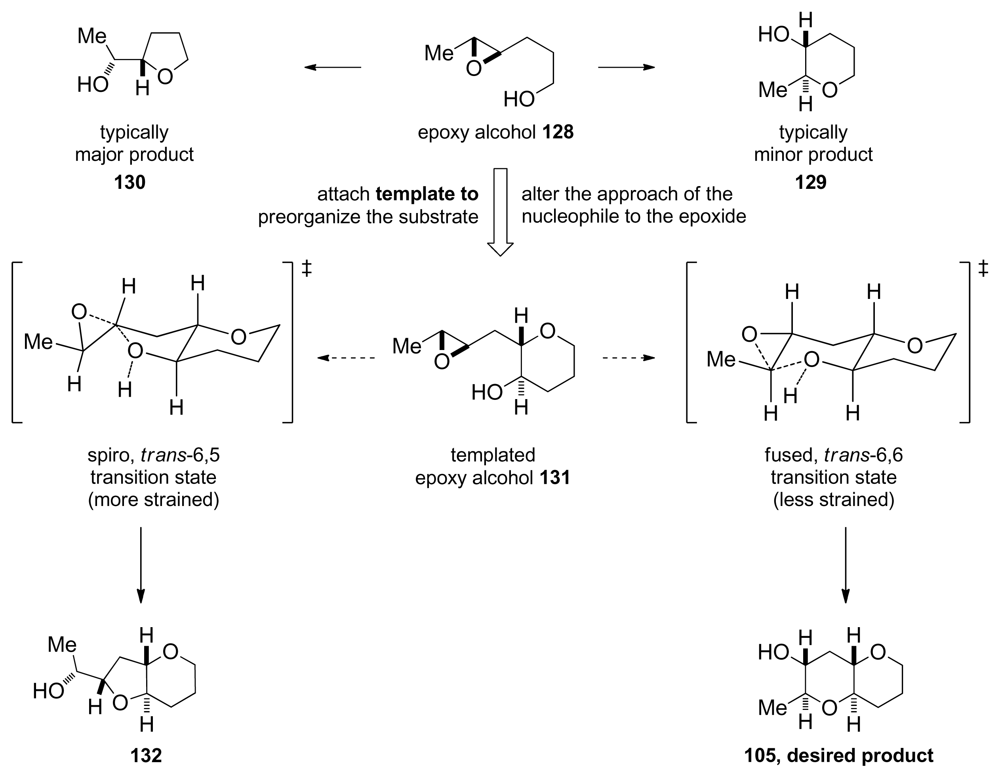
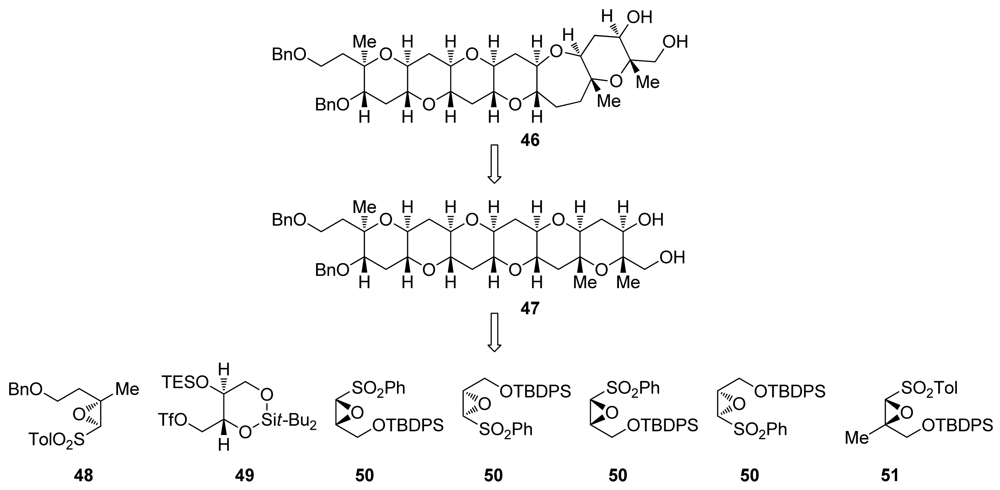
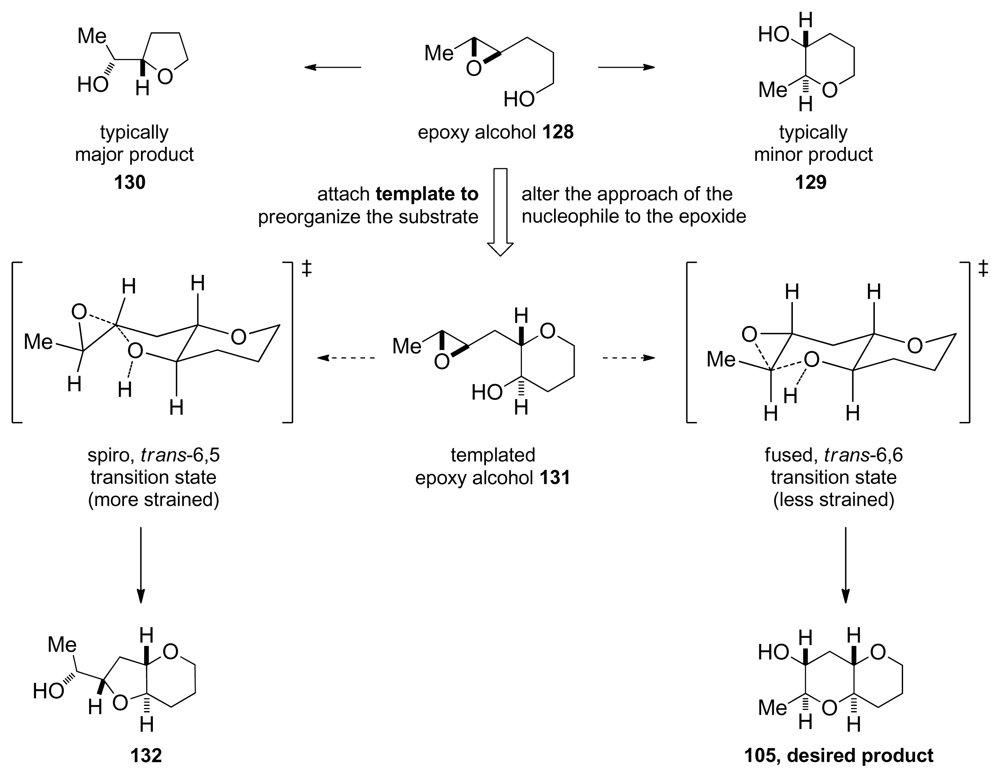
5.2. Reactions of disubstituted epoxides
6. Construction of Oxepanes via Endo-Selective Epoxide-Opening Reactions
6.1. Reactions of electronically biased epoxides
6.2. Reactions of disubstituted epoxides
7. Future Directions
Acknowledgements
References and Notes
- Gallimore, AR. The biosynthesis of polyketide-derived polycyclic ethers. Nat Prod Rep 2009, 26, 266–280. [Google Scholar]
- Fernandez, JJ; Souto, ML; Norte, M. Marine polyether triterpenes. Nat Prod Rep 2000, 17, 235–246. [Google Scholar]
- Vilotijevic, I; Jamison, TF. Epoxide-opening cascades in the synthesis of polycyclic polyether natural products. Angew Chem Int Ed 2009, 48, 5250–5281. [Google Scholar]
- Sasaki, M; Matsumori, N; Maruyama, T; Nonomura, T; Murata, M; Tachibana, K; Yasumoto, T. The complete structure of maitotoxin. Part I. Configuration of the C1–14 side chain. Angew Chem Int Ed 1996, 35, 1672–1675. [Google Scholar]
- Nonomura, T; Sasaki, M; Matsumori, N; Murata, M; Tachibana, K; Yasumoto, T. The complete structure of maitotoxin, Part II. Configuration of the C135–142 side chain and absolute configuration of the entire molecule. Angew Chem Int Ed 1996, 35, 1675–1678. [Google Scholar]
- Lin, Y.-Y; Risk, M; Ray, SM; Van Engen, D; Clardy, J; Golik, J; James, JC; Nakanishi, K. Isolation and structure of brevetoxin B from the red tide dinoflagellate Ptychodiscus brevis (Gymnodinium breve). J Am Chem Soc 1981, 103, 6773–6775. [Google Scholar]
- Murata, M; Legrand, AM; Ishibashi, Y; Yasumoto, T. Structures of ciguatoxin and its congener. J Am Chem Soc 1989, 111, 8929–8931. [Google Scholar]
- Yasumoto, T; Murata, M. Marine toxins. Chem Rev 1993, 93, 1897–1909. [Google Scholar]
- Murata, M; Naoki, H; Iwashita, T; Matsunaga, S; Sasaki, M; Yokoyama, A; Yasumoto, T. Structure of maitotoxin. J Am Chem Soc 1993, 115, 2060–2062. [Google Scholar]
- Inoue, M. Convergent strategies for syntheses of trans-fused polycyclic ethers. Chem Rev 2005, 105, 4379–4405. [Google Scholar]
- Nakata, T. Total synthesis of marine polycyclic ethers. Chem Rev 2005, 105, 4314–4347. [Google Scholar]
- Nicolaou, KC; Frederick, MO; Aversa, RJ. The continuing saga of the marine polyether biotoxins. Angew Chem Int Ed 2008, 47, 7182–7225. [Google Scholar]
- Sasaki, M; Fuwa, H. Convergent strategies for the total synthesis of polycyclic ether marine metabolites. Nat Prod Rep 2008, 25, 401–426. [Google Scholar]
- Sellner, KG; Doucette, GJ; Kirkpatrick, GJ. Harmful algal blooms: causes, impacts and detection. J Ind Microbiol Biotechnol 2003, 30, 383–406. [Google Scholar]
- Murata, M; Legrand, AM; Ishibashi, Y; Fukui, M; Yasumoto, T. Structures and configurations of ciguatoxin from the moray eel Gymnothorax javanicus and its likely precursor from the dinoflagellate Gambierdiscus toxicus. J Am Chem Soc 1990, 112, 4380–4386. [Google Scholar]
- Flewelling, LJ; Naar, JP; Abbott, JP; Baden, DG; Barros, NB; Bossart, GD; Bottein, M-YD; Hammond, DG; Haubold, EM; Heil, CA; Henry, MS; Jacocks, HM; Leighfield, TA; Pierce, RH; Pitchford, TD; Rommel, SA; Scott, PS; Steidinger, KA; Truby, EW; Van Dolah, FM; Landsberg, JH. Brevetoxicosis: red tides and marine mammal mortalities. Nature 2005, 435, 755–756. [Google Scholar]
- Lewis, RJ. The changing face of ciguatera. Toxicon 2000, 39, 97–106. [Google Scholar]
- Poli, MA; Mende, TJ; Baden, DG. Brevetoxins, unique activators of voltage-sensitive sodium channels, bind to specific sites in rat brain synaptosomes. Mol Pharmacol 1986, 30, 129–135. [Google Scholar]
- Cuypers, E; Abdel-Mottaleb, Y; Kopljar, I; Rainier, JD; Raes, AL; Snyders, DJ; Tytgat, J. Gambierol, a toxin produced by the dinoflagellate Gambierdiscus toxicus, is a potent blocker of voltage-gated potassium channels. Toxicon 2008, 51, 974–983. [Google Scholar]
- Sinkins, WG; Estacion, M; Prasad, V; Goel, M; Shull, GE; Kunze, DL; Schilling, WP. Maitotoxin converts the plasmalemmal Ca2+ pumpinto a Ca2+-permeable nonselective cation channel. Am J Physiol Cell Physiol 2009, 297, 1533–1543. [Google Scholar]
- Ferrari, S; Ciminiello, P; Dell’Aversano, C; Forino, M; Malaguti, C; Tubaro, A; Poletti, R; Yasumoto, T; Fattorusso, E; Rossini, GP. Structure-activity relationships of yessotoxins in cultured cells. Chem Res Toxicol 2004, 17, 1251–1257. [Google Scholar]
- Ronzitti, G; Callegari, F; Malaguti, C; Rossini, GP. Selective disruption of the E-cadherin-catenin system by an algal toxin. Br J Cancer 2004, 90, 1100–1107. [Google Scholar]
- Nagai, H; Murata, M; Torigoe, K; Satake, M; Yasumoto, T. Gambieric acids, new potent antifungal substances with unprecedented polyether structures from a marine dinoflagellate Gambierdiscus toxicus. J Org Chem 1992, 57, 5448–5453. [Google Scholar]
- Nagai, H; Mikami, Y; Yazawa, K; Gonoi, T; Yasumoto, T. Biological activities of novel polyether antifungals, gambieric acids A and B from a marine dinoflagellate Gambierdiscus toxicus. J Antibiot 1993, 46, 520–522. [Google Scholar]
- Bourdelais, AJ; Campbell, S; Jacocks, H; Naar, J; Wright, JLC; Carsi, J; Baden, DG. Brevenal is a natural inhibitor of brevetoxin action in sodium channel receptor binding assays. Cell Mol Neurobiol 2004, 24, 553–563. [Google Scholar]
- Abraham, WM; Bourdelais, AJ; Sabater, JR; Ahmed, A; Lee, TA; Serebriakov, I; Baden, DG. Airway responses to aerosolized brevetoxins in an animal model of asthma. Am J Respir Crit Care Med 2005, 171, 26–34. [Google Scholar]
- Baden, DG; Abraham, WM; Bourdelais, AJ; Michelliza, S. Fused pentacyclic polyethers. US Patent 7,638,500, 29 December 2009. [Google Scholar]
- Dutton, CJ; Banks, BJ; Cooper, CB. Polyether ionophores. Nat Prod Rep 1995, 12, 165–181. [Google Scholar]
- Cane, DE; Celmer, WD; Westley, JW. Unified stereochemical model of polyether antibiotic structure and biogenesis. J Am Chem Soc 1983, 105, 3594–3600. [Google Scholar]
- Nakanishi, K. The chemistry of brevetoxins: A review. Toxicon 1985, 23, 473–479. [Google Scholar]
- Lee, MS; Repeta, DJ; Nakanishi, K; Zagorski, MG. Biosynthetic origins and assignments of carbon 13 NMR peaks of brevetoxin B. J Am Chem Soc 1986, 108, 7855–7856. [Google Scholar]
- Chou, HN; Shimizu, Y. Biosynthesis of brevetoxins. Evidence for the mixed origin of the backbone carbon chain and possible involvement of dicarboxylic acids. J Am Chem Soc 1987, 109, 2184–2185. [Google Scholar]
- Lee, MS; Qin, G; Nakanishi, K; Zagorski, MG. Biosynthetic studies of brevetoxins, potent neurotoxins produced by the dinoflagellate Gymnodinium breve. J Am Chem Soc 1989, 111, 6234–6241. [Google Scholar]
- Gallimore, AR; Spencer, JB. Stereochemical uniformity in marine polyether ladders-implications for the biosynthesis and structure of maitotoxin. Angew Chem Int Ed 2006, 45, 4406–4413. [Google Scholar]
- Baldwin, JE. Rules for ring closure. J Chem Soc Chem Commun 1976, 734–736. [Google Scholar]
- Satake, M. Biosynthesis of the marine polyether toxin, yessotoxin. Tennen Yuki Kagobutsu Toronkai Koen Yoshishu 2000, 42, 259–264. [Google Scholar]
- Snyder, RV; Gibbs, PDL; Palacios, A; Abiy, L; Dickey, R; Lopez, JV; Rein, KS. Polyketide synthase genes from marine dinoflagellates. Mar Biotechnol 2003, 5, 1–12. [Google Scholar]
- Snyder, RV; Guerrero, MA; Sinigalliano, CD; Winshell, J; Perez, R; Lopez, JV; Rein, KS. Localization of polyketide synthase encoding genes to the toxic dinoflagellate Karenia brevis. Phytochemistry 2005, 66, 1767–1780. [Google Scholar]
- Perez, R; Liu, L; Lopez, J; An, T; Rein, KS. Diverse bacterial PKS sequences derived from okadaic acid-producing dinoflagellates. Mar Drugs 2008, 6, 164–179. [Google Scholar]
- Murata, M; Izumikawa, M; Tachibana, K; Fujita, T; Naoki, H. Labeling pattern of okadaic acid from 18O2 and [18O2]acetate elucidated by collisionally induced dissociation tandem mass spectrometry. J Am Chem Soc 1998, 120, 147–151. [Google Scholar]
- Izumikawa, M; Murata, M; Tachibana, K; Tujita, T; Naoki, H. 18O-labelling pattern of okadaic acid from H2 18O in dinoflagellate Prorocentrum lima elucidated by tandem mass spectrometry. Eur J Biochem 2000, 267, 5179–5183. [Google Scholar]
- Chou, H.-N; Shimizu, Y; Van Duyne, G; Clardy, J. Isolation and structures of two new polycyclic ethers from Gymnodinium breve davis (Ptychodiscus brevis). Tetrahedron Lett 1985, 26, 2865–2868. [Google Scholar]
- Satake, M; Campbell, A; Van Wagoner, RM; Bourdelais, AJ; Jacocks, H; Baden, DG; Wright, JLC. Brevisin: an aberrant polycyclic ether structure from the dinoflagellate Karenia brevis and its implications for polyether assembly. J Org Chem 2009, 74, 989–994. [Google Scholar]
- Satake, M; Bourdelais, AJ; Van Wagoner, RM; Baden, DG; Wright, JLC. Brevisamide: an unprecedented monocyclic ether alkaloid from the dinoflagellate Karenia brevis that provides a potential model for ladder-frame initiation. Org Lett 2008, 10, 3465–3468. [Google Scholar]
- Fuwa, H; Ebine, M; Bourdelais, AJ; Baden, DG; Sasaki, M. Total synthesis, structure revision, and absolute configuration of (−)-brevenal. J Am Chem Soc 2006, 128, 16989–16999. [Google Scholar]
- Fuwa, H; Ebine, M; Sasaki, M. Total synthesis of the proposed structure of brevenal. J Am Chem Soc 2006, 128, 9648–9650. [Google Scholar]
- Nicolaou, KC; Frederick, MO. On the structure of maitotoxin. Angew Chem Int Ed 2007, 46, 5278–5282. [Google Scholar]
- Nicolaou, KC; Frederick, MO; Burtoloso, ACB; Denton, RM; Rivas, F; Cole, KP; Aversa, RJ; Gibe, R; Umezawa, T; Suzuki, T. Chemical synthesis of the GHIJKLMNO ring system of maitotoxin. J Am Chem Soc 2008, 130, 7466–7476. [Google Scholar]
- Townsend, CA; Basak, A. Experiments and speculations on the role of oxidative cyclization chemistry in natural product biosynthesis. Tetrahedron 1991, 47, 2591–2602. [Google Scholar]
- McDonald, FE; Towne, TB. syn-Oxidative polycyclization of hydroxy polyenes: A new approach to polyether synthesis. J Am Chem Soc 1994, 116, 7921–7922. [Google Scholar]
- McDonald, FE; Towne, TB; Schultz, CC. Metal-oxo induced syn-oxidative polycyclizations of hydroxypolyenes: biomimetic synthesis of polycyclic ether natural products. Pure Appl Chem 1998, 70, 355–358. [Google Scholar]
- Giner, J.-L; Li, X; Mullins, JJ. Mechanistic studies of the biomimetic epoxy ester-orthoester and orthoester-cyclic ether rearrangements. J Org Chem 2003, 68, 10079–10086. [Google Scholar]
- Giner, J.-L. Tetrahydropyran formation by rearrangement of an epoxy ester: a model for the biosynthesis of marine polyether toxins. J Org Chem 2005, 70, 721–724. [Google Scholar]
- Sakemi, S; Higa, T; Jefford, CW; Bernardinelli, G. Venustatriol: a new antiviral triterpene tetracyclic ether from Laurencia venusta. Tetrahedron Lett 1986, 27, 4287–4290. [Google Scholar]
- Matsuo, Y; Suzuki, M; Masuda, M. Enshuol, a Novel Squalene-derived Pentacyclic Triterpene Alcohol from a New Species of the Red Algal Genus Laurencia. Chem Lett 1995, 24, 1043–1044. [Google Scholar]
- Hashimoto, M; Kan, T; Nozaki, K; Yanagiya, M; Shirahama, H; Matsumoto, T. Total syntheses of (+)-thyrsiferol, (+)-thyrsiferyl 23-acetate, and (+)-venustatriol. J Org Chem 1990, 55, 5088–5107. [Google Scholar]
- Manriquez, CP; Souto, ML; Gavin, JA; Norte, M; Fernandez, JJ. Several new squalene-derived triterpenes from Laurencia. Tetrahedron 2001, 57, 3117–3123. [Google Scholar]
- Kashman, Y; Rudi, A. On the biogenesis of marine isoprenoids. Phytochem Rev 2005, 3, 309–323. [Google Scholar]
- Domingo, V; Arteaga, JF; Moral, JFQd; Barrero, AF. Unusually cyclized triterpenes: occurrence, biosynthesis and chemical synthesis. Nat Prod Rep 2009, 26, 115–134. [Google Scholar]
- Tanuwidjaja, J; Ng, S.-S; Jamison, TF. Total synthesis of ent-dioxepandehydrothyrsiferol via a bromonium-initiated epoxide-opening cascade. J Am Chem Soc 2009, 131, 12084–12085. [Google Scholar]
- Johnson, CD. Stereoelectronic effects in the formation of 5- and 6-membered rings: the role of Baldwin’s rules. Acc Chem Res 1993, 26, 476–482. [Google Scholar]
- Danishefsky, S; Dynak, J; Hatch, E; Yamamoto, M. Ring construction through transpositions of activated cyclopropanes. J Am Chem Soc 1974, 96, 1256–1259. [Google Scholar]
- Katsuki, T; Sharpless, KB. The first practical method for asymmetric epoxidation. J Am Chem Soc 1980, 102, 5974–5976. [Google Scholar]
- Johnson, RA; Sharpless, KB. Catalytic asymmetric epoxidation of allylic alcohols. In Catalytic Asymmetric Synthesis, 1st ed; Ojirna, I, Ed.; VCH: New York, NY, USA, 1993; pp. 103–158. [Google Scholar]
- Katsuki, T; Martin, VS. Asymmetric epoxidation of allylic alcohols: The Katsuki-Sharpless epoxidation reaction. Org React 1996, 48, 1–299. [Google Scholar]
- Zhang, W; Loebach, JL; Wilson, SR; Jacobsen, EN. Enantioselective epoxidation of unfunctionalized olefins catalyzed by salen manganese complexes. J Am Chem Soc 1990, 112, 2801–2803. [Google Scholar]
- Jacobsen, EN; Zhang, W; Muci, AR; Ecker, JR; Deng, L. Highly enantioselective epoxidation catalysts derived from 1,2-diaminocyclohexane. J Am Chem Soc 1991, 113, 7063–7064. [Google Scholar]
- Brandes, BD; Jacobsen, EN. Highly enantioselective, catalytic epoxidation of trisubstituted olefins. J Org Chem 1994, 59, 4378–4380. [Google Scholar]
- Chang, S; Galvin, JM; Jacobsen, EN. Effect of chiral quaternary ammonium salts on (salen)Mn-catalyzed epoxidation of cis-olefins. A highly enantioselective, catalytic route to trans-epoxides. J Am Chem Soc 1994, 116, 6937–6938. [Google Scholar]
- Tu, Y; Wang, Z.-X; Shi, Y. An efficient asymmetric epoxidation method for trans-olefins mediated by a fructose-derived ketone. J Am Chem Soc 1996, 118, 9806–9807. [Google Scholar]
- Wang, Z.-X; Tu, Y; Frohn, M; Zhang, J.-R; Shi, Y. An efficient catalytic asymmetric epoxidation method. J Am Chem Soc 1997, 119, 11224–11235. [Google Scholar]
- Shi, Y. Organocatalytic asymmetric epoxidation of olefins by chiral ketones. Acc Chem Res 2004, 37, 488–496. [Google Scholar]
- Wong, OA; Shi, Y. Organocatalytic oxidation. asymmetric epoxidation of olefins catalyzed by chiral ketones and iminium salts. Chem Rev 2008, 108, 3958–3987. [Google Scholar]
- Nicolaou, KC; Duggan, ME; Hwang, CK; Somers, PK. Activation of 6-endo over 5-exo epoxide openings. Ring-selective formation of tetrahydropyran systems and stereocontrolled synthesis of the ABC ring framework of brevetoxin B. J Chem Soc Chem Commun 1985, 1359–1362. [Google Scholar]
- Nicolaou, KC; Prasad, CVC; Somers, PK; Hwang, CK. Activation of 6-endo over 5-exo hydroxy epoxide openings. Stereoselective and ring selective synthesis of tetrahydrofuran and tetrahydropyran systems. J Am Chem Soc 1989, 111, 5330–5334. [Google Scholar]
- Nicolaou, KC; Reddy, KR; Skokotas, G; Sato, F; Xiao, XY. Total synthesis of hemibrevetoxin B. J Am Chem Soc 1992, 114, 7935–7936. [Google Scholar]
- Kadota, I; Jung-Youl, P; Koumura, N; Pollaud, G; Matsukawa, Y; Yamamoto, Y. Total synthesis of hemibrevetoxin B. Tetrahedron Lett 1995, 36, 5777–5780. [Google Scholar]
- Morimoto, M; Matsukura, H; Nakata, T. Total synthesis of hemibrevetoxin B. Tetrahedron Lett 1996, 37, 6365–6368. [Google Scholar]
- Mori, Y; Yaegashi, K; Furukawa, H. Oxiranyl anions in organic synthesis: application to the synthesis of hemibrevetoxin B. J Am Chem Soc 1997, 119, 4557–4558. [Google Scholar]
- Nicolaou, KC; Hwang, CK; Duggan, ME; Nugiel, DA; Abe, Y; Reddy, KB; DeFrees, SA; Reddy, DR; Awartani, RA. Total synthesis of brevetoxin B. 1. First generation strategies and new approaches to oxepane systems. J Am Chem Soc 1995, 117, 10227–10238. [Google Scholar]
- Nicolaou, KC; Rutjes, FPJT; Theodorakis, EA; Tiebes, J; Sato, M; Untersteller, E. Total synthesis of brevetoxin B. 3. Final strategy and completion. J Am Chem Soc 1995, 117, 10252–10263. [Google Scholar]
- Nicolaou, KC; Rutjes, FPJT; Theodorakis, EA; Tiebes, J; Sato, M; Untersteller, E. Total synthesis of brevetoxin B. 2. Completion. J Am Chem Soc 1995, 117, 1173–1174. [Google Scholar]
- Nicolaou, KC; Theodorakis, EA; Rutjes, FPJT; Sato, M; Tiebes, J; Xiao, XY; Hwang, CK; Duggan, ME; Yang, Z. Total synthesis of brevetoxin B. 2. Second generation strategies and construction of the dioxepane region [DEFG]. J Am Chem Soc 1995, 117, 10239–10251. [Google Scholar]
- Nicolaou, KC; Theodorakis, EA; Rutjes, FPJT; Tiebes, J; Sato, M; Untersteller, E; Xiao, XY. Total synthesis of brevetoxin B. 1. CDEFG framework. J Am Chem Soc 1995, 117, 1171–1172. [Google Scholar]
- Matsuo, G; Hori, N; Matsukura, H; Nakata, T. Synthetic studies on brevetoxin B. Part 2. Stereoselective synthesis of the EFG-ring system. Tetrahedron Lett 2000, 41, 7677–7680. [Google Scholar]
- Matsuo, G; Matsukura, H; Hori, N; Nakata, T. Synthetic studies on brevetoxin B. Part 1. Stereoselective synthesis of the ABC-ring system. Tetrahedron Lett 2000, 41, 7673–7676. [Google Scholar]
- Matsuo, G; Kawamura, K; Hori, N; Matsukura, H; Nakata, T. Total synthesis of brevetoxin B. J Am Chem Soc 2004, 126, 14374–14376. [Google Scholar]
- Nicolaou, KC; Yang, Z; Shi, G; Gunzner, JL; Agrios, KA; Gartner, P. Total synthesis of brevetoxin A. Nature 1998, 392, 264–269. [Google Scholar]
- Nicolaou, KC; Bunnage, ME; McGarry, DG; Shi, S; Somers, PK; Wallace, PA; Chu, X- J; Agrios, KA; Gunzner, JL; Yang, Z. Total synthesis of brevetoxin A: Part 1. First generation strategy and construction of BCD ring system. Chem Eur J 1999, 5, 599–617. [Google Scholar]
- Nicolaou, KC; Wallace, PA; Shi, S; Ouellette, MA; Bunnage, ME; Gunzner, JL; Agrios, KA; Shi, G.-Q; Gartner, P; Yang, Z. Total synthesis of brevetoxin A: Part 2. Second generation strategy and construction of EFGH model system. Chem Eur J 1999, 5, 618–627. [Google Scholar]
- Nicolaou, KC; Shi, G.-Q; Gunzner, JL; Gartner, P; Wallace, PA; Ouellette, MA; Shi, S; Bunnage, ME; Agrios, KA; Veale, CA; Hwang, C.-K; Hutchinson, J; Prasad, CVC; Ogilvie, WW; Yang, Z. Total synthesis of brevetoxin A: Part 3. Construction of GHIJ and BCDE ring systems. Chem Eur J 1999, 5, 628–645. [Google Scholar]
- Nicolaou, KC; Gunzner, JL; Shi, G.-Q; Agrios, KA; Gartner, P; Yang, Z. Total synthesis of brevetoxin A: Part 4. Final stages and completion. Chem Eur J 1999, 5, 646–658. [Google Scholar]
- Fuwa, H; Kainuma, N; Tachibana, K; Sasaki, M. Total synthesis of (−)-gambierol. J Am Chem Soc 2002, 124, 14983–14992. [Google Scholar]
- Kadota, I; Takamura, H; Sato, K; Ohno, A; Matsuda, K; Satake, M; Yamamoto, Y. Convergent total syntheses of gambierol and 16-epi-gambierol and their biological activities. J Am Chem Soc 2003, 125, 11893–11899. [Google Scholar]
- Kadota, I; Takamura, H; Sato, K; Ohno, A; Matsuda, K; Yamamoto, Y. Total synthesis of gambierol. J Am Chem Soc 2003, 125, 46–47. [Google Scholar]
- Furuta, H; Hasegawa, Y; Mori, Y. Total synthesis of gambierol. Org Lett 2009, 11, 4382–4385. [Google Scholar]
- Nicolaou, KC; Duggan, ME; Hwang, CK. Synthesis of the FG ring system of brevetoxin B. J Am Chem Soc 1989, 111, 6676–6682. [Google Scholar]
- Mori, Y; Yaegashi, K; Furukawa, H. A new strategy for the reiterative synthesis of trans-fused tetrahydropyrans via alkylation of oxiranyl anion and 6-endo cyclization. J Am Chem Soc 1996, 118, 8158–8159. [Google Scholar]
- Mori, Y; Furuta, H; Takase, T; Mitsuoka, S; Furukawa, H. Synthesis of methyl-substituted trans-fused tetrahydropyrans via 6-endo cyclization. Tetrahedron Lett 1999, 40, 8019–8022. [Google Scholar]
- Mori, Y; Yaegashi, K; Furukawa, H. Stereoselective synthesis of the 6,7,6- and 6,7,7-ring systems of polycyclic ethers by 6-endo cyclization and ring expansion. Tetrahedron 1997, 53, 12917–12932. [Google Scholar]
- Mori, Y; Yaegashi, K; Furukawa, H. Formal total synthesis of hemibrevetoxin B by an oxiranyl anion strategy. J Org Chem 1998, 63, 6200–6209. [Google Scholar]
- Mori, Y; Takase, T; Noyori, R. Iterative synthesis of the ABCDEF-ring system of yessotoxin and adriatoxin. Tetrahedron Lett 2003, 44, 2319–2322. [Google Scholar]
- Furuta, H; Takase, T; Hayashi, H; Noyori, R; Mori, Y. Synthesis of trans-fused polycyclic ethers with angular methyl groups using sulfonyl-stabilized oxiranyl anions. Tetrahedron 2003, 59, 9767–9777. [Google Scholar]
- Fujiwara, K; Tokiwano, T; Murai, A. La(OTf)3-catalyzed 6-endo epoxide opening of 4,5- epoxy-4-methoxymethyl-1-hexanols. Tetrahedron Lett 1995, 36, 8063–8066. [Google Scholar]
- Tokiwano, T; Fujiwara, K; Murai, A. Effect of molecular sieves and methanol on endo-selectivity in the La(OTf)3-catalyzed cyclization of 5-hydroxy-2-methoxymethyl-1,2-epoxides. Chem Lett 2000, 272–273. [Google Scholar]
- Tokiwano, T; Fujiwara, K; Marai, A. Biomimetic construction of fused tricyclic ether by cascaded endo-cyclization of a hydroxy triepoxide. Synlett 2000, 335–338. [Google Scholar]
- Bravo, F; McDonald, FE; Neiwert, WA; Do, B; Hardcastle, KI. Biomimetic synthesis of fused polypyrans: oxacyclization stereo- and regioselectivity is a function of the nucleophile. Org Lett 2003, 5, 2123–2126. [Google Scholar]
- Tarselli, MA; Zuccarello, JL; Lee, SJ; Gagne, MR. Gold(I)-catalyzed cascade cyclization of allenyl epoxides. Org Lett 2009, 11, 3490–3492. [Google Scholar]
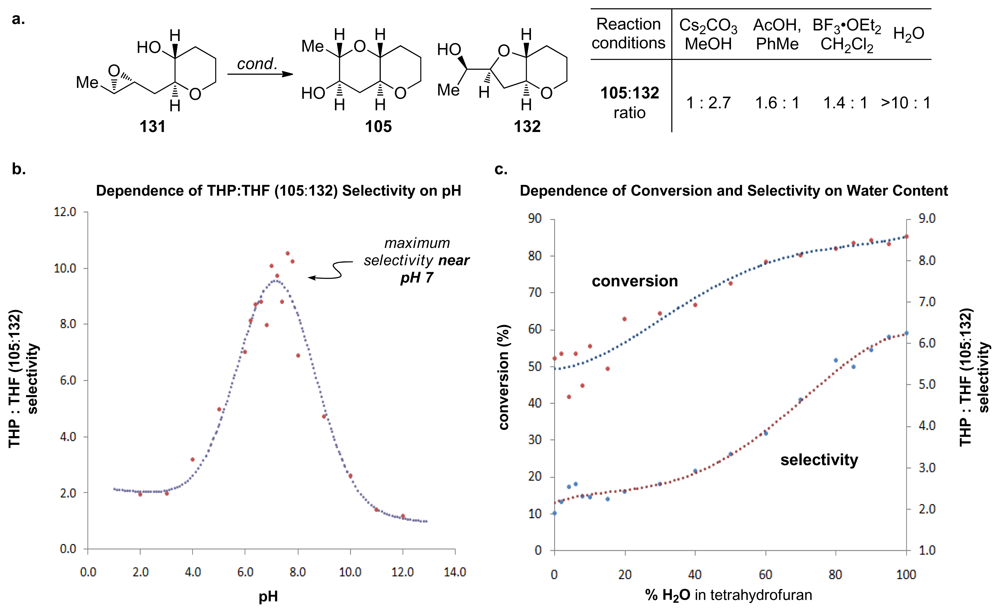
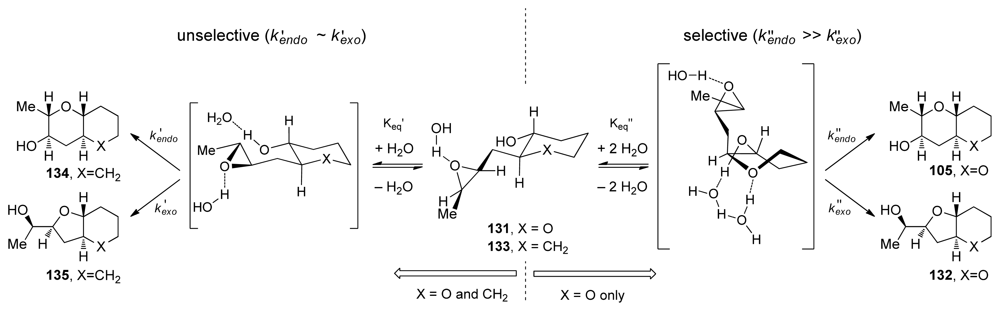
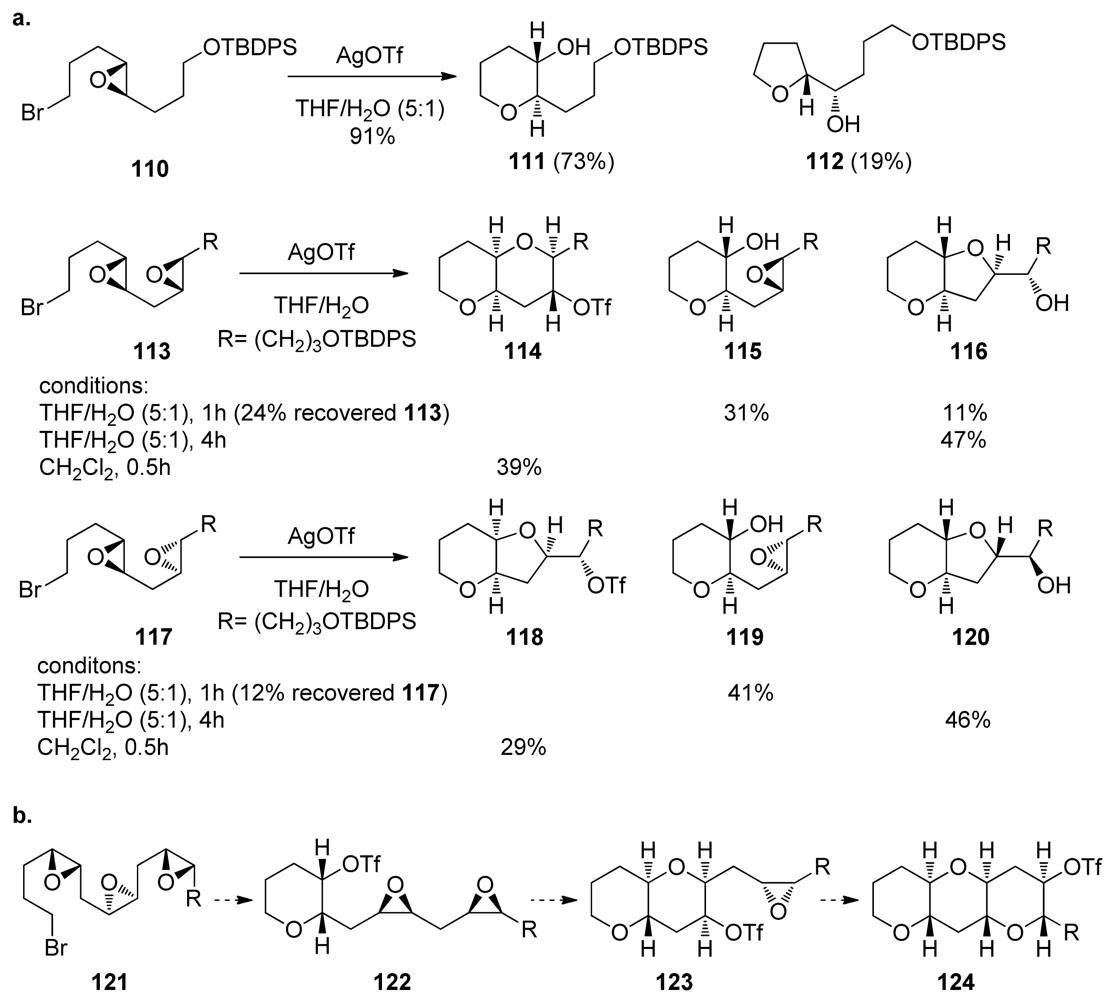
- Morten, CJ; Byers, JA; Van Dyke, AR; Vilotijevic, I; Jamison, TF. The development of endo-selective epoxide-opening cascades in water. Chem Soc Rev 2009, 38, 3175–3192. [Google Scholar]
- Morten, CJ; Jamison, TF. New synthetic strategies for the stereocontrolled synthesis of substituted ‘skipped’ diepoxides. Tetrahedron 2009, 65, 6648–6655. [Google Scholar]
- Morten, CJ; Jamison, TF. Water overcomes methyl group directing effects in epoxide-opening cascades. J Am Chem Soc 2009, 131, 6678–6679. [Google Scholar]
- Zakarian, A; Batch, A; Holton, RA. A convergent total synthesis of hemibrevetoxin B. J Am Chem Soc 2003, 125, 7822–7824. [Google Scholar]
- Na, J; Houk, KN; Shevlin, CG; Janda, KD; Lerner, RA. The energetic advantage of 5-exo versus 6-endo epoxide openings: a preference overwhelmed by antibody catalysis. J Am Chem Soc 1993, 115, 8453–84. [Google Scholar]
- Na, J; Houk, KN. Predicting antibody catalyst selectivity from optimum binding of catalytic groups to a hapten. J Am Chem Soc 1996, 118, 9204–9205. [Google Scholar]
- Wan, S; Gunaydin, H; Houk, KN; Floreancig, PE. An experimental and computational approach to defining structure/reactivity relationships for intramolecular addition reactions to bicyclic epoxonium ions. J Am Chem Soc 2007, 129, 7915–7923. [Google Scholar]
- Morimoto, Y; Nishikawa, Y; Ueba, C; Tanaka, T. Reagent-controlled switching of 5-exo to 6-endo cyclizations in epoxide openings. Angew Chem Int Ed 2006, 45, 810–812. [Google Scholar]
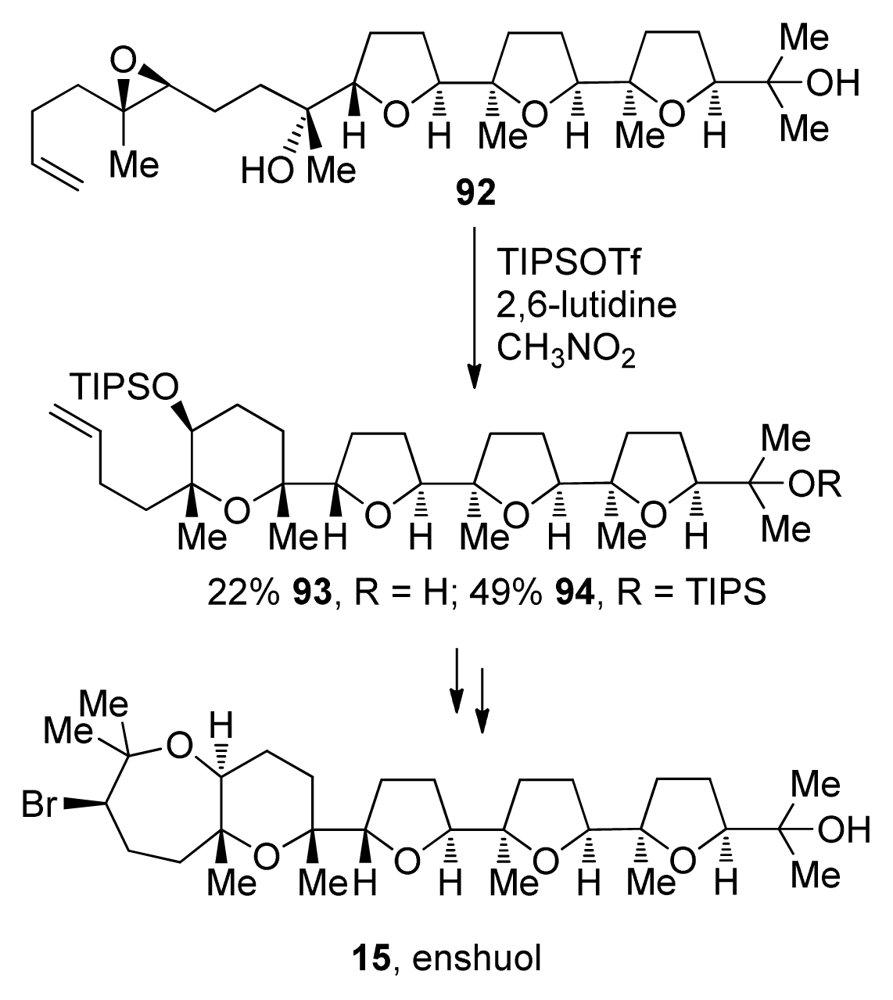
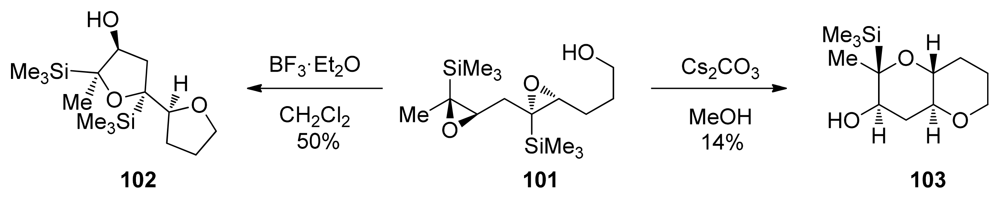
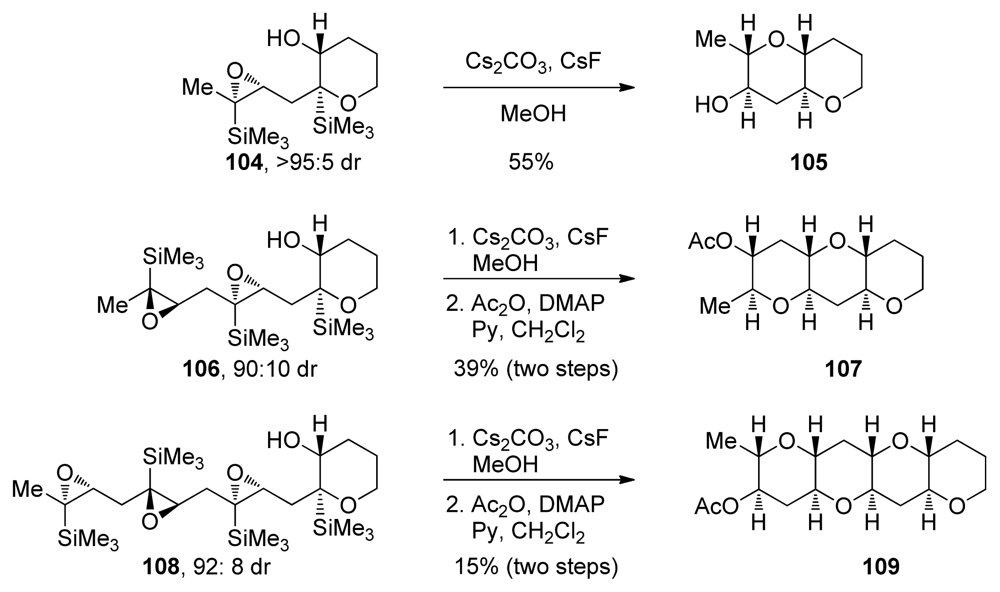
- Morimoto, Y; Yata, H; Nishikawa, Y. Assignment of the absolute configuration of the marine pentacyclic polyether (+)-enshuol by total synthesis. Angew Chem Int Ed 2007, 46, 6481–6484. [Google Scholar]
- Morimoto, Y; Nishikawa, Y; Takaishi, M. Total synthesis and complete assignment of the stereostructure of a cytotoxic bromotriterpene polyether (+)-aurilol. J Am Chem Soc 2005, 127, 5806–5807. [Google Scholar]
- Hudrlik, PF; Arcoleo, JP; Schwartz, RH; Misra, RN; Rona, RJ. Hydrolytic ring-opening of α,β-epoxysilanes to α,β-dihydroxysilanes. Tetrahedron Lett 1977, 18, 591–594. [Google Scholar]
- Hudrlik, PF; Hudrlik, AM; Kulkarni, AK. Protodesilylation reactions of simple β-hydroxysilanes (and α-hydroxysilanes). Homo-Brook rearrangements. J Am Chem Soc 1982, 104, 6809–6811. [Google Scholar]
- Hudrlik, PF; Holmes, PE; Hudrlik, AM. Protodesilylation reactions of α- and β-hydroxysilanes: deuterium labeling and silicon-directed epoxide openings. Tetrahedron Lett 1988, 29, 6395–6398. [Google Scholar]
- Hudrlik, PF; Hudrlik, AM. α,β-Epoxysilanes. In Advances in Silicon Chemistry, 1st ed; Larson, GL, Ed.; JAI Press: Greenwich, CT, USA, 1993; Volume 2, pp. 1–89. [Google Scholar]
- Fristad, WE; Bailey, TR; Paquette, LA; Gleiter, R; Boehm, MC. Regiospecific photosensitized oxygenation of vinylsilanes. A method for converting saturated ketones to 1,2-transposed allylic alcohols. Possible role of silicon in directing the regioselectivity of epoxysilane cleavage reactions. J Am Chem Soc 1979, 101, 4420–4423. [Google Scholar]
- Heffron, TP; Jamison, TF. SiMe3-based homologation-epoxidation-cyclization strategy for ladder thp synthesis. Org Lett 2003, 5, 2339–2342. [Google Scholar]
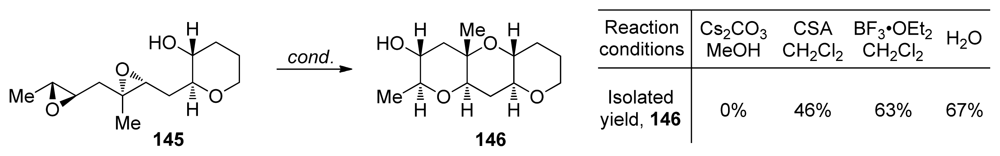
- Simpson, GL; Heffron, TP; Merino, E; Jamison, TF. Ladder polyether synthesis via epoxide-opening cascades using a disappearing directing group. J Am Chem Soc 2006, 128, 1056–1057. [Google Scholar]
- Hayashi, N; Fujiwara, K; Murai, A. The biomimetic construction of fused cyclic polyethers. Tetrahedron 1997, 53, 12425–12468. [Google Scholar]
- Heffron, TP; Jamison, TF. Concerning; Lewis; acid promoted, directing-group-free epoxide-ring- opening cascades. In Synlett; 2006; pp. 2329–2333. [Google Scholar]
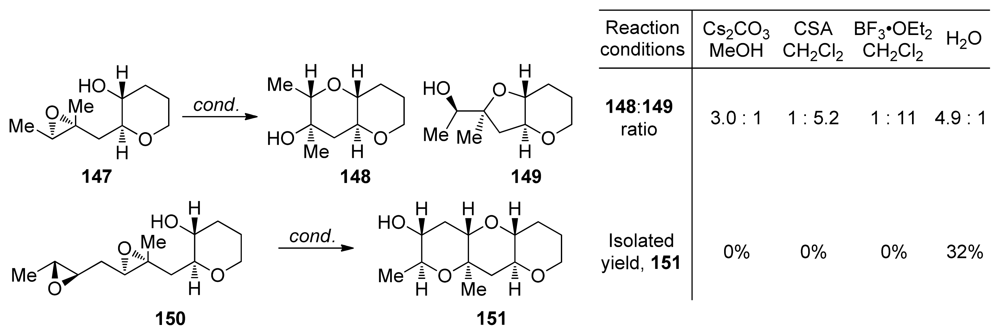
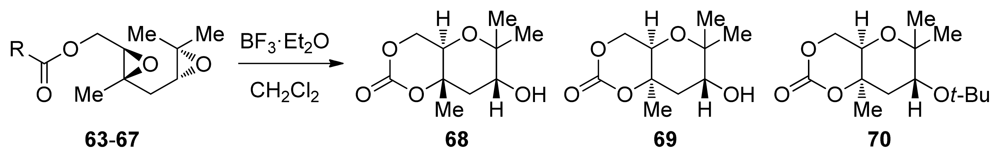
- Tong, R; McDonald, FE; Fang, X; Hardcastle, KI. Biomimetic synthesis of fused bispyran: Lewis acid effects on oxacyclizations of polyepoxides. Synthesis 2007, 2337–2342. [Google Scholar]
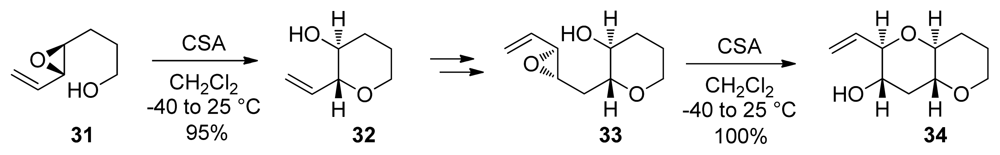
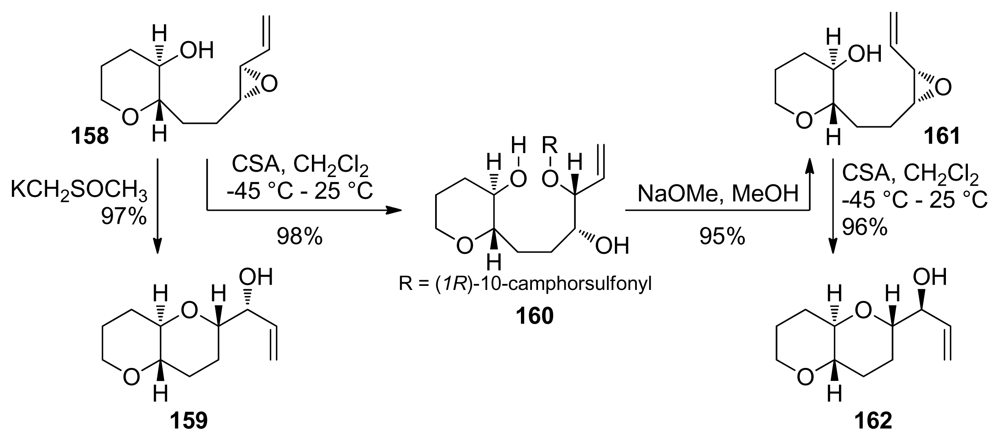
- Vilotijevic, I; Jamison, TF. Epoxide-opening cascades promoted by water. Science 2007, 317, 1189–1192. [Google Scholar]
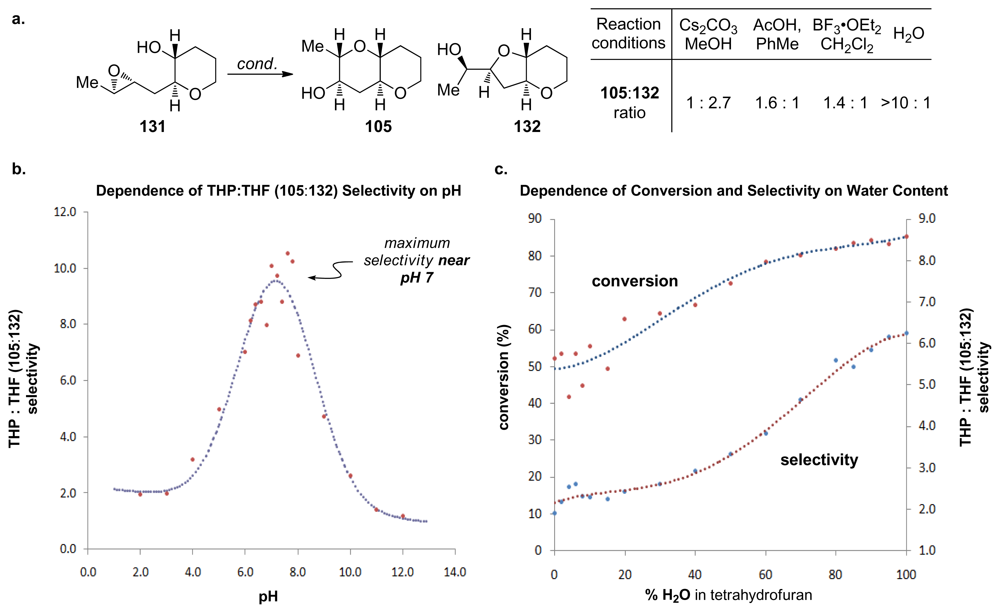
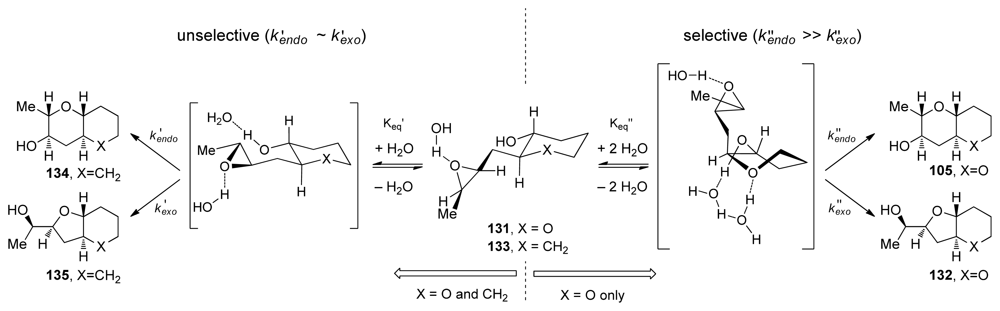
- Byers, JA; Jamison, TF. On the synergism between H2O and a tetrahydropyran template in the regioselective cyclization of an epoxy alcohol. J Am Chem Soc 2009, 131, 6383–6385. [Google Scholar]
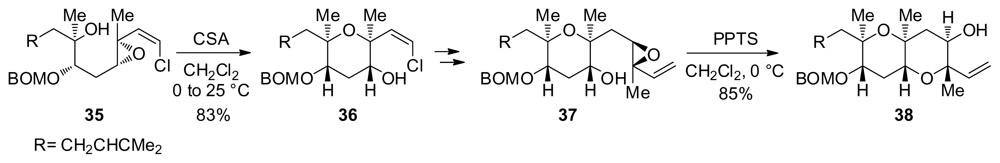
- Van Dyke, AR; Jamison, TF. Functionalized templates for the convergent assembly of polyethers: synthesis of the HIJK rings of gymnocin A. Angew Chem Int Edit 2009, 48, 4430–4432. [Google Scholar]
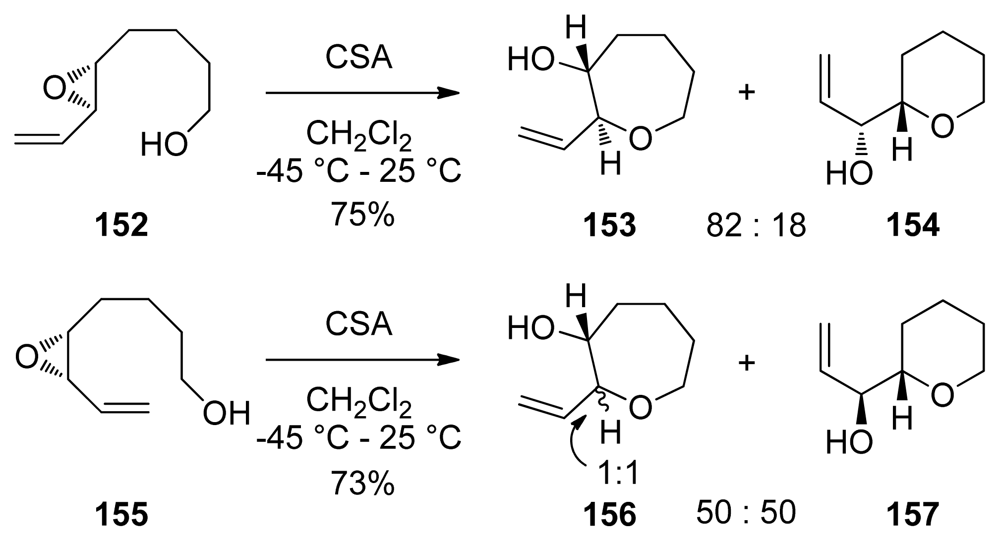
- Nicolaou, KC; Prasad, CVC; Somers, PK; Hwang, CK. Activation of 7-endo over 6-exo epoxide openings. Synthesis of oxepane and tetrahydropyran systems. J Am Chem Soc 1989, 111, 5335–5340. [Google Scholar]
- Matsukura, H; Morimoto, M; Koshino, H; Nakata, T. Stereoselective synthesis of tetrahydropyran and oxepane systems by the endo-cyclization of hydroxy styrylepoxides. Tetrahedron Lett 1997, 38, 5545–5548. [Google Scholar]
- Mukai, C; Sugimoto, Y-i; Ikeda, Y; Hanaoka, M. A new procedure for highly stereoselective and regioselective synthesis of 2-ethynyl-3-hydroxytetrahydropyran derivatives based on alkyne- Co2(CO)6 complex. Tetrahedron 1998, 54, 823–850. [Google Scholar]
- Mukai, C; Yamaguchi, S; Kim, IJ; Hanaoka, M. Co2(CO)8-mediated endo mode cyclization of epoxy alcohols: synthesis of 2-ethynyl-3-hydroxy-2-methyltetrahydropyran and 2-ethynyl-3- hydroxy-3-methyltetrahydropyran derivatives. Chem Pharm Bull 2001, 49, 613–618. [Google Scholar]
- Sasaki, M; Inoue, M; Takamatsu, K; Tachibana, K. Stereocontrolled synthesis of the JKLM ring fragment of ciguatoxin. J Org Chem 1999, 64, 9399–9415. [Google Scholar]
- Fujiwara, K; Mishima, H; Amano, A; Tokiwano, T; Murai, A. La(OTf)3-catalyzed 7-endo and 8-endo selective cyclizations of hydroxy epoxides. Tetrahedron Lett 1998, 39, 393–396. [Google Scholar]
- Cookson, RC; Liverton, NJ. A total synthesis of zoapatanol. J Chem Soc Perkin Trans 1 1985, 1589–1595. [Google Scholar]
- Whitby, R; Yeates, C; Kocienski, P; Costello, G. A synthetic approach to zoapatanol and related bicyclic analogs. J Chem Soc Chem Commun 1987, 429–430. [Google Scholar]
- Kocienski, P; Love, C; Whitby, R; Roberts, DA. Synthesis of zoapatanol. Tetrahedron Lett 1988, 29, 2867–2870. [Google Scholar]
- Kocienski, PJ; Love, CJ; Whitby, RJ; Costello, G; Roberts, DA. A total synthesis of (±)- zoapatanol and demethyl-ORF13811. Tetrahedron 1989, 45, 3839–3848. [Google Scholar]
- McDonald, FE; Wang, X; Do, B; Hardcastle, KI. Synthesis of oxepanes and trans-fused bisoxepanes via biomimetic, endo-regioselective tandem oxacyclizations of polyepoxides. Org Lett 2000, 2, 2917–2919. [Google Scholar]
- McDonald, FE; Bravo, F; Wang, X; Wei, X; Toganoh, M; Rodriguez, JR; Do, B; Neiwert, WA; Hardcastle, KI. Endo-oxacyclizations of polyepoxides: biomimetic synthesis of fused polycyclic ethers. J Org Chem 2002, 67, 2515–2523. [Google Scholar]
- Bravo, F; McDonald, FE; Neiwert, WA; Hardcastle, KI. Alkene substituents for selective activation of endo-regioselective polyepoxide oxacyclizations. Org Lett 2004, 6, 4487–4489. [Google Scholar]
- Kumar, VS; Aubele, DL; Floreancig, PE. Electron transfer initiated heterogenerative cascade cyclizations: polyether synthesis under nonacidic conditions. Org Lett 2002, 4, 2489–2492. [Google Scholar]
- Kumar, VS; Wan, S; Aubele, DL; Floreancig, PE. Oxidatively generated electrophiles as initiators of epoxide cascade cyclization processes. Tetrahedron: Asymmetry 2005, 16, 3570–3578. [Google Scholar]
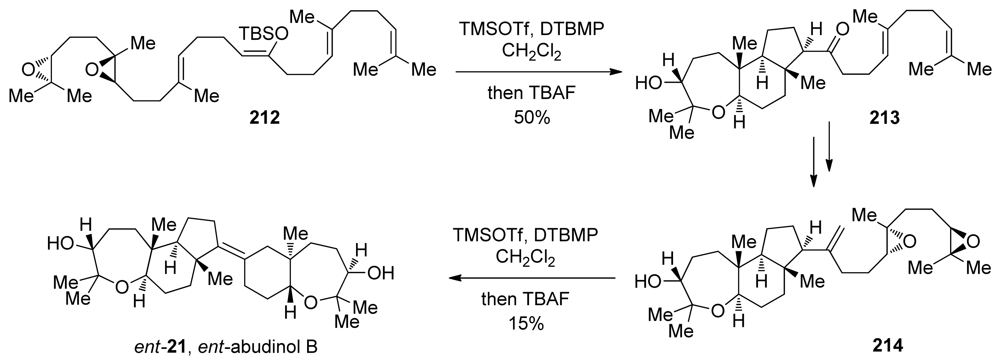
- Tong, R; Valentine, JC; McDonald, FE; Cao, R; Fang, X; Hardcastle, KI. Total syntheses of durgamone, nakorone, and abudinol B via biomimetic oxa- and carbacyclizations. J Am Chem Soc 2007, 129, 1050–1051. [Google Scholar]
- Tong, R; McDonald, FE. Mimicking biosynthesis: total synthesis of the triterpene natural product abudinol B from a squalene-like precursor. Angew Chem Int Ed 2008, 47, 4377–4379. [Google Scholar]
- Tong, RB; Boone, MA; McDonald, FE. Stereo- and regioselective synthesis of squalene tetraepoxide. J Org Chem 2009, 74, 8407–8409. [Google Scholar]
- Morimoto, Y; Okita, T; Takaishi, M; Tanaka, T. Total synthesis and determination of the absolute configuration of (+)-intricatetraol. Angew Chem Int Ed 2007, 46, 1132–1135. [Google Scholar]
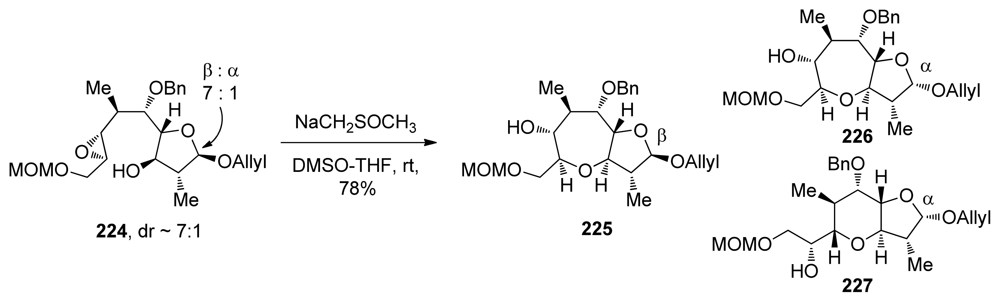
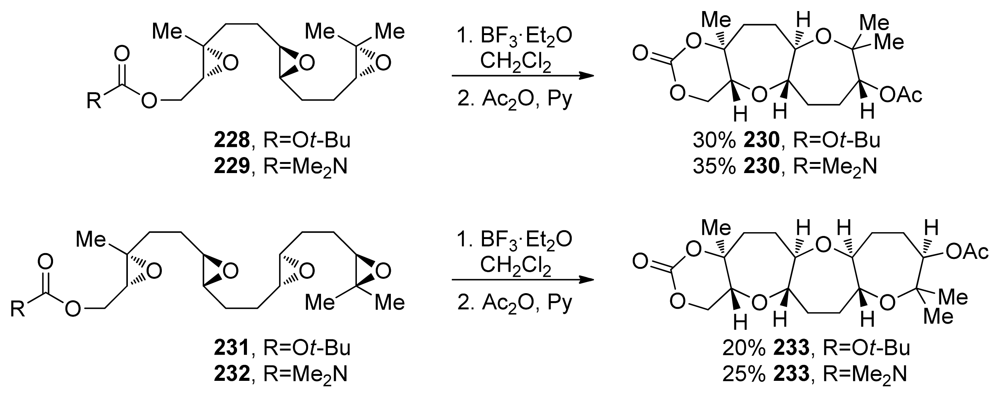
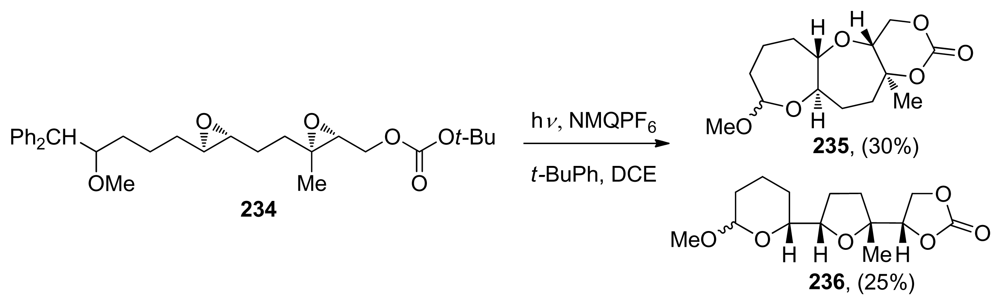
- Valentine, JC; McDonald, FE; Neiwert, WA; Hardcastle, KI. Biomimetic synthesis of trans, syn, trans-fused polyoxepanes: remarkable substituent effects on the endo-regioselective oxacyclization of polyepoxides. J Am Chem Soc 2005, 127, 4586–4587. [Google Scholar]
- Tokunaga, M; Larrow, JF; Kakiuchi, F; Jacobsen, EN. Asymmetric catalysis with water: efficient kinetic resolution of terminal epoxides by means of catalytic hydrolysis. Science 1997, 277, 936–938. [Google Scholar]
- Wu, MH; Hansen, KB; Jacobsen, EN. Regio- and enantioselective cyclization of epoxy alcohols catalyzed by a [CoIII(salen)] complex. Angew Chem Int Ed 1999, 38, 2012–2014. [Google Scholar]
- Wu, B; Parquette, JR; RajanBabu, TV. Regiodivergent ring opening of chiral aziridines. Science 2009, 326, 1662. [Google Scholar]
- Janda, KD; Shevlin, CG; Lerner, RA. Antibody catalysis of a disfavored chemical transformation. Science 1993, 259, 490–493. [Google Scholar]
- Janda, KD; Shevlin, CG; Lerner, RA. Oxepane synthesis along a disfavored pathway: the rerouting of a chemical reaction using a catalytic antibody. J Am Chem Soc 1995, 117, 2659–2660. [Google Scholar]
- Gallimore, AR; Stark, CBW; Bhatt, A; Harvey, BM; Demydchuk, Y; Bolanos-Garcia, V; Fowler, DJ; Staunton, J; Leadlay, PF; Spencer, JB. Evidence for the role of the monB genes in polyether ring formation during monensin biosynthesis. Chem Biol 2006, 13, 453–460. [Google Scholar]
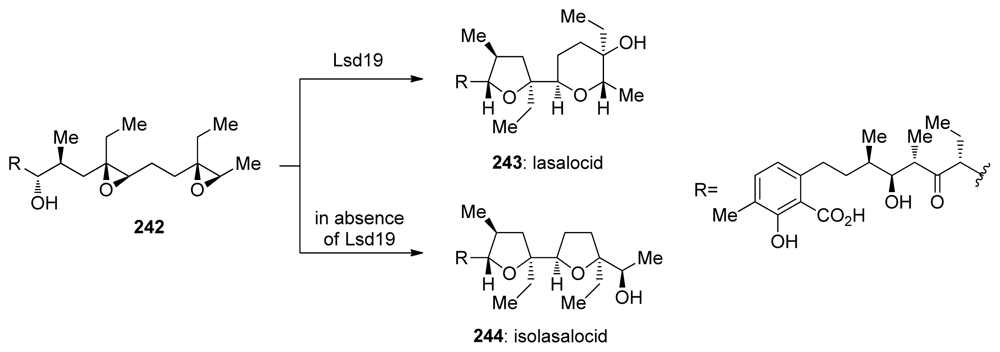
- Shichijo, Y; Migita, A; Oguri, H; Watanabe, M; Tokiwano, T; Watanabe, K; Oikawa, H. Epoxide hydrolase Lsd19 for polyether formation in the biosynthesis of lasalocid A: direct experimental evidence on polyene-polyepoxide hypothesis in polyether biosynthesis. J Am Chem Soc 2008, 130, 12230–122. [Google Scholar]
- Smith, L; Hong, H; Spencer, JB; Leadlay, PF. Analysis of specific mutants in the lasalocid gene cluster: Evidence for enzymatic catalysis of a disfavoured polyether ring closure. ChemBioChem 2008, 9, 2967–2975. [Google Scholar]
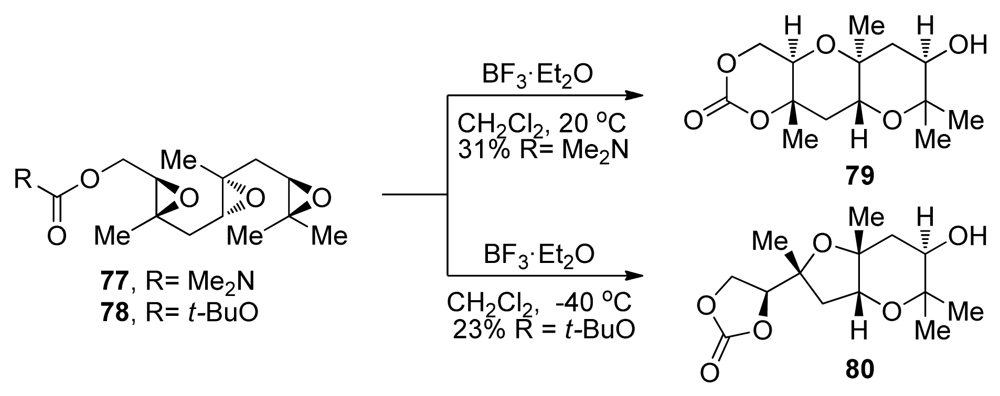
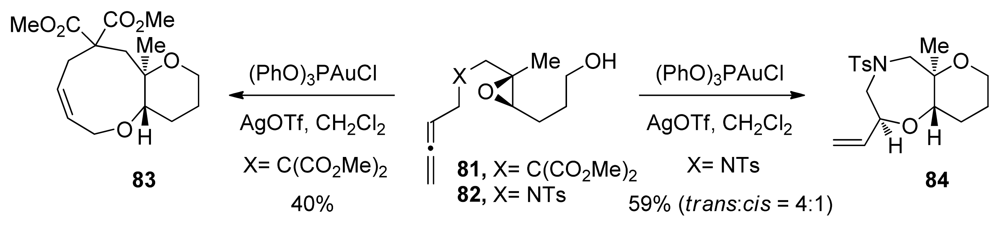
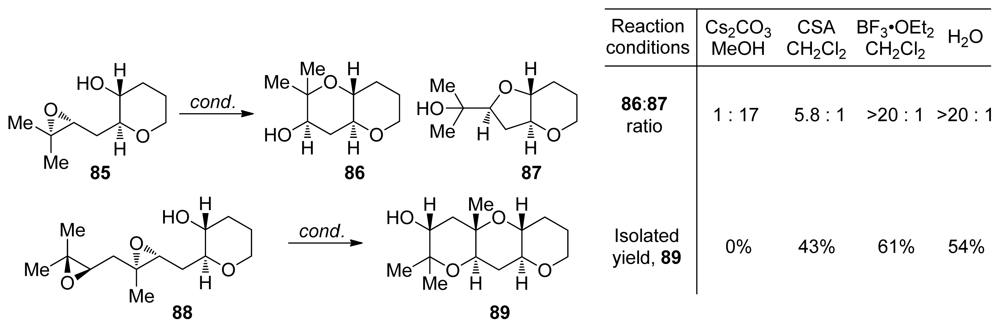
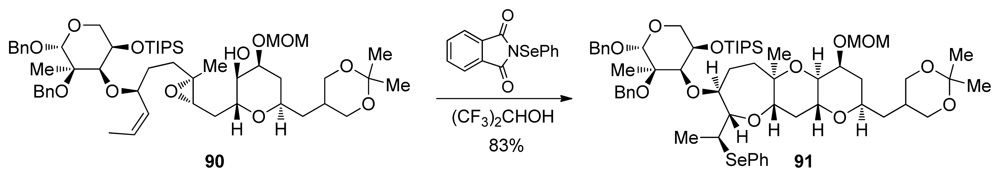

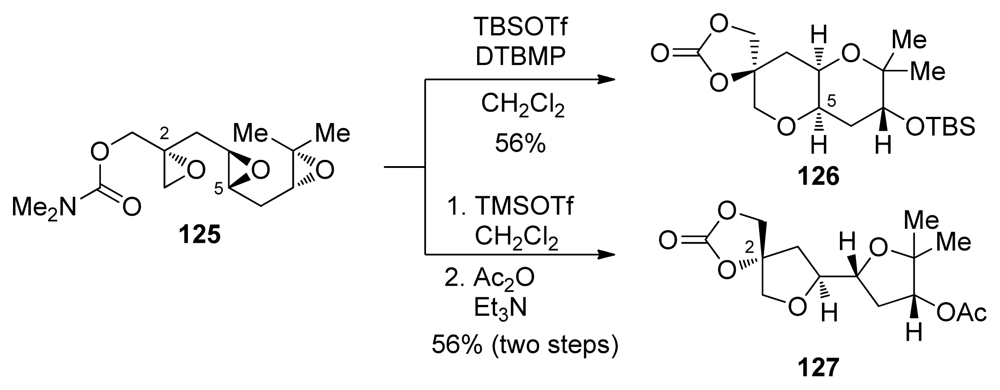
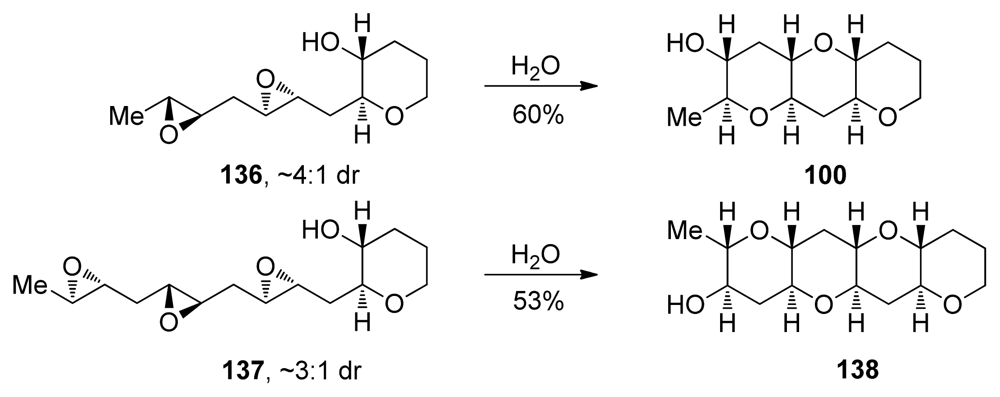
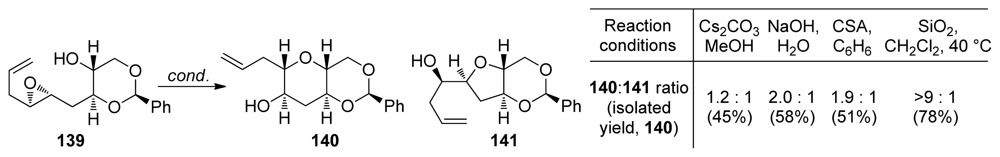
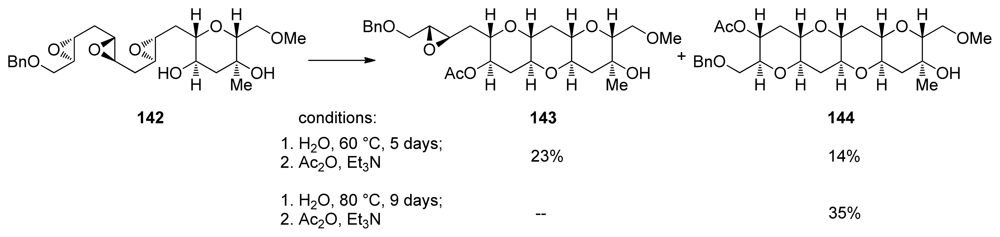


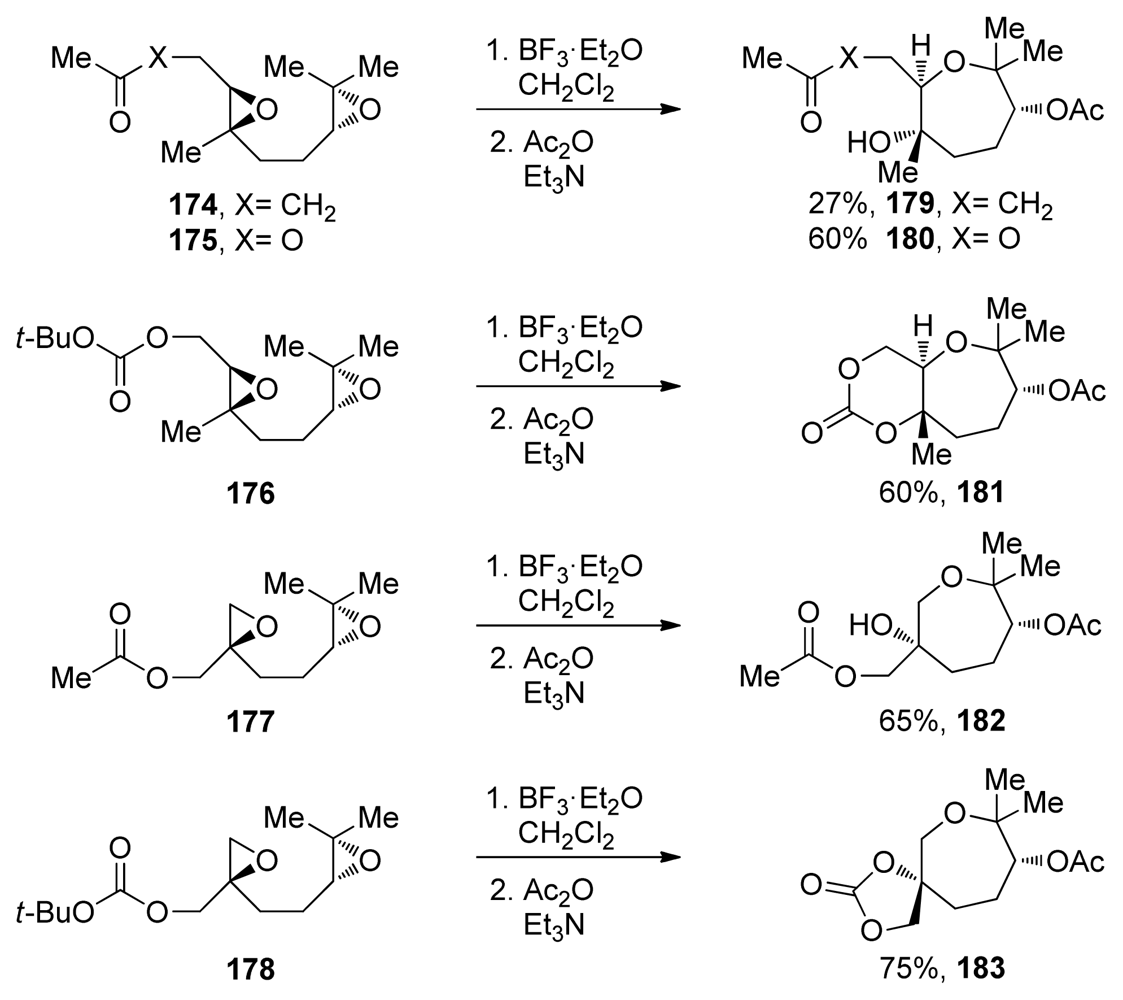
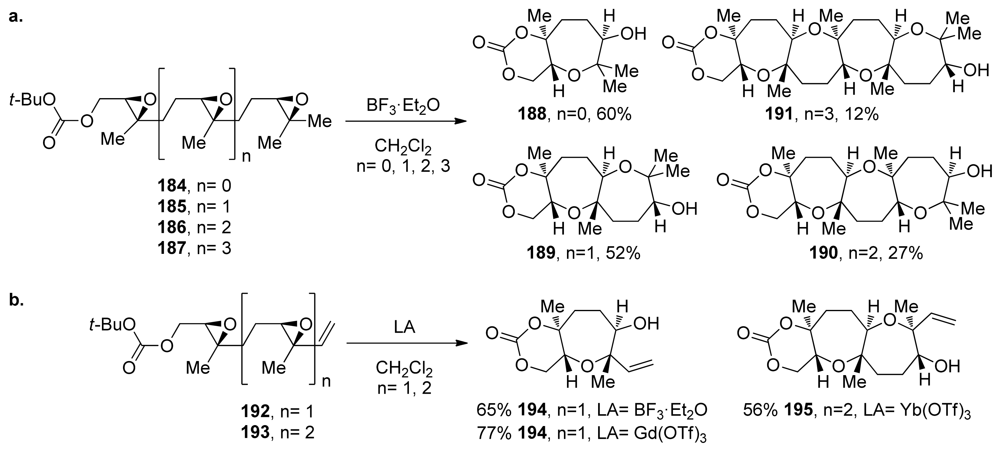
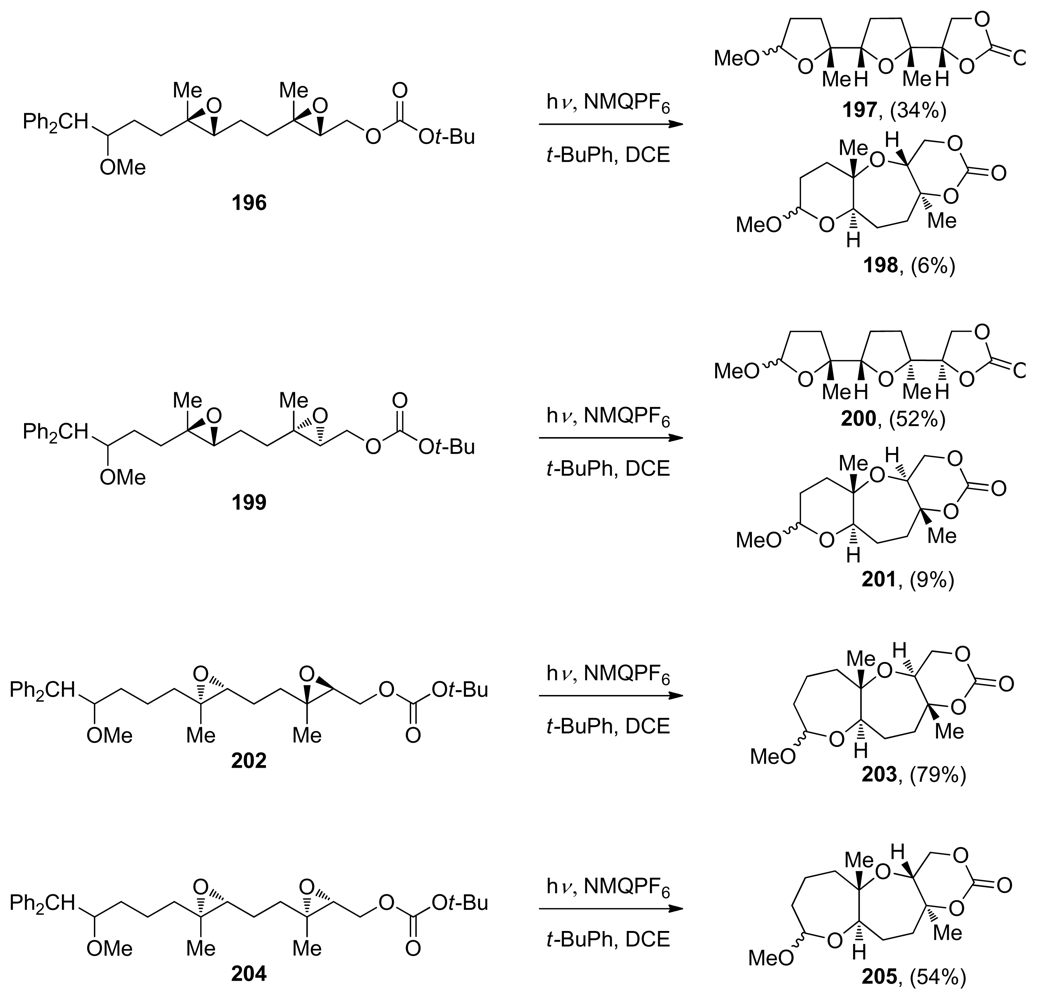
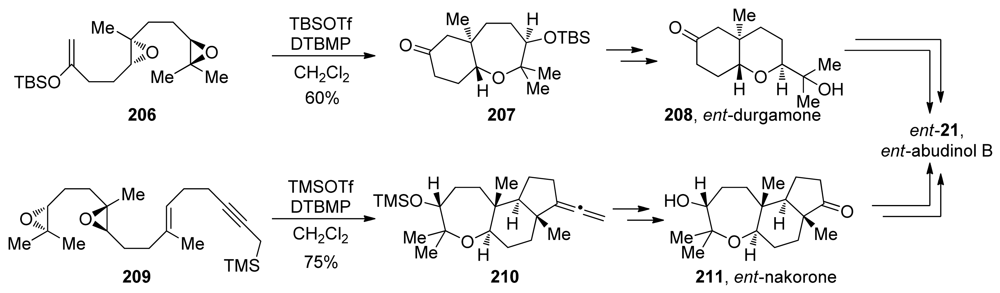


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | R | Concentration | T (°C) | Yield of 68 (%) | Yield of 69 (%) | Yield of 70 (%) |
| 1 | 63 | t-BuO | 0.05 M | −40 | <4 | 56 | 12 |
| 2 | 63 | t-BuO | 0.05 M | 40 | – | 65 | 4 |
| 3 | 63 | t-BuO | 0.5 M | −40 | – | 42 | 10 |
| 4 | 64 | PhNH | 0.05 M | −40 | – | 70 | – |
| 5 | 65 | Me2N | 0.05 M | −40 | 35 | 10 | – |
| 6 | 65 | Me2N | 0.05 M | 20 | 55 | 21 | – |
| 7 | 66 | (CH2)4N | 0.05 M | −40 | 32 | 9 | – |
| 8 | 67 | O(CH2CH2)2N | 0.05 M | −40 | 34 | 13 | – |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vilotijevic, I.; Jamison, T.F. Synthesis of Marine Polycyclic Polyethers via Endo-Selective Epoxide-Opening Cascades. Mar. Drugs 2010, 8, 763-809. https://doi.org/10.3390/md8030763
Vilotijevic I, Jamison TF. Synthesis of Marine Polycyclic Polyethers via Endo-Selective Epoxide-Opening Cascades. Marine Drugs. 2010; 8(3):763-809. https://doi.org/10.3390/md8030763
Chicago/Turabian StyleVilotijevic, Ivan, and Timothy F. Jamison. 2010. "Synthesis of Marine Polycyclic Polyethers via Endo-Selective Epoxide-Opening Cascades" Marine Drugs 8, no. 3: 763-809. https://doi.org/10.3390/md8030763