ω-Conotoxins GVIA, MVIIA and CVID: SAR and Clinical Potential


Abstract
:Introduction
ω-Conotoxins
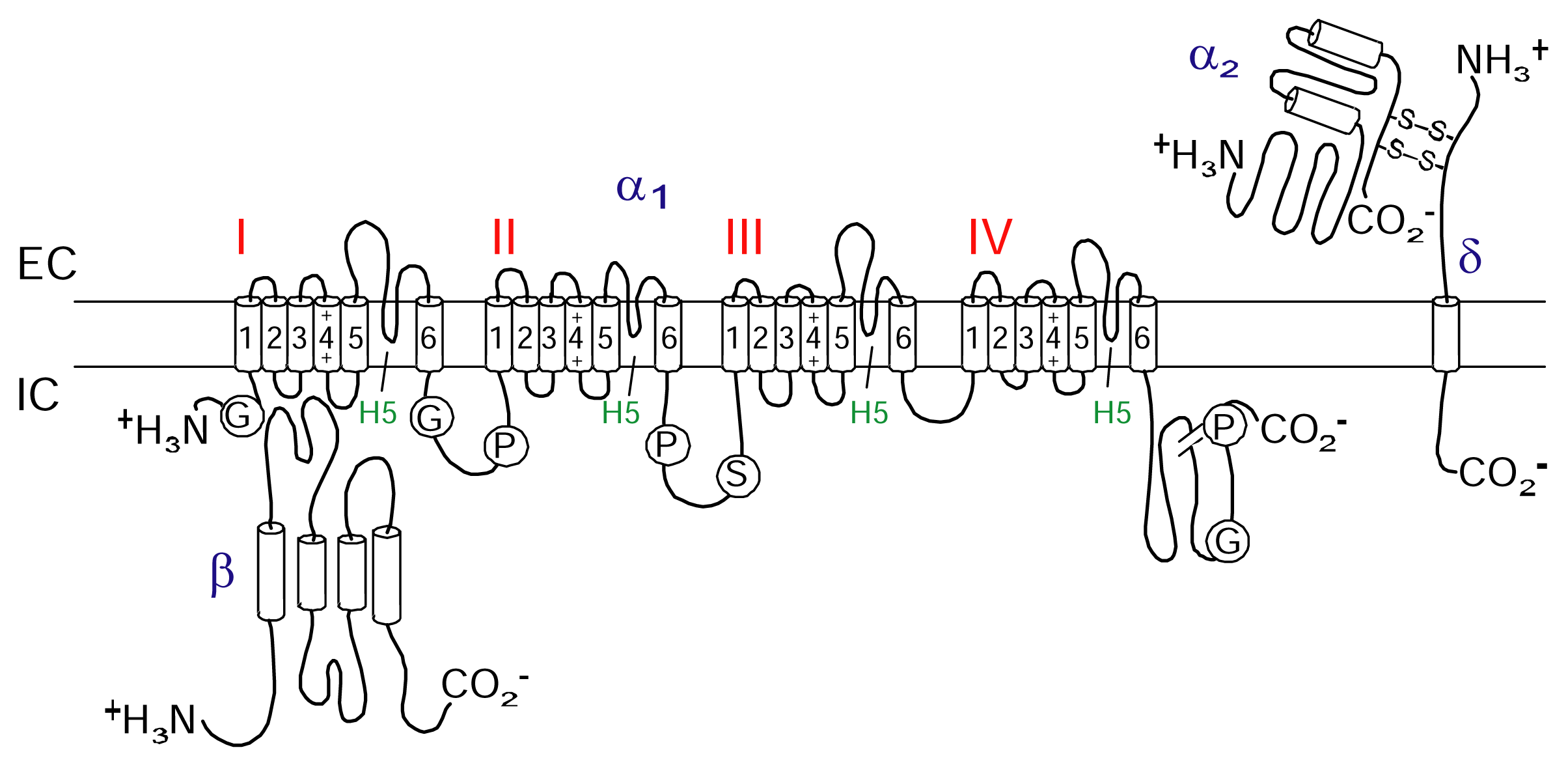
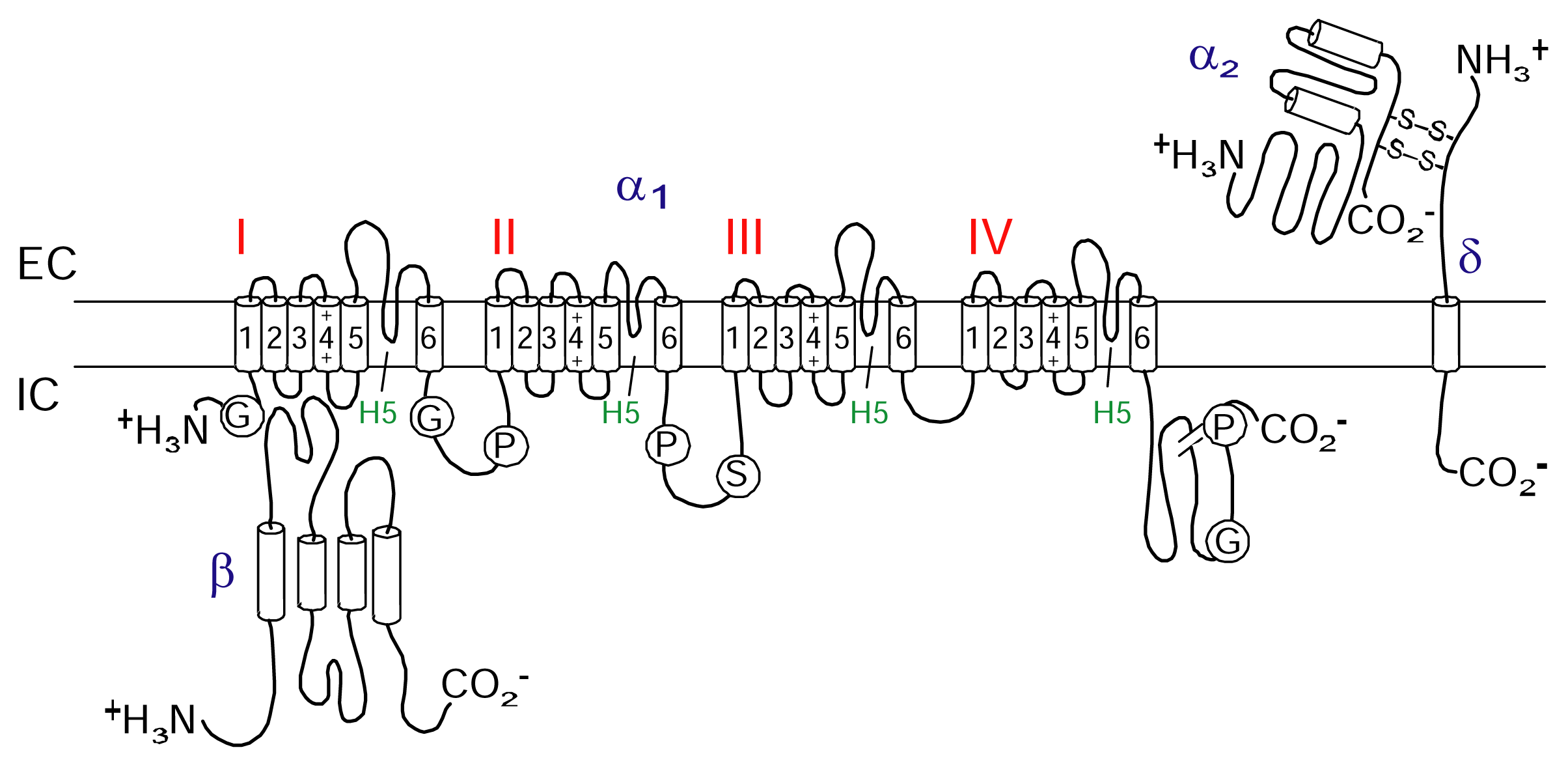
Voltage-gated calcium channels
CaV Channel as a therapeutic target
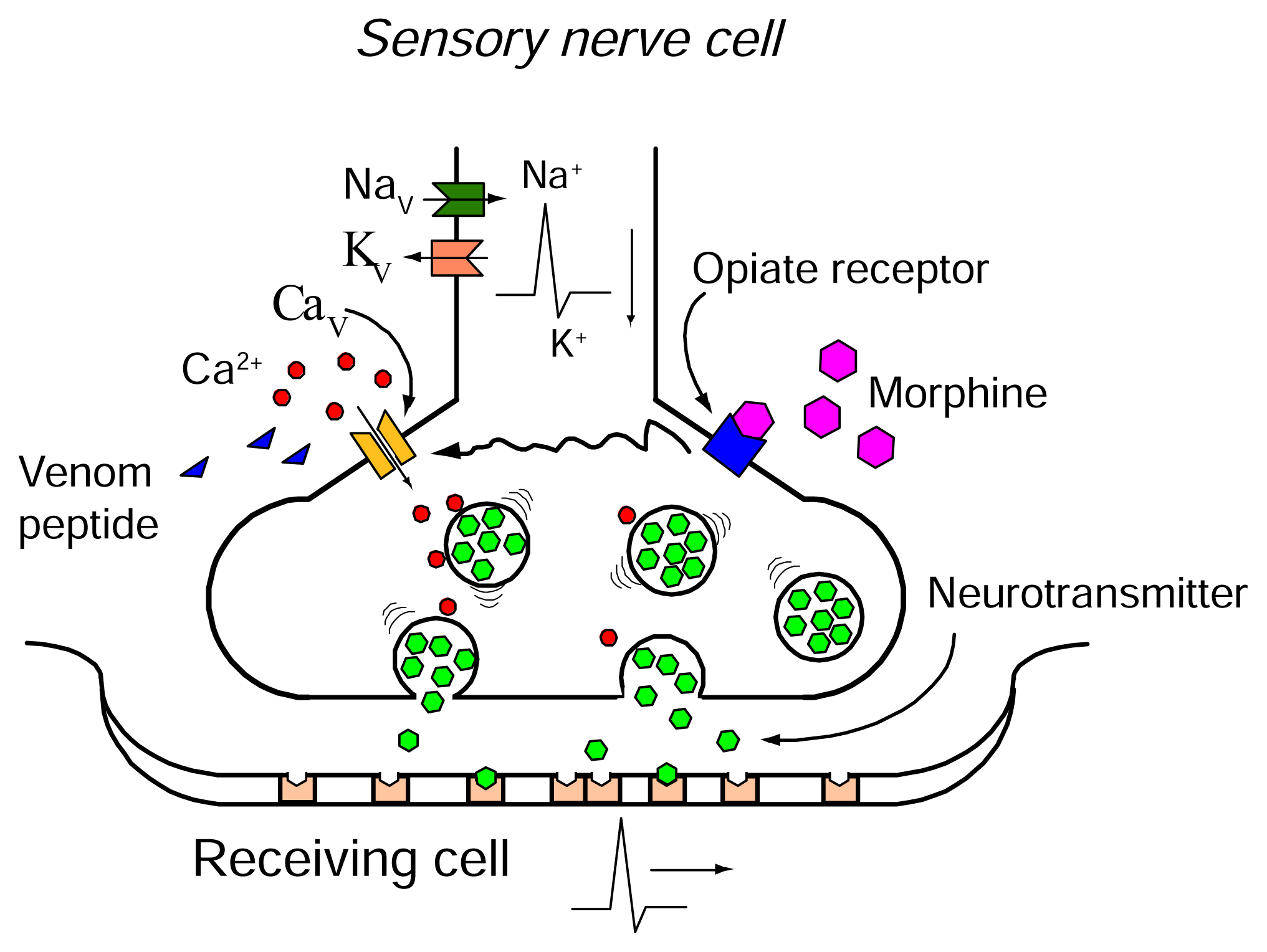
ω-Conotoxins as therapeutics
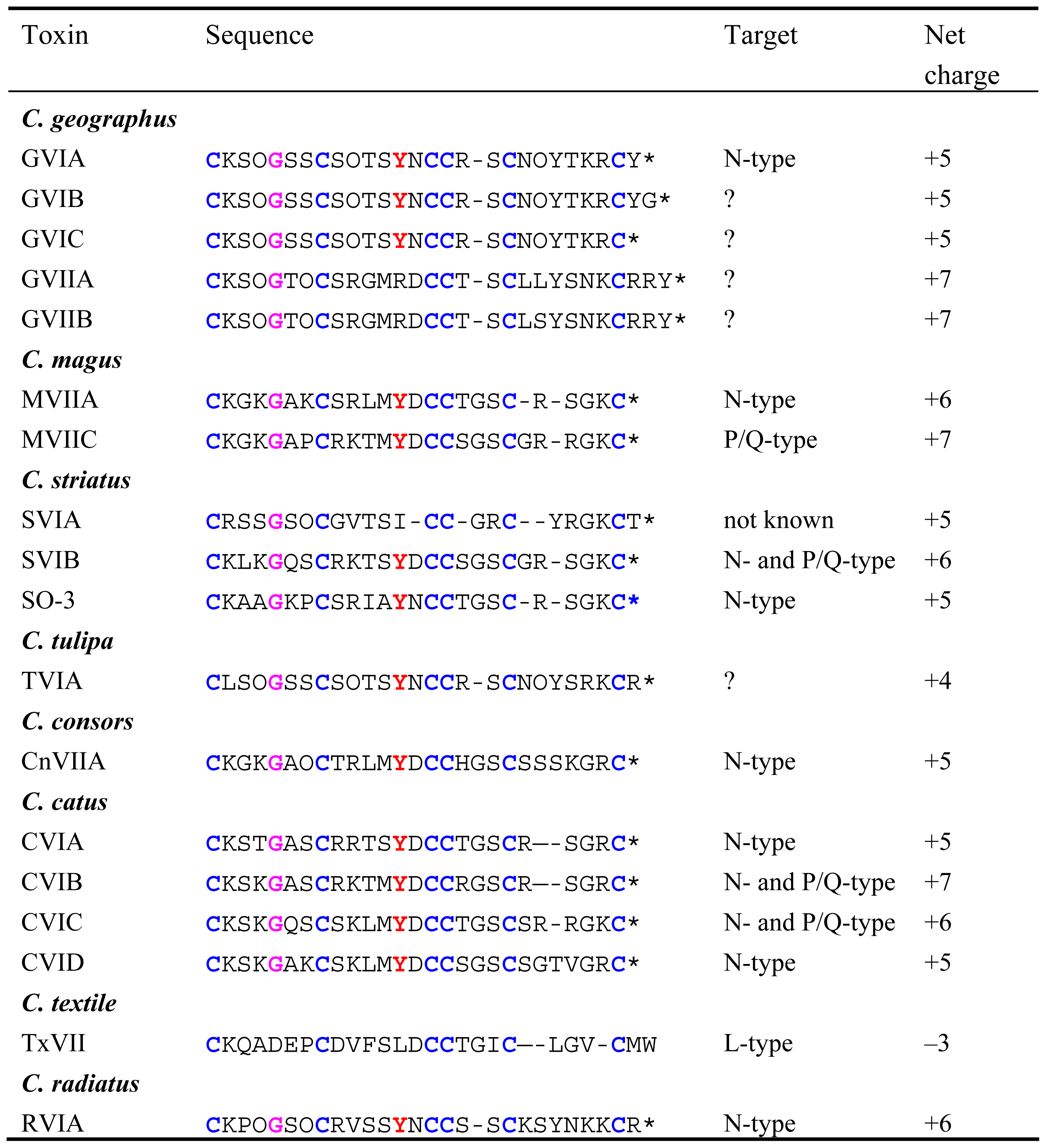
GVIA
MVIIC and SVIB
MVIIA (Ziconotide or Prialt)
CVID (AM336)
Opioids and ω-conotoxins
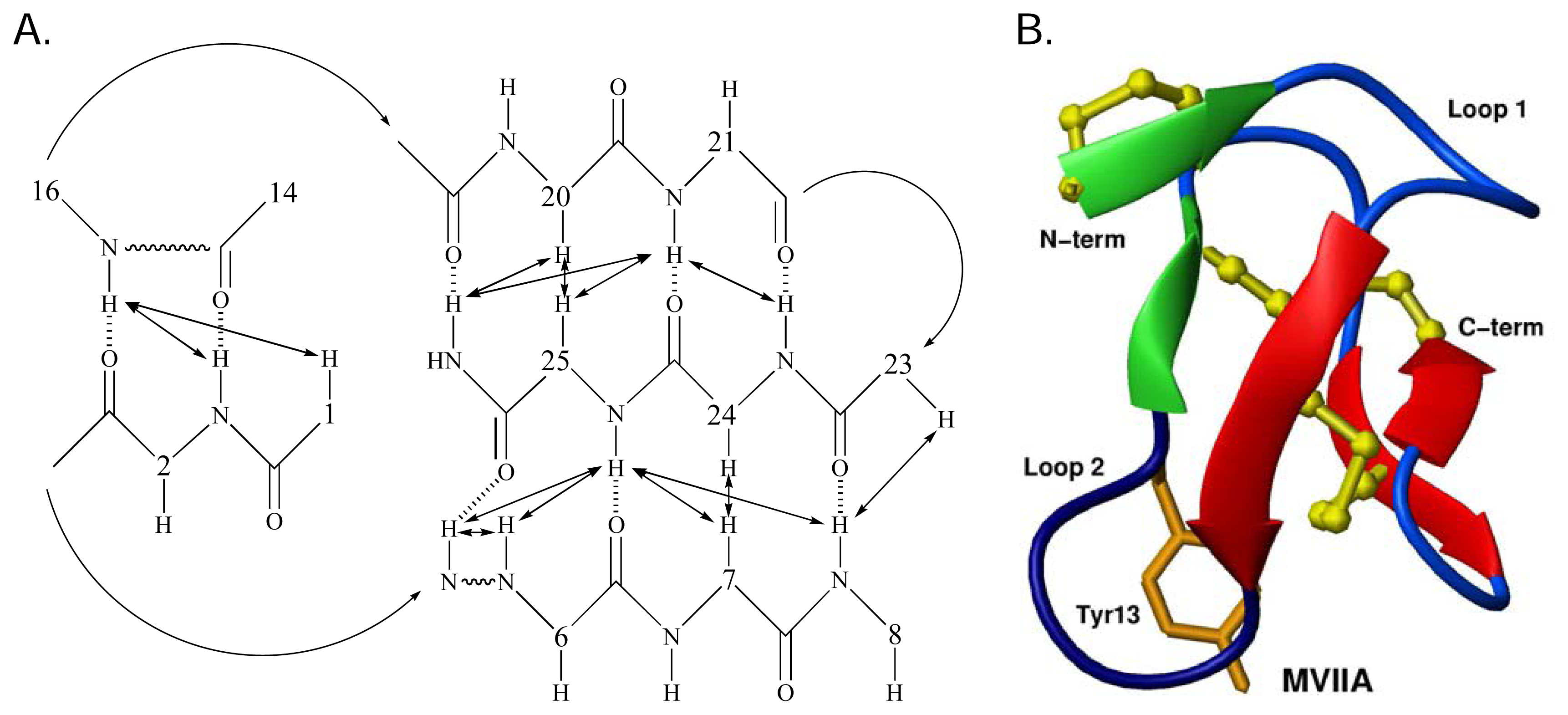
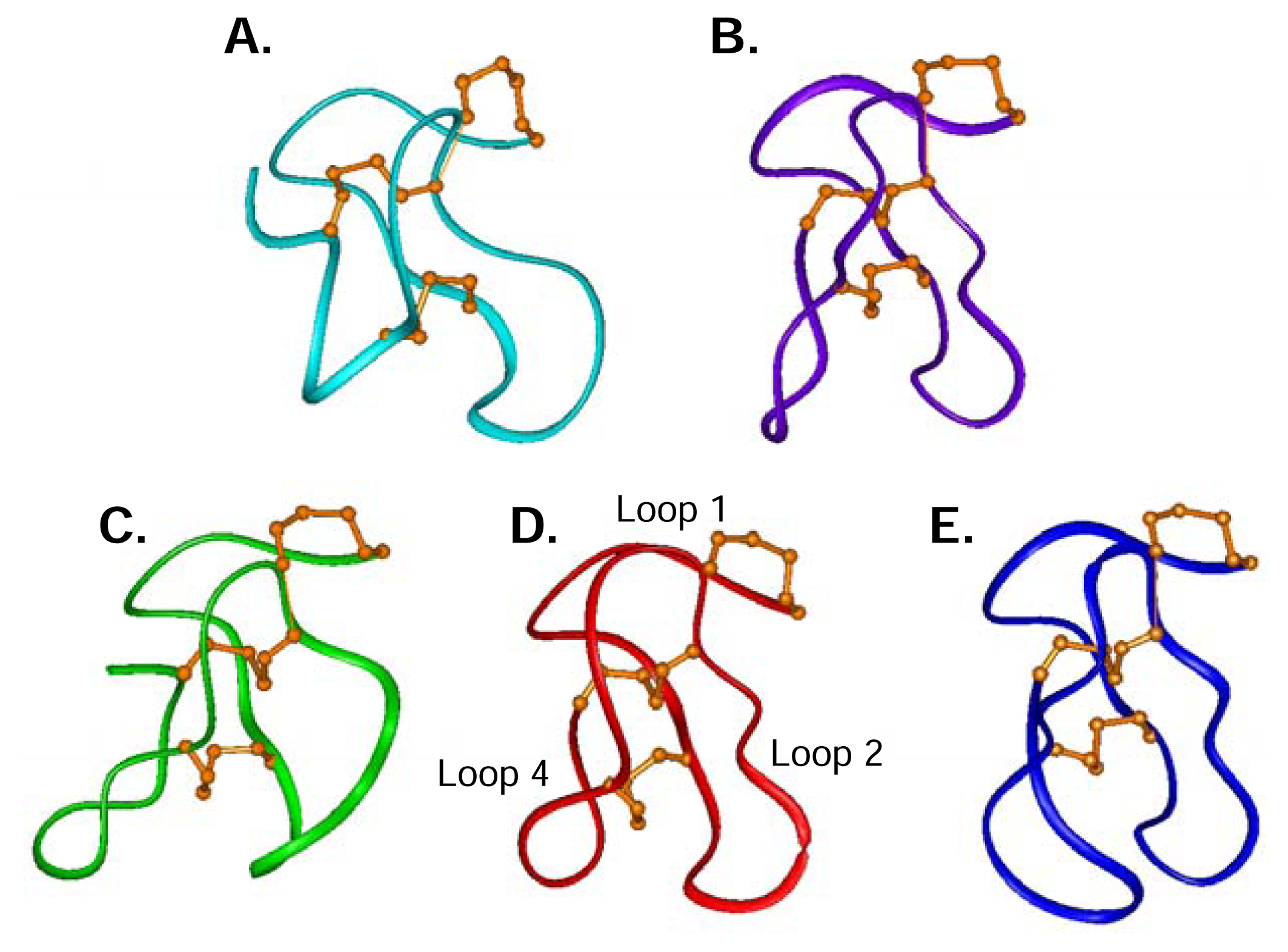
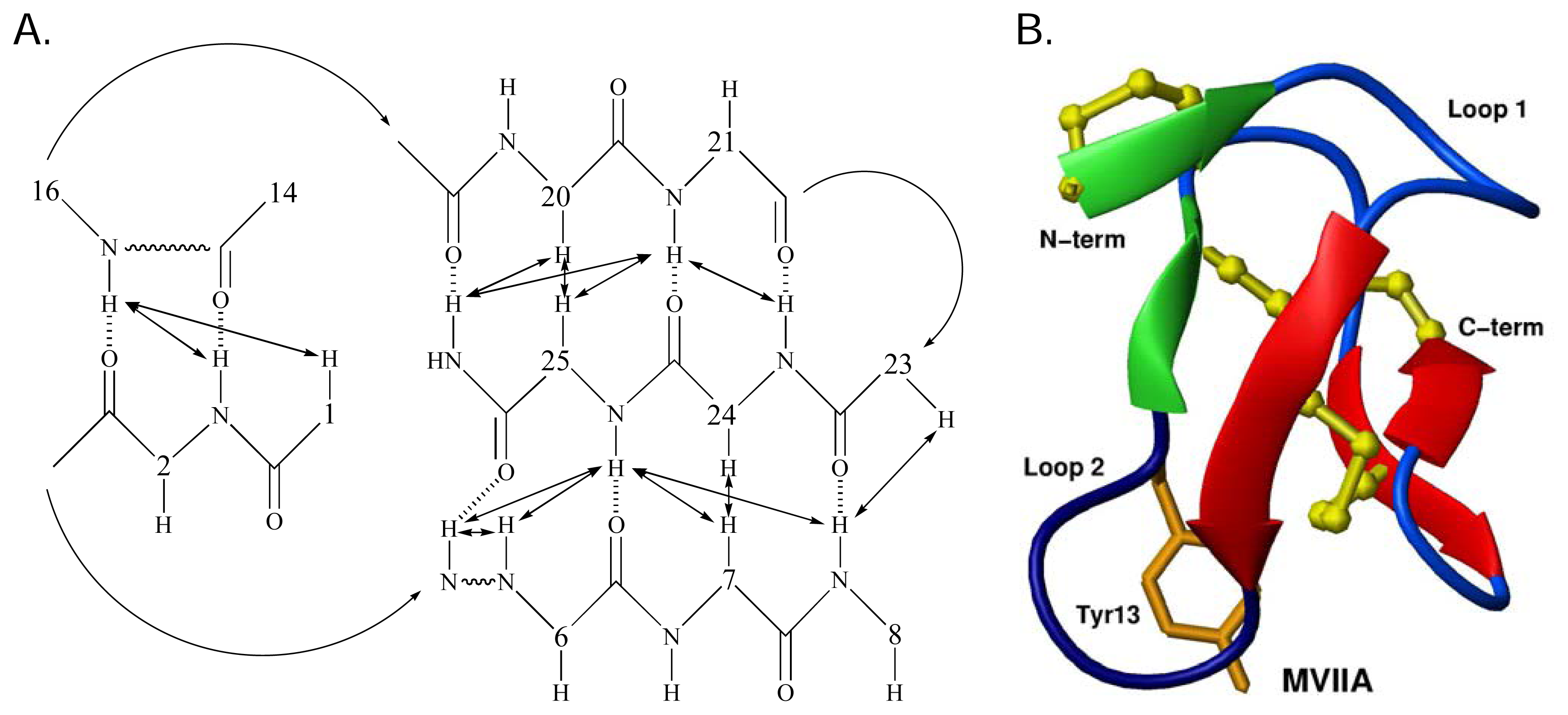
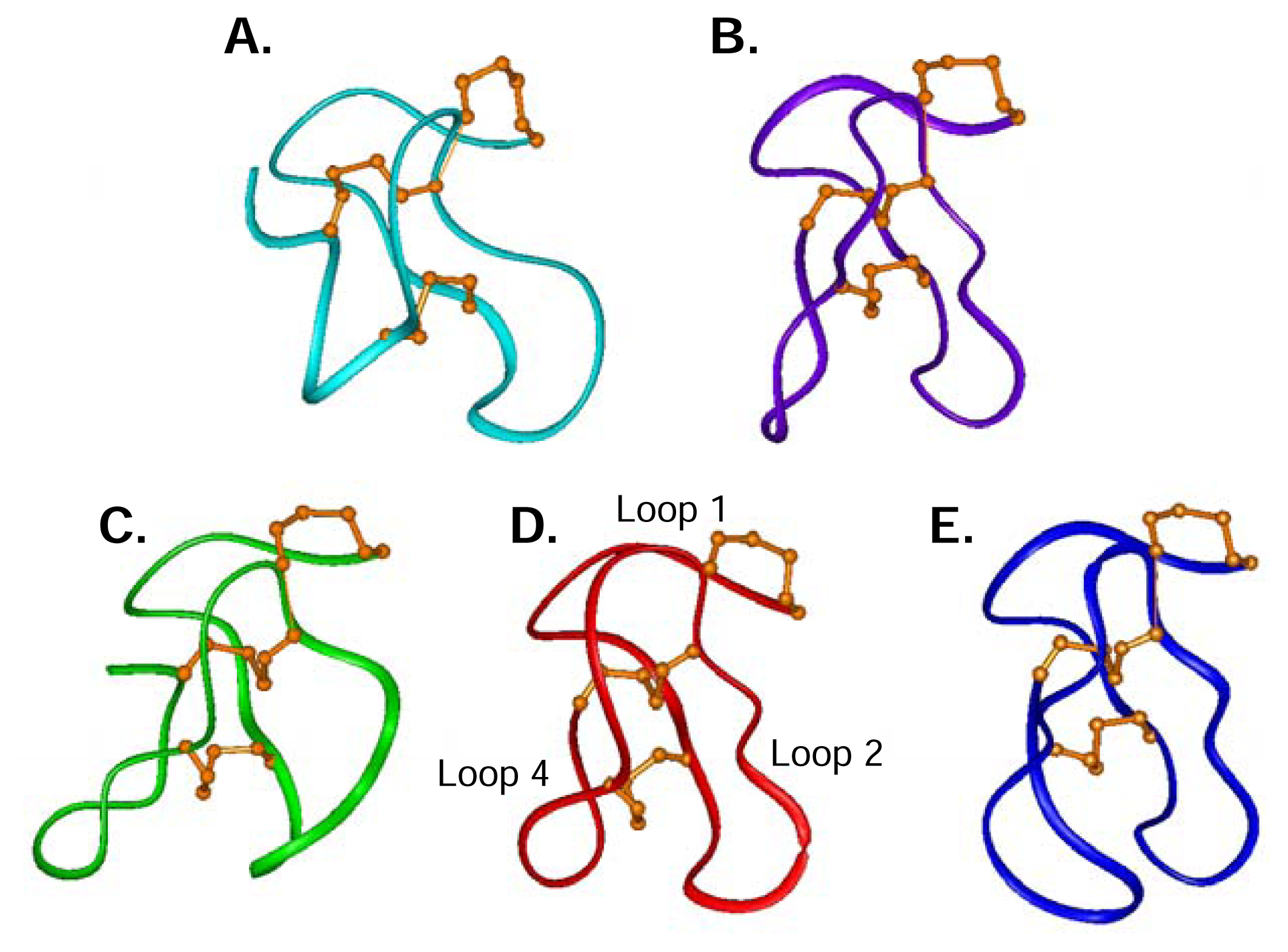
Structural studies of ω-conotoxins
The role of structure in structure-activity relationships
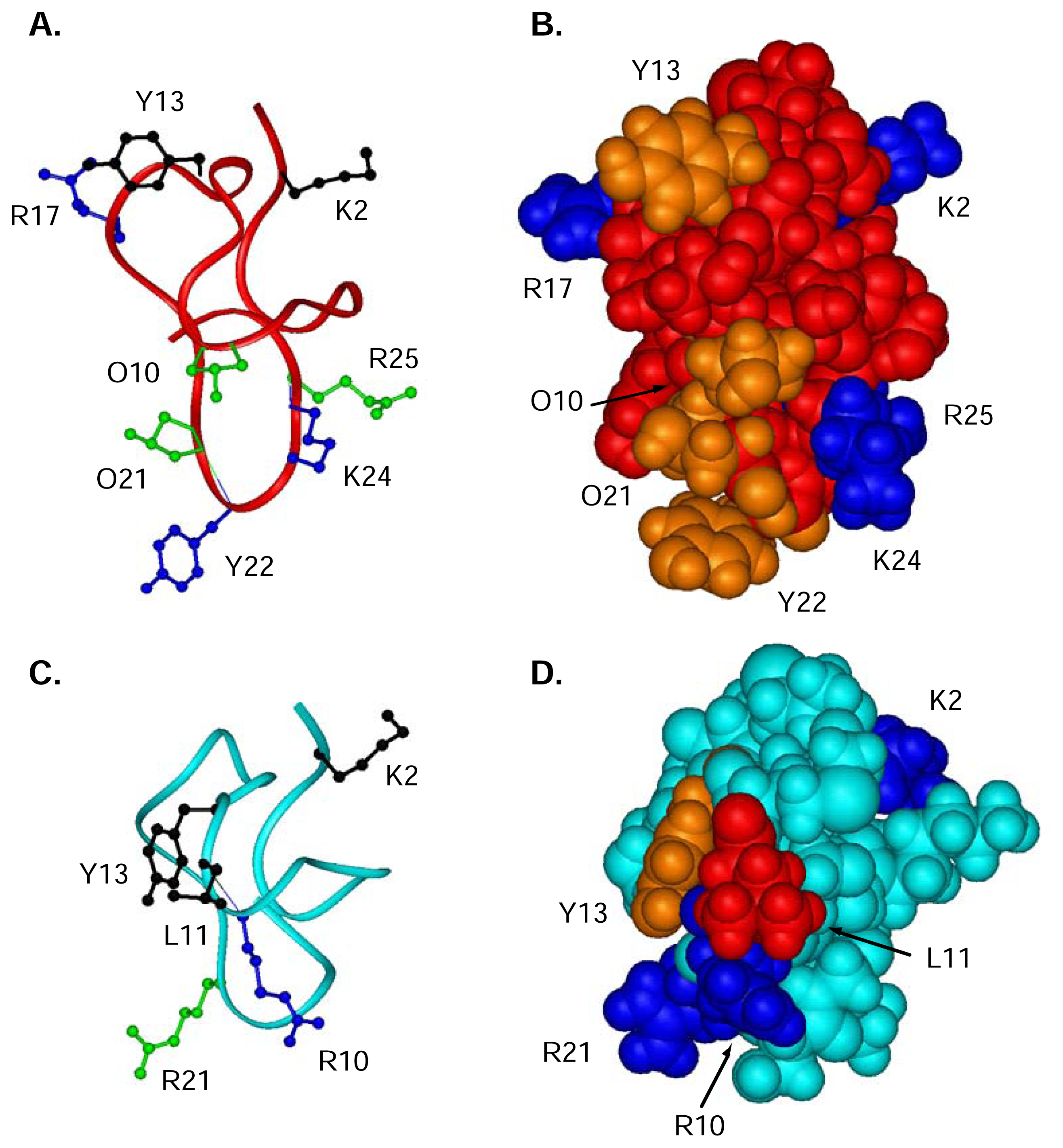
ω-Conotoxin residues important for binding to the N-type CaV channel
Abbreviations
| FDA | Food and Drug Administration |
| AChR | nicotinic acetylcholine receptor |
| NaV | voltage-gated sodium channel |
| KV | voltage-gated potassium channel |
| CaV | voltage-gated calcium channel |
| CNS | central nervous system |
| PNS | peripheral nervous system |
| IT | intrathecal |
| ED50 | dose causing 50% effect |
| TD50 | dose causing toxicity in 50% of animals |
| IC50 | dose causing 50% inhibition |
| NMR | nuclear magnetic resonance spectroscopy |
| 3D | three dimensional |
| SAR | structure-activity relationships |
| CD | circular dichroism |
- Samples Availability: Not available.
References
- Olivera, B. M.; Gray, W. R.; Zeikus, R.; McIntosh, J. M.; Varga, J.; Rivier, J.; de Santos, V.; Cruz, L. J. Peptide neurotoxins from fish-hunting cone snails. Science 1985, 230, 1338–1343. [Google Scholar]
- Olivera, B. M.; Rivier, J.; Clark, C.; Ramilo, C. A.; Corpuz, G. P.; Abogadie, F. C.; Mena, E. E.; Woodward, S. R.; Hillyard, D. R.; Cruz, L. J. Diversity of Conus neuropeptides. Science 1990, 249, 257–263. [Google Scholar]
- Olivera, B. M.; Rivier, J.; Scott, J. K.; Hillyard, D. R.; Cruz, L. J. Conotoxins. J. Biol. Chem 1991, 266, 22067–22070. [Google Scholar]
- McIntosh, J. M.; Jones, R. M. Cone venom-from accidental stings to deliberate injection. Toxicon 2001, 39, 1447–1451. [Google Scholar]
- Miljanich, G. P.; Ramachandran, J. Antagonists of neuronal calcium channels: structure, function, and therapeutic implications. Annu. Rev. Pharmacol. Toxicol 1995, 35, 707–734. [Google Scholar]
- Fainzilber, M.; Lodder, J. C.; van der Schors, R. C.; Li, K. W.; Yu, Z.; Burlingame, A. L.; Geraerts, W. P.; Kits, K. S. A novel hydrophobic omega-conotoxin blocks molluscan dihydropyridine-sensitive calcium channels. Biochemistry 1996, 35, 8748–8752. [Google Scholar]
- Cruz, L. J.; Gray, W. R.; Olivera, B. M.; Zeikus, R. D.; Kerr, L.; Yoshikami, D.; Moczydlowski, E. Conus geographus toxins that discriminate between neuronal and muscle sodium channels. J. Biol. Chem 1985, 260, 9280–9288. [Google Scholar]
- Takahashi, T.; Momiyama, A. Different types of calcium channels mediate central synaptic transmission. Nature 1993, 366, 156–158. [Google Scholar]
- Hillyard, D. R.; Monje, V. D.; Mintz, I. M.; Bean, B. P.; Nadasdi, L.; Ramachandran, J.; Miljanich, G.; Azimi-Zoonooz, A.; McIntosh, J. M.; Cruz, L. J.; Imperial, J. S.; Olivera, B. M. A new Conus peptide ligand for mammalian presynaptic Ca2+ channels. Neuron 1992, 9, 69–77. [Google Scholar]
- Lewis, R. J.; Nielsen, K. J.; Craik, D. J.; Loughnan, M. L.; Adams, D. A.; Sharpe, I. A.; Luchian, T.; Adams, D. J.; Bond, T.; Thomas, L.; Jones, A.; Matheson, J. L.; Drinkwater, R.; Andrews, P. R.; Alewood, P. F. Novel omega-conotoxins from Conus catus discriminate among neuronal calcium channel subtypes. J. Biol. Chem 2000, 275, 35335–35344. [Google Scholar]
- Myers, R. A.; Cruz, L. J.; Rivier, J. E.; Olivera, B. M. Conus Peptides as Chemical Probes for Receptors and Ion Channels. Chem. Rev 1993, 93, 1923–1936. [Google Scholar]
- Olivera, B. M.; McIntosh, J. M.; Cruz, L. J.; Luque, F. A.; Gray, W. R. Purification and sequence of a presynaptic peptide toxin from Conus geographus venom. Biochemistry 1984, 23, 5087–5090. [Google Scholar]
- Abe, T.; Saisu, H. Identification of the receptor for omega-conotoxin in brain. Probable components of the calcium channel. J. Biol. Chem 1987, 262, 9877–9882. [Google Scholar]
- Olivera, B. M.; Cruz, L. J.; de Santos, V.; LeCheminant, G. W.; Griffin, D.; Zeikus, R.; McIntosh, J. M.; Galyean, R.; Varga, J.; Gray, W. R.; Rivier, J. Neuronal calcium channel antagonists. Discrimination between calcium channel subtypes using omega-conotoxin from Conus magus venom. Biochemistry 1987, 26, 2086–2090. [Google Scholar]
- Ramilo, C. A.; Zafaralla, G. C.; Nadasdi, L.; Hammerland, L. G.; Yoshikami, D.; Gray, W. R.; Kristipati, R.; Ramachandran, J.; Miljanich, G.; Olivera, B. M.; Cruz, L. J. Novel alpha- and omega-conotoxins from Conus striatus venom. Biochemistry 1992, 31, 9919–9926. [Google Scholar]
- Wen, L.; Yang, S.; Qiao, H.; Liu, Z.; Zhou, W.; Zhang, Y.; Huang, P. SO-3, a new O-superfamily conopeptide derived from Conus striatus, selectively inhibits N-type calcium currents in cultutured hipppcampal neurons. Br. J. Pharmacol 2005, 145, 728–739. [Google Scholar]
- Fox, J. A. Novel omega-conopeptides reduced field potential amplitudes in the rat hippocampal slice. Neurosci. Lett 1994, 165, 157–160. [Google Scholar]
- Favreau, P.; Gilles, N.; Lamthanh, H.; Bournaud, R.; Shimahara, T.; Bouet, F.; Laboute, P.; Letourneux, Y.; Menez, A.; Molgo, J.; Le Gall, F. A new omega-conotoxin that targets N-type voltage-sensitive calcium channels with unusual specificity. Biochemistry 2001, 40, 14567–14575. [Google Scholar]
- Abbott, J. R.; Litzinger, M. J. Different omega-conotoxins mark the development of Swiss Webster mouse cortex suggesting N-type voltage sensitive calcium channel subtypes. Int. J. Dev. Neurosci 1994, 12, 43–47. [Google Scholar]
- Currie, K. P.; Fox, A. P. Comparison of N- and P/Q-type voltage-gated calcium channel current inhibition. J. Neurosci 1997, 17, 4570–4579. [Google Scholar]
- McDonough, S. I.; Swartz, K. J.; Mintz, I. M.; Boland, L. M.; Bean, B. P. Inhibition of calcium channels in rat central and peripheral neurons by omega-conotoxin MVIIC. J. Neurosci 1996, 16, 2612–2623. [Google Scholar]
- Seabrook, G. R.; Adams, D. J. Inhibition of neurally-evoked transmitter release by calcium channel antagonists in rat parasympathetic ganglia. Br. J. Pharmacol 1989, 97, 1125–1136. [Google Scholar]
- Wu, L. G.; Saggau, P. Presynaptic inhibition of elicited neurotransmitter release. Trends. Neurosci 1997, 20, 204–212. [Google Scholar]
- Fern, R.; Ransom, B. R.; Waxman, S. G. Voltage-gated calcium channels in CNS white matter: role in anoxic injury. J. Neurophysiol 1995, 74, 369–377. [Google Scholar]
- Yamada, K.; Teraoka, T.; Morita, S.; Hasegawa, T.; Nabeshima, T. Omega-conotoxin GVIA protects against ischemia-induced neuronal death. Neuropharmacology 1994, 33, 251–254. [Google Scholar]
- Bowersox, S. S.; Gadbois, T.; Singh, T.; Pettus, M.; Wang, Y. X.; Luther, R. R. Selective N-type neuronal voltage-sensitive calcium channel blocker, SNX-111, produces spinal antinociception in rat models of acute, persistent and neuropathic pain. J. Pharmacol. Exp. Ther 1996, 279, 1243–1249. [Google Scholar]
- Elmslie, K. S. Calcium channel blockers in the treatment of disease. J. Neurosci. Res 2004, 75, 733–741. [Google Scholar]
- Malmberg, A. B.; Yaksh, T. L. Effect of continuous intrathecal infusion of omega-conopeptides, N-type calcium-channel blockers, on behavior and antinociception in the formalin and hot-plate tests in rats. Pain 1995, 60, 83–90. [Google Scholar]
- Nebe, J.; Vanegas, H.; Schaible, H. G. Spinal application of omega-conotoxin GVIA, an N-type calcium channel antagonist, attenuates enhancement of dorsal spinal neuronal responses caused by intra-articular injection of mustard oil in the rat. Exp. Brain. Res 1998, 120, 61–69. [Google Scholar]
- Neugebauer, V.; Vanegas, H.; Nebe, J.; Rumenapp, P.; Schaible, H. G. Effects of N- and L-type calcium channel antagonists on the responses of nociceptive spinal cord neurons to mechanical stimulation of the normal and the inflamed knee joint. J. Neurophysiol 1996, 76, 3740–3749. [Google Scholar]
- Omote, K.; Kawamata, M.; Satoh, O.; Iwasaki, H.; Namiki, A. Spinal antinociceptive action of an N-Type voltage-dependent calcium channel blocker and the synergistic interaction with morphine. Anesthesiology 1996, 84, 636–643. [Google Scholar]
- Scott, D.; Wright, C.; Angus, J. Actions of intrathecal omega-conotoxins CVID, GVIA, MVIIA, and morphine in acute and neuropathic pain in the rat. Eur. J. Pharmacol 2002, 451, 279–286. [Google Scholar]
- Sluka, K. A. Blockade of calcium channels can prevent the onset of secondary hyperalgesia and allodynia induced by intradermal injection of capsaicin in rats. Pain 1997, 71, 157–164. [Google Scholar]
- Smith, M. T.; Cabot, P. J.; Ross, F. B.; Robertson, A. D.; Lewis, R. J. The novel N-type calcium channel blocker, AM336, produces potent dose-dependent antinociception after intrathecal dosing in rats and inhibits substance P release in rat spinal cord slices. Pain 2002, 96, 119–127. [Google Scholar]
- White, D. M.; Cousins, M. J. Effect of subcutaneous administration of calcium channel blockers on nerve injury-induced hyperalgesia. Brain Res 1998, 801, 50–58. [Google Scholar]
- Catterall, W. A. Structure and function of voltage-gated ion channels. Annu. Rev. Biochem 1995, 64, 493–531. [Google Scholar]
- Wheeler, D. B.; Randall, A.; Tsien, R. W. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science 1994, 264, 107–111. [Google Scholar]
- Arikkath, J.; Cambell, K. P. Auxiliary subunits: essential components of the voltage-gated calcium channel complex. Curr. Opin. Neurobiol 2003, 13, 298–307. [Google Scholar]
- Augustine, G. J.; Charlton, M. P.; Smith, S. J. Calcium action in synaptic transmitter release. Annu. Rev. Neurosci 1987, 10, 633–693. [Google Scholar]
- Miller, R. J. Multiple calcium channels and neuronal function. Science 1987, 235, 46–52. [Google Scholar]
- Altier, C.; Zamponi, G. W. Targeting Ca2+ channels to treat pain: T-type versus N-type. Trends. Pharmacol. Sci 2004, 25, 465–470. [Google Scholar]
- McGivern, J. G.; McDonough, S. I. Voltage-gated calcium channels as targets for the treatment of chronic pain. Curr. Drug Targets CNS Neurol. Disord 2004, 3, 457–478. [Google Scholar]
- Gohil, K.; Bell, J. R.; Ramachandran, J.; Miljanich, G. P. Neuroanatomical distribution of receptors for a novel voltage-sensitive calcium-channel antagonist, SNX-230 (omega-conopeptide MVIIC). Brain Res 1994, 653, 258–266. [Google Scholar]
- Kent, S. B. H. Chemical synthesis of peptides and proteins. Ann. Rev. Biochem 1988, 57, 957–989. [Google Scholar]
- Takemura, M.; Kiyama, H.; Fukui, H.; Tohyama, M.; Wada, H. Autoradiographic visualization in rat brain of receptors for omega-conotoxin GVIA, a newly discovered calcium antagonist. Brain Res 1988, 451, 386–389. [Google Scholar]
- Westenbroek, R. E.; Hell, J. W.; Warner, C.; Dubel, S. J.; Snutch, T. P.; Catterall, W. A. Biochemical properties and subcellular distribution of an N-type calcium channel alpha 1 subunit. Neuron 1992, 9, 1099–1115. [Google Scholar]
- Westenbroek, R. E.; Hoskins, L.; Catterall, W. A. Localization of Ca2+ channel subtypes on rat spinal motor neurons, interneurons, and nerve terminals. J. Neurosci 1998, 18, 6319–6330. [Google Scholar]
- Malmberg, A. B.; Yaksh, T. L. Voltage-sensitive calcium channels in spinal nociceptive processing: blockade of N- and P-type channels inhibits formalin-induced nociception. J. Neurosci 1994, 14, 4882–4890. [Google Scholar]
- Chaplan, S. R.; Pogrel, J. W.; Yaksh, T. L. Role of voltage-dependent calcium channel subtypes in experimental tactile allodynia. J. Pharmacol. Exp. Ther 1994, 269, 1117–1123. [Google Scholar]
- Cox, B. N-type Ca2+ channel blockers in pain and stroke. Exp. Opin. Ther. Patents 1998, 8, 1237–1250. [Google Scholar]
- Williams, M.; Kowaluk, E. A.; Arneric, S. P. Emerging molecular approaches to pain therapy. J. Med. Chem 1999, 42, 1481–1500. [Google Scholar]
- Winquist, R. J.; Qian Pan, J.; Gribkoff, V. K. Use-dependent blockade of Cav2.2 voltage-gated calcium channels for neuopathic pain. Biochem. J 2005, 70, 489–499. [Google Scholar]
- Bridges, D.; Thompson, S. W. N.; Rice, A. S. C. Mechanisms of neuropathic pain. Br. J. Anaesth 2001, 87, 12–26. [Google Scholar]
- Prado, W. A. Involvement of calcium in pain and nociception. J. Med. Biol. Res 2001, 34, 449–461. [Google Scholar]
- Vanegas, H.; Schaible, H. Effects of antagonists to high-threshold calcium channels upon spinal mechanisms of pain, hyperalgesia and allodynia. Pain 2000, 85, 9–18. [Google Scholar]
- Stix, G. A toxin against pain. Sci. Am 2005, 292, 70–75. [Google Scholar]
- Nielsen, K. J.; Thomas, L.; Lewis, R. J.; Alewood, P. F.; Craik, D. J. A consensus structure for omega-conotoxins with different selectivities for voltage-sensitive calcium channel subtypes: comparison of MVIIA, SVIB and SNX-202. J. Mol. Biol 1996, 263, 297–310. [Google Scholar]
- Ellinor, P. T.; Zhang, J. F.; Horne, W. A.; Tsien, R. W. Structural determinants of the blockade of N-type calcium channels by a peptide neurotoxin. Nature 1994, 372, 272–275. [Google Scholar]
- Small, D. L.; Monette, R.; Buchan, A. M.; Morley, P. Identification of calcium channels involved in neuronal injury in rat hippocampal slices subjected to oxygen and glucose deprivation. Brain Res 1997, 753, 209–218. [Google Scholar]
- Adams, D. J.; Smith, A. B.; Schroeder, C. I.; Yasuda, T.; Lewis, R. J. ω-Conotoxin CVID inhibit a pharmacologically distinct voltage-sensitive calcium channel associated with transmitter release from preganglionic nerve terminals. J. Biol. Chem. 2003, 278. [Google Scholar]
- Lewis, R. J.; Garcia, M. G. Therapeutic potential of venom peptides. Nat. Rev. Drug Disc 2003, 2, 790–802. [Google Scholar]
- Bowersox, S. S.; Valentino, K. L.; Luther, R. R. Neuronal voltage-sensitive calcium channels. Drugs News Perspect 1994, 7, 261–268. [Google Scholar]
- Minami, K.; Raymond, C.; Martin-Moutot, N.; Ohtake, A.; Van Renterghem, C.; Takahashi, M; Seagar, M. J.; Mori, Y.; Sato, K. Role of Thr11 in the binding of ω-conotoxin MVIIC to the N-type Ca2+ channels. FEBS Lett. 2001, 491, 127–130. [Google Scholar]
- Nadasdi, L.; Yamashiro, D.; Chung, D.; Tarczy-Hornoch, K.; Adriaenssens, P.; Ramachandran, J. Structure-activity analysis of a Conus peptide blocker of N-type neuronal calcium channels. Biochemistry 1995, 34, 8076–8081. [Google Scholar]
- Nielsen, K. J.; Adams, D.; Thomas, L.; Bond, T.; Alewood, P. F.; Craik, D. J.; Lewis, R. J. Structure-activity relationships of omega-conotoxins MVIIA, MVIIC and 14 loop splice hybrids at N and P/Q-type calcium channels. J. Mol. Biol 1999, 289, 1405–1421. [Google Scholar]
- Sasaki, T.; Kobayashi, K.; Kohno, T.; Sato, K. Combinatorial synthesis of ω-conotoxin MVIIC analogues and their binding with N- and P/Q-type calcium channels. FEBS Lett 2000, 466, 125–129. [Google Scholar]
- Sato, K.; Park, N.-G.; Kohno, T.; Maeda, T.; Kim, J.-I.; Kato, R.; Takahashi, M. Role of basic residues for the binding of ω-conotoxin GVIA to N-type calcium channels. Biochem. Biophys. Res. Commun 1993, 194, 1292–1296. [Google Scholar]
- Sato, K.; Raymond, C.; Martin-Moutot, N.; Sasaki, T.; Omori, A.; Ohtake, A.; Kim, J. I.; Kohno, T.; Takahashi, M.; Seagar, M. Binding of chimeric analogs of omega-conotoxin MVIIA and MVIIC to the N- and P/Q-type calcium channels. FEBS Lett 1997, 414, 480–484. [Google Scholar]
- Sato, K.; Raymond, C.; Martin-Moutot, N.; Sasaki, T.; Ohtake, A.; Minami, K.; van Renterghem, C.; Kim, J. I.; Takahashi, M.; Seagar, M. J. Binding of Ala-scanning analogs of ω-conotoxin MVIIC to N- and P/Q-type calcium channels. FEBS Lett 2000, 469, 147–150. [Google Scholar]
- Sato, K.; Raymond, C.; Martin-Moutot, N.; Sasaki, T.; Ohtake, A.; Minami, K.; van Renterghem, C.; Takahasi, M.; Seager, M. J. Binding of six chimeric analogs of ω-conotoxin MVIIA and MVIIC to N- and P/Q-type calcium channels. Biochem. Biophys. Res. Commun 2000, 269, 254–256. [Google Scholar]
- Dunlap, K.; Luebke, J. I.; Turner, T. J. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci 1995, 18, 89–98. [Google Scholar]
- Olivera, B. M.; Miljanich, G. P.; Ramachandran, J.; Adams, M. E. Calcium Channel Diversity and Neurotransmitter Release: The ω–Conotoxins and the ω-Agatoxins. Ann. Rev. Biochem 1994, 63, 823–867. [Google Scholar]
- Tsien, R. W.; Lipscombe, D.; Madison, D. V.; Bley, K. R.; Fox, A. P. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci 1988, 11, 431–438. [Google Scholar]
- Vega, T.; De Pascual, R.; Bulbena, O.; Garcia, A. G. Effects of omega-toxins on noradrenergic neurotransmission in beating guinea pig atria. Eur. J. Pharmacol 1995, 276, 231–238. [Google Scholar]
- Hirata, H.; Albillos, A.; Fernandez, F.; Medrano, J.; Jurkiewicz, A.; Garcia, A. G. Omega- Conotoxins block neurotransmission in the rat vas deferens by binding to different presynaptic sites on the N-type Ca2+ channel. Eur. J. Pharmacol 1997, 321, 217–223. [Google Scholar]
- Kristipati, R.; Nadasdi, L.; Tarczy-Hornoch, K.; Lau, K.; Miljanich, G. P.; Ramachandran, J.; Bell, J. R. Characterization of the binding of omega-conopeptides to different classes of non-L-type neuronal calcium channels. Mol. Cell. Neurosci 1994, 5, 219–228. [Google Scholar]
- Lin, Z.; Haus, S.; Edgerton, J.; Lipscombe, D. Identification of functionally distinct isoforms of the N-type Ca2+ channel in rat sympathetic ganglia and brain. Neuron 1997, 18, 153–166. [Google Scholar]
- Wright, C. E.; Robertson, A. D.; Whorlow, S. L.; Angus, J. A. Cardiovascular and autonomic effects of omega-conotoxins MVIIA and CVID in conscious rabbits and isolated tissue assays. Br. J. Pharmacol 2000, 131, 1325–1336. [Google Scholar]
- Pin, J. P.; Bockaert, J. Omega-conotoxin GVIA and dihydropyridines discriminate two types of Ca2+ channels involved in GABA release from striatal neurons in culture. Eur. J. Pharmacol 1990, 188, 81–84. [Google Scholar]
- Llinas, R.; Sugimori, M.; Hillman, D. E.; Cherksey, B. Distribution and functional significance of the P-type, voltage- dependent Ca2+ channels in the mammalian central nervous system. Trends Neurosci 1992, 15, 351–355. [Google Scholar]
- Valentino, K.; Newcomb, R.; Gadbois, T.; Singh, T.; Bowersox, S.; Bitner, S.; Justice, A.; Yamashiro, D.; Hoffman, B. B.; Ciaranello, R.; Miljanich, G.; Ramachandran, J. A selctive N-type calcium channel antagonist protects against neuronal loss after global cerebral ischemia. Proc. Natl. Acad. Sci. USA 1993, 90, 7894–7897. [Google Scholar]
- Miljanich, G. P.; Approved!!!, Prialt. Ziconotide intrathecal infusion), a conopeptide for treating severe chronic pain. In Venoms to Drugs 2005; 2005; Heron Island, Australia. [Google Scholar]
- Wermeling, D. P. Ziconotide, an intrathecally administered N-type calcium channle antagonsit for the treatment of chronic pain. Pharmacotherapy 2005, 25, 1084–1094. [Google Scholar]
- Wang, Y. X.; Bezprozvannaya, S.; Bowersox, S. S.; Nadasdi, L.; Miljanich, G.; Mezo, G.; Silva, D.; Tarczy-Hornoch, K.; Luther, R. R. Peripheral versus central potencies of N-type voltage-sensitive calcium channel blockers. Naunyn Schmiedebergs Arch. Pharmacol 1998, 357, 159–168. [Google Scholar]
- Jain, K. K. An evaluation of intrathecal ziconotide for the treatment of chronic pain. Expert Opin. Investig. Drugs 2000, 9, 2403–2410. [Google Scholar]
- Smith, M. L.; Siesjo, B. K. Postischemic treatment of omega-conopeptide SNX-111 protects the rat brain against ischemic damage. In Pharmacology of Cerebral Ischemia; Krieglstein, J., Ed.; 1992; pp. 161–166. [Google Scholar]
- Zhao, Q.; Smith, M. L.; Siesjo, B. K. The omega-conopeptide SNX-111, an N-type calcium-channel blocker, dramatically ameliorates brain damage due to transient focal ischemia. Acta. Physiol. Scand 1994, 150, 459–461. [Google Scholar]
- Gaur, S.; Newcomb, R.; Rivnay, B.; Bell, J. R.; Yamashiro, D.; Ramachandran, J.; Miljanich, G. P. Calcium channel antagonist peptides define several components of transmitter release in the hippocampus. Neuropharmacology 1994, 33, 1211–1219. [Google Scholar]
- MacPherson, R. D. The pharmacologcial basis of contemporary pain management. Pharmacol. Ther 2000, 88, 163–185. [Google Scholar]
- Chaplan, S. R.; Bach, F. W.; Pogrel, J. W.; Chung, J. M.; Yaksh, T. L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 1994, 53, 55–63. [Google Scholar]
- Yamamoto, T.; Sakashita, Y. Differential effects of intrathecally administered N- and P-type voltage-sensitive calcium channel blockers upon two models of experimental mononeuropathy in the rat. Brain Res 1998, 794, 329–232. [Google Scholar]
- Miljanich, G. P. Ziconotide: neuronal calcium channel blocker for the treating severe chronic pain. Curr. Med. Chem 2004, 11, 1715–1723. [Google Scholar]
- Atanassoff, P. G.; Hartmannsgruber, M. W. B.; Thrasher, J.; Wermeling, D.; Longton, W.; Gaete, R.; Singh, R.; Mayo, M.; McGuire, D.; Luther, R. R. Ziconotide, a new N-type calcium channel blocker, administrered intracthecally for acute postoperative pain. Reg. Anesth. Pain Med 2000, 25, 274–8. [Google Scholar]
- McGuire, D.; Bowersox, S.; Fellmann, J. D.; Luther, R. R. Sympatholysis after neuron-specific, N-type, voltage-sensitive calcium channel blockade: first demonstration of N-channel function in humans. J. Cardiovasc. Pharmacol 1997, 30, 400–403. [Google Scholar]
- Penn, R. D.; Paice, J. A. Adverse effects associated with the intrathecal administration of ziconotide. Pain 2000, 85, 291–296. [Google Scholar]
- Levin, T.; Petrides, G.; Weiner, J.; Saravay, S. Intractable delerium associated with ziconotide succesfully treated with electoconvulsive therapy. Psychosomatics 2002, 43, 63–66. [Google Scholar]
- Brose, W. G.; Gutlove, D. P.; Luther, R. R.; Bowersox, S. S.; McGuire, D. Use of intrathecal SNX-111, a novel, N-type, voltage-sensitive, calcium channel blocker, in the management of intractable brachial plexus avulsion pain. Clin. J. Pain 1997, 13, 256–259. [Google Scholar]
- Wang, Y.-X.; Gao, D.; Pettus, M.; Phillips, C.; Bowersox, S. S. Interactions of intrathecally administrated ziconotide, a selective blocker of neuronal N-type voltage-sensitive calcium channels, with morphine on nociception in rats. Pain 2000, 84. [Google Scholar]
- Cousins, M. J.; Cjoucke, R. C.; Cher, C. M.; Brooker, C. D.; Amor, P. E.; Crump, D. E. A phase I clinical trial of AM336, a novel N-type calcium channel blocker. 10th World Congress of Pain, IASP Press.
- Abram, S. E.; Yaksh, T. L. Morphine, but not inhalation anesthesia, blocks post-injury facilitation. The role of preemptive suppression of afferent transmission. Anesthesiology 1993, 78, 713–721. [Google Scholar]
- Bourinet, E.; Soong, T. W.; Stea, A.; Snutch, T. P. Determinants of the G-protein dependent opioid modulation of neuronal calcium channels. Proc. Natl. Acad. Sci. U S A 1996, 93, 1486–1491. [Google Scholar]
- De Waard, M.; Liu, H.; Walker, D.; Scott, V. E.; Gurnett, C. A.; Campbell, K. P. Direct binding of G-protein betagamma complex to voltage-dependent calcium channels. Nature 1997, 385, 446–450. [Google Scholar]
- Seward, E.; Hammond, C.; Henderson, G. Mu-opioid-receptor-mediated inhibition of the N-type calcium-channel current. Proc. R. Soc. Lond. B. Biol. Sci 1991, 244, 129–135. [Google Scholar]
- Toth, P. T.; Shekter, L. R.; Ma, G. H.; Philipson, L. H.; Miller, R. J. Selective G-protein regulation of neuronal calcium channels. J. Neurosci 1996, 16, 4617–4624. [Google Scholar]
- Zhang, J. F.; Ellinor, P. T.; Aldrich, R. W.; Tsien, R. W. Multiple structural elements in voltage-dependent Ca2+ channels support their inhibition by G proteins. Neuron 1996, 17, 991–1003. [Google Scholar]
- Basbaum, A. I. Insights into the development of opioid tolerance. Pain 1995, 61, 349–352. [Google Scholar]
- Kennedy, C.; Henderson, G. Mu-opioid receptor inhibition of calcium current: development of homologous tolerance in single SH-SY5Y cells after chronic exposure to morphine in vitro. Mol. Pharmacol 1991, 40, 1000–1005. [Google Scholar]
- Kennedy, C.; Henderson, G. Chronic exposure to morphine does not induce dependence at the level of the calcium channel current in human SH-SY5Y cells. Neuroscience 1992, 49, 937–944. [Google Scholar]
- Tao, P. L.; Lee, C. R.; Law, P. Y.; Loh, H. H. The interaction of the mu-opioid receptor and G protein is altered after chronic morphine treatment in rats. Naunyn Schmiedebergs Arch. Pharmacol 1993, 348, 504–508. [Google Scholar]
- Suh, H. W.; Song, D. K.; Choi, S. R.; Huh, S. O.; Kim, Y. H. Effects of intrathecal injection of nimodipine, omega-conotoxin GVIA, calmidazolium, and KN-62 on the antinociception induced by cold water swimming stress in the mouse. Brain Res 1997, 767, 144–147. [Google Scholar]
- Basilico, L.; Parolaro, D.; Rubino, T.; Gori, E.; Giagnoni, G. Influence of omega-conotoxin on morphine analgesia and withdrawal syndrome in rats. Eur. J. Pharmacol 1992, 218, 75–81. [Google Scholar]
- Pirec, V.; Laurito, C. E.; Lu, Y.; Yeomans, D. C. The combined effects of N-type calcium channel blockers and morphine on A delta versus C fiber mediated nociception. Anesth. Analg 2001, 92, 239–243. [Google Scholar]
- Suematsu, M.; Ohnishi, T.; Shinno, E.; Maeda, S.; Matsumoto, K.; Sakuda, M.; Saito, K. Effect of prolonged administration of clonidine on [3H]PN 200-110 and [125I]omega-conotoxin binding in mouse brain. Neurosci. Lett 1993, 163, 193–196. [Google Scholar]
- Davis, J. H.; Bradley, E. K.; Miljanich, G. P.; Nadasdi, L.; Ramachandran, J.; Basus, V. J. Solution structure of omega-conotoxin GVIA using 2-D NMR spectroscopy and relaxation matrix analysis. Biochemistry 1993, 32, 7396–7405. [Google Scholar]
- Pallaghy, P. K.; Duggan, B. M.; Pennington, M. W.; Norton, R. S. Three-dimensional structure in solution of the calcium channel blocker omega-conotoxin. J. Mol. Biol 1993, 234, 405–420. [Google Scholar]
- Pallaghy, P. K.; Norton, R. S. Refined solution structure of omega-conotoxin GVIA: implications for calcium channel binding. J. Pept. Res 1999, 53, 343–351. [Google Scholar]
- Sevilla, P.; Bruix, M.; Santoro, J.; Gago, F.; Garcia, A. G.; Rico, M. Three-dimensional structure of omega-conotoxin GVIA determined by 1H NMR. Biochem. Biophys. Res. Commun 1993, 192, 1238–1244. [Google Scholar]
- Skalicky, J. J.; Metzler, W. J.; Ciesla, D. J.; Galdes, A.; Pardi, A. Solution structure of the calcium channel antagonist omega-conotoxin GVIA. Protein Sci 1993, 2, 1591–1603. [Google Scholar]
- Atkinson, R. A.; Kieffer, B.; Dejaegere, A.; Sirockin, F.; Lefevre, J. F. Structural and dynamic characterization of omega-conotoxin MVIIA: the binding loop exhibits slow conformational exchange. Biochemistry 2000, 39, 3908–3919. [Google Scholar]
- Basus, V. J.; Nadasdi, L.; Ramachandran, J.; Miljanich, G. P. Solution structure of omega-conotoxin MVIIA using 2D NMR spectroscopy. FEBS Lett 1995, 370, 163–169. [Google Scholar]
- Kohno, T.; Kim, J. I.; Kobayashi, K.; Kodera, Y.; Maeda, T.; Sato, K. Three-dimensional structure in solution of the calcium channel blocker omega-conotoxin MVIIA. Biochemistry 1995, 34, 10256–10265. [Google Scholar]
- Farr-Jones, S.; Miljanich, G. P.; Nadasdi, L.; Ramachandran, J.; Basus, V. J. Solution structure of omega-conotoxin MVIIC, a high affinity ligand of P-type calcium channels, using 1H NMR spectroscopy and complete relaxation matrix analysis. J. Mol. Biol 1995, 248, 106–124. [Google Scholar]
- Nemoto, N.; Kubo, S.; Yoshida, T.; Chino, N.; Kimura, T.; Sakakibara, S.; Kyogoku, Y.; Kobayashi, Y. Solution structure of omega-conotoxin MVIIC determined by NMR. Biochem. Biophys. Res. Commun 1995, 207, 695–700. [Google Scholar]
- Pallaghy, P. K.; Nielsen, K. J.; Craik, D. J.; Norton, R. S. A common structural motif incorporating a cystine knot and a triple- stranded beta-sheet in toxic and inhibitory polypeptides. Prot. Sci 1994, 3, 1833–1839. [Google Scholar]
- Nielsen, K. J.; Adams, D. A.; Alewood, P. F.; Lewis, R. J.; Thomas, L.; Schroeder, T.; Craik, D. J. Effects of chirality at Tyr13 on the structure-activity relationships of omega-conotoxins from Conus magus. Biochemistry 1999, 38, 6741–6751. [Google Scholar]
- Nielsen, K. J.; Schroeder, T.; Lewis, R. J. Structure-activity relationship of ω-conotoxins at the N-type voltage-sensitive calcium channels. J. Mol. Recogn 2000, 13, 1–16. [Google Scholar]
- Kobayashi, K.; Sasaki, T.; Sato, K.; Kohno, T. Three-dimensional solution structure of omega-conotoxin TxVII, an L- type calcium channel blocker. Biochemistry 2000, 39, 14761–14767. [Google Scholar]
- Author Methods for producing analgesia. International Patent Classification, A61K 038/00, C07K 005/00, C07K 007/00 United States Patent 5, 859, 186, 1999.
- Author Compositions for delayed treatement of ischemia-related neuronal damage. International patent application no PCT/US96 09766; Classification, C07K 7/10, A61K 37/02, 1993.
- Kim, J. I.; Takahashi, M.; Ogura, A.; Kohno, T.; Kudo, Y.; Sato, K. Hydroxyl group of Tyr13 is essential for the activity of omega- conotoxin GVIA, a peptide toxin for N-type calcium channel. J. Biol. Chem 1994, 269, 23876–23978. [Google Scholar]
- Lew, M. J.; Flinn, J. P.; Pallaghy, P. K.; Murphy, R.; Whorlow, S. L.; Wright, C. E.; Norton, R. S.; Angus, J. A. Structure-function relationships of omega-conotoxin GVIA. Synthesis, structure, calcium channel binding, and functional assay of alanine- substituted analogues. J. Biol. Chem 1997, 272, 12014–12023. [Google Scholar]
- Kim, J. I.; Takahashi, M.; Martin-Moutot, N.; Seagar, M. J.; Ohtake, A.; Sato, K. Tyr13 is essential for the binding of omega-conotoxin MVIIC to the P/Q- type calcium channel. Biochem. Biophys. Res. Commun 1995, 214, 305–309. [Google Scholar]
- Flinn, J. P.; Pallaghy, P. K.; Lew, M. J.; Murphy, R.; Angus, J. A.; Norton, R. S. Roles of key functional groups in omega-conotoxin GVIA synthesis, structure and functional assay of selected peptide analogues. Eur. J. Biochem 1999, 262, 447–455. [Google Scholar]
- Schroeder, C. I.; Smythe, M. L.; Lewis, R. J. Development of small molecules that mimic the binding of ω-conotoxin at the N-type voltage-gated calcium channel. Molec. Diversity 2004, 8, 127–134. [Google Scholar]
- Baell, J. B.; Duggan, P. J.; Forsyth, S. A.; Lewis, R. J.; Lok, Y. P.; Schroeder, C. I. Synthesis and biological evaluation of non-peptidic mimetics of ω-conotoxin GVIA. Bioorg. Med. Chem 2004, 12, 4025–4037. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Superfamily | Cysteine Arrangement | Family | Molecular target | Example |
|---|---|---|---|---|
| A | CC-C-C | α | AChR* (antagonist) | α-Vc1.1 |
| CC-C-C | ρ | α1-adrenoreceptor (antagonist) | ρ-TIA | |
| CC-C-C-C-C | αA | AChR (antagonist) | αA-EIVA | |
| CC-C-C-C-C | κA | K+ channel (antagonist) | κA-SVIA | |
| M | CC-C-C-CC | μ | Na+ channel (blocker) | μ-PIIIA |
| CC-C-C-CC | ψ | AChR (non-competitive antagonist) | ψ-PIIIE | |
| O | C-C-CC-C-C | δ | Na+ channel (delays inactivation) | δ-TxVIA |
| C-C-CC-C-C | μO | Na+ channel (blocker) | μO-MrVIB | |
| C-C-CC-C-C | ω | Ca2+ channel (blocker) | ω-MVIIA | |
| C-C-CC-C-C | κ | K+ channel (blocker) | κ-PVIIA | |
| C-C-CC-C-C | γ | Pacemaker channels (blocker) | γ-PnVIIA | |
| P | C-C-C-C-C-C | Spastic | Unknown | Tx9a |
| S | C-C-C-C-C-C-C-C | σ | 5-HT3* (antagonist) | σ-GVIIIA |
| T | CC-CC | τ | Presynaptic Ca2+ channels (blocker) | ɛ-TxIX |
| CC-C-C | χ | Noraderenalin transporter (inhibitor) | χ-MrIA | |
| N/A* | C-C | Conopressin | Vasopressin receptors (antagonist) GPCR* | Conopressin-S |
| N/A* | C-C | Contryphan | Unknown | Contryphan-R |
| No Cysteines | Helical | Conantokin | NMDA* channel (antagonist) | Conantokin-G |
| No Cysteines | Contulakin | NTR* (agonist) | Contulakin-G |
| Calcium channel | α1 subunit | Ca2+ current | Peptide antagonist |
|---|---|---|---|
| Cav1.1–1.4 | α1S, C, D, F | L | calciseptine, ω-agatoxin IIIA (α1C) |
| Cav2.1 | α1A | P/Q | ω-agatoxin, MVIIC |
| Cav2.2 | α1B | N | ω-GVIA, ω-MVIIA, ω-CVID |
| Cav2.3 | α1E | R | SNX-482 |
| Cav3.1–3.3 | α1G, H, I | T | Kurtoxin (α1G) |
© 2006 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
Schroeder, C.I.; Lewis, R.J. ω-Conotoxins GVIA, MVIIA and CVID: SAR and Clinical Potential. Mar. Drugs 2006, 4, 193-214. https://doi.org/10.3390/md403193
Schroeder CI, Lewis RJ. ω-Conotoxins GVIA, MVIIA and CVID: SAR and Clinical Potential. Marine Drugs. 2006; 4(3):193-214. https://doi.org/10.3390/md403193
Chicago/Turabian StyleSchroeder, Christina I., and Richard J. Lewis. 2006. "ω-Conotoxins GVIA, MVIIA and CVID: SAR and Clinical Potential" Marine Drugs 4, no. 3: 193-214. https://doi.org/10.3390/md403193
APA StyleSchroeder, C. I., & Lewis, R. J. (2006). ω-Conotoxins GVIA, MVIIA and CVID: SAR and Clinical Potential. Marine Drugs, 4(3), 193-214. https://doi.org/10.3390/md403193