Drugs and Cosmetics from the Sea


1
ICBAS-Instituto de Ciências Biomédicas de Abel Salazar and CIIMAR, Universidade do Porto, 4099-003 Porto, Portugal
2
Department of Aquatic Science, Faculty of Science, Burapha University, Bangsaen, 20131 Chonburi, Thailand
*
Author to whom correspondence should be addressed.
Mar. Drugs 2004, 2(2), 73-82; https://doi.org/10.3390/md202073
Submission received: 27 April 2004
/
Accepted: 13 May 2004
/
Published: 25 May 2004
Abstract
:The marine environment is a rich source of both biological and chemical diversity. This diversity has been the source of unique chemical compounds with the potential for industrial development as pharmaceuticals, cosmetics, nutritional supplements, molecular probes, fine chemicals and agrochemicals. In recent years, a significant number of novel metabolites with potent pharmacological properties has been discovered from the marine organisms. Although there are only a few marine-derived products currently on the market, several robust new compounds derived from marine natural products are now in the clinical pipeline, with more clinical development. While the marine world offers an extremely rich resource for novel compounds, it also represents a great challenge that requires inputs from various scientific areas to bring the marine chemical diversity up to its therapeutic potential.
Introduction
Research into the pharmacological properties of marine natural products has led to the discovery of many potently active agents considered worthy of clinical application. The marine environment is an exceptional reservoir of bioactive natural products, many of which exhibit structural/chemical features not found in terrestrial natural products [1]. Marine organisms have evolved biochemical and physiological mechanisms that include the production of bioactive compounds for such purposes as reproduction, communication, and protection against predation, infection and competition [2]. Because of the physical and chemical conditions in the marine environment, almost every class of marine organism exhibits a variety of molecules with unique structural features.
But beyond the chemical diversity, the sea also provides amazing biological diversity. Among 34 fundamental phyla of life, 17 occur on land whereas 32 occur in the sea [with some overlap]. From the fundamental point of view of biodiversity, the ocean is far more diverse and really would have been the better place to start to develop a natural Pharmacy.
To date, researchers have isolated approximately 7000 marine natural products, 25 percent of which are from algae, 33 percent from sponges, 18 percent from coelenterates (sea whips, sea fans and soft corals), and 24 percent from representatives of other invertebrate phyla such as ascidians (also called tunicates), opisthobranch molluscs (nudibranchs, sea hares etc), echinoderms (starfish, sea cucumbers etc) and bryozoans (moss animals). A simplistic analysis of these data reveals that as the search for “Drugs from the Sea” progresses at the rate of a 10 percent increase in new compounds per year, researchers are concentrating their efforts on slow-moving or sessile invertebrate phyla that have soft bodies, and lack of spines or a shell, i.e. animals that require a chemical defence mechanism [3].
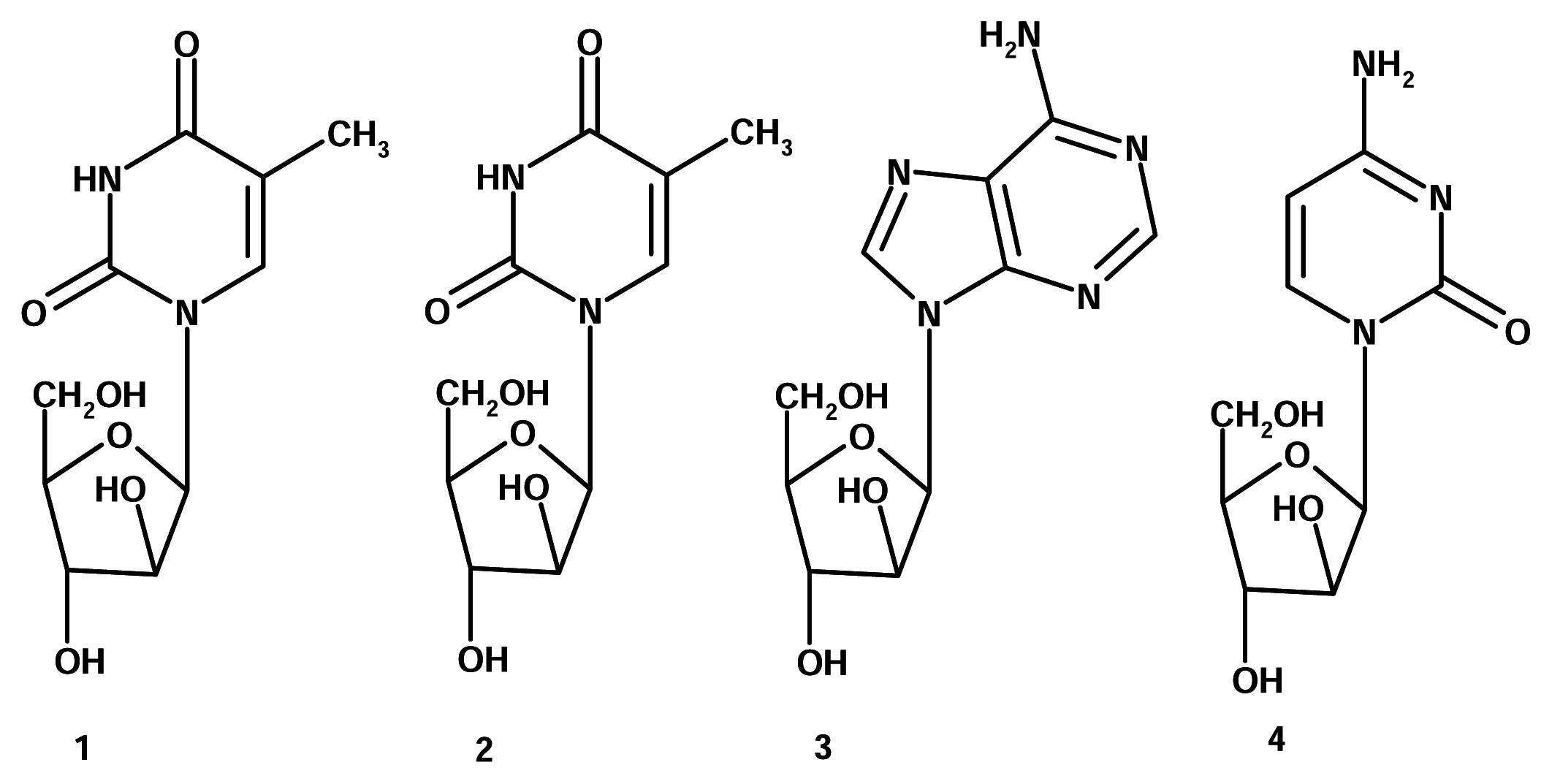
Actually, there was a first, albeit very small, wave of marine-derived drugs that resulted in several products currently on drugstore shelves. Back in the 1950s, Bergmann et al. isolated several nucleosides from the Caribbean sponge Tethya crypta (Tethylidae). Two of these, spongothymidine (1) and spongouridine (2) contained a rare arabinose sugar rather than ribose, which is a quite ubiquitous sugar in nucleosides. This discovery led researchers to synthesize analogues, Ara-A (3, Vidarabine®, Vidarabin Thilo®) and Ara-C (4, Cytarabine, Alexan®, Udicil®), which improved antiviral activity. Currently, these are the only marine related compounds in clinical use [4].
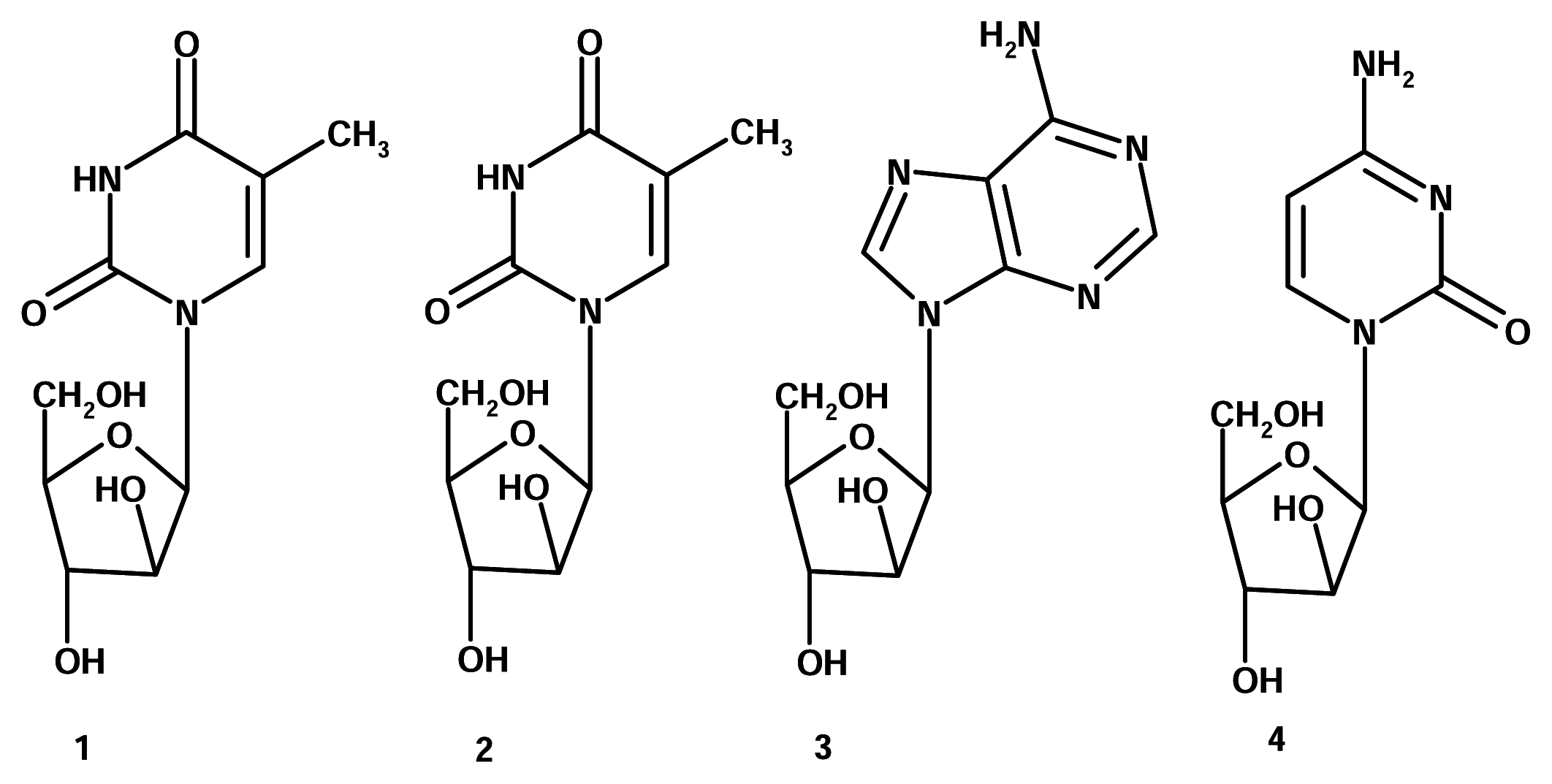
Considering the importance of nucleoside-analogues in antiviral and anticancer therapy, e.g., 3’-azido-3’-deoxythymidine (AZT, Zidovudin), the original discovery of Bergmann can be considered one of immense significance.
Compounds in Preclinical and Clinical Evaluation
In recent years many marine natural products which are promising candidates for new drugs have been discovered (Table 1):
However, as the relevant clinical data were not available for most of the compounds indicated in Table 1, only some of them shall be mentioned in detail in this review
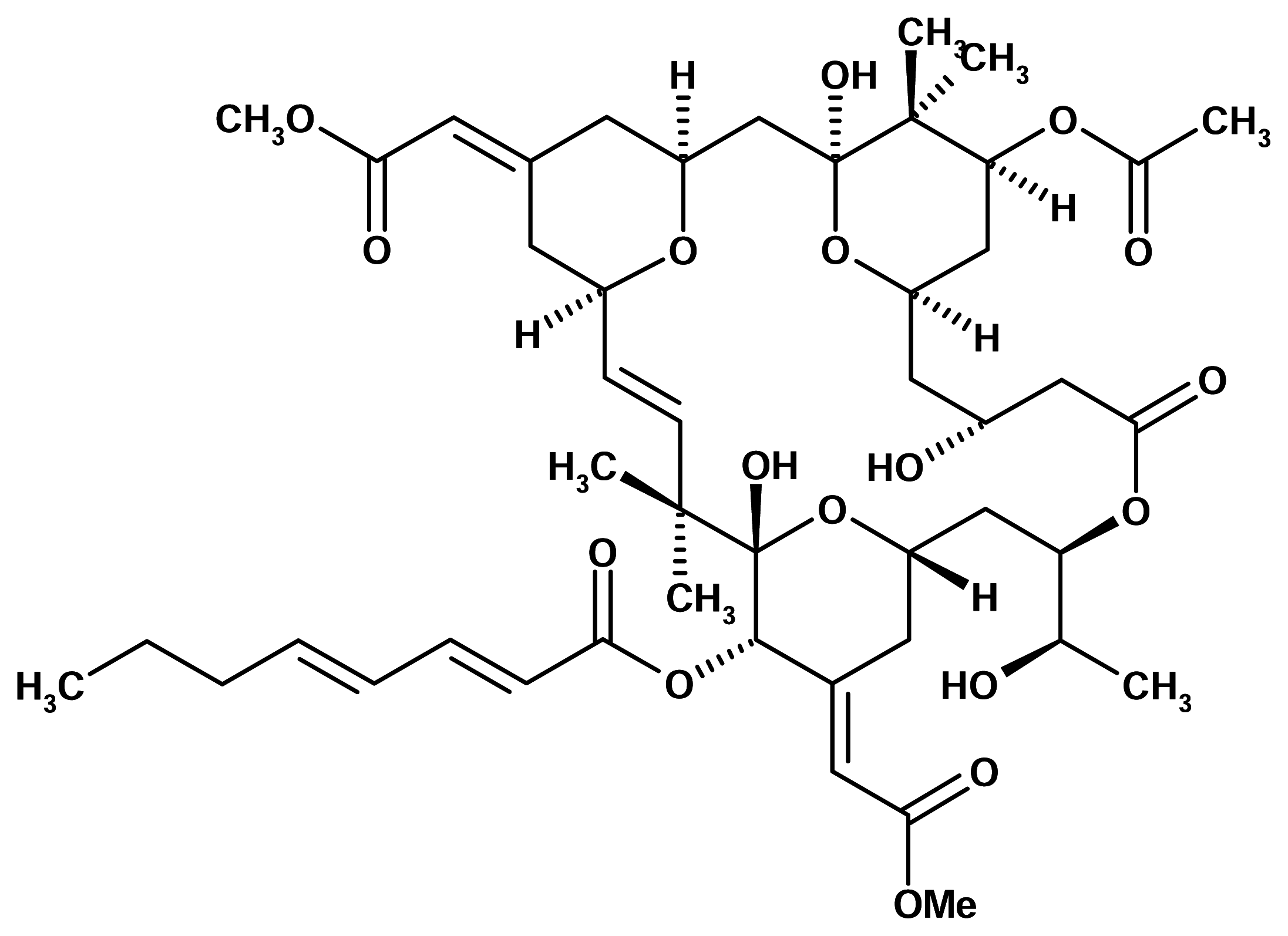
Bryostatin 1
The bryostatins are macrocyclic lactones isolated from the marine bryozoan Bugula neritina (Bugulidae). Bryostatin 1 is one of the most abundant and best studied compounds of this series. It was originally described on the basis of inhibiting growth in murine P388 lymphocytic leukemia cells at subnanomolar concentrations. A range of properties have subsequently been described including activation of T-cells, immunomodulation and stimulation of haematopoietic progenitor cells. However, only many years after its discovery was the molecular site of action of this compound identified.
Bryostatin 1 was found to bind to protein kinase C with high affinity, which may be the mechanistic basis for both observed anticancer and immunostimulating activities [5].
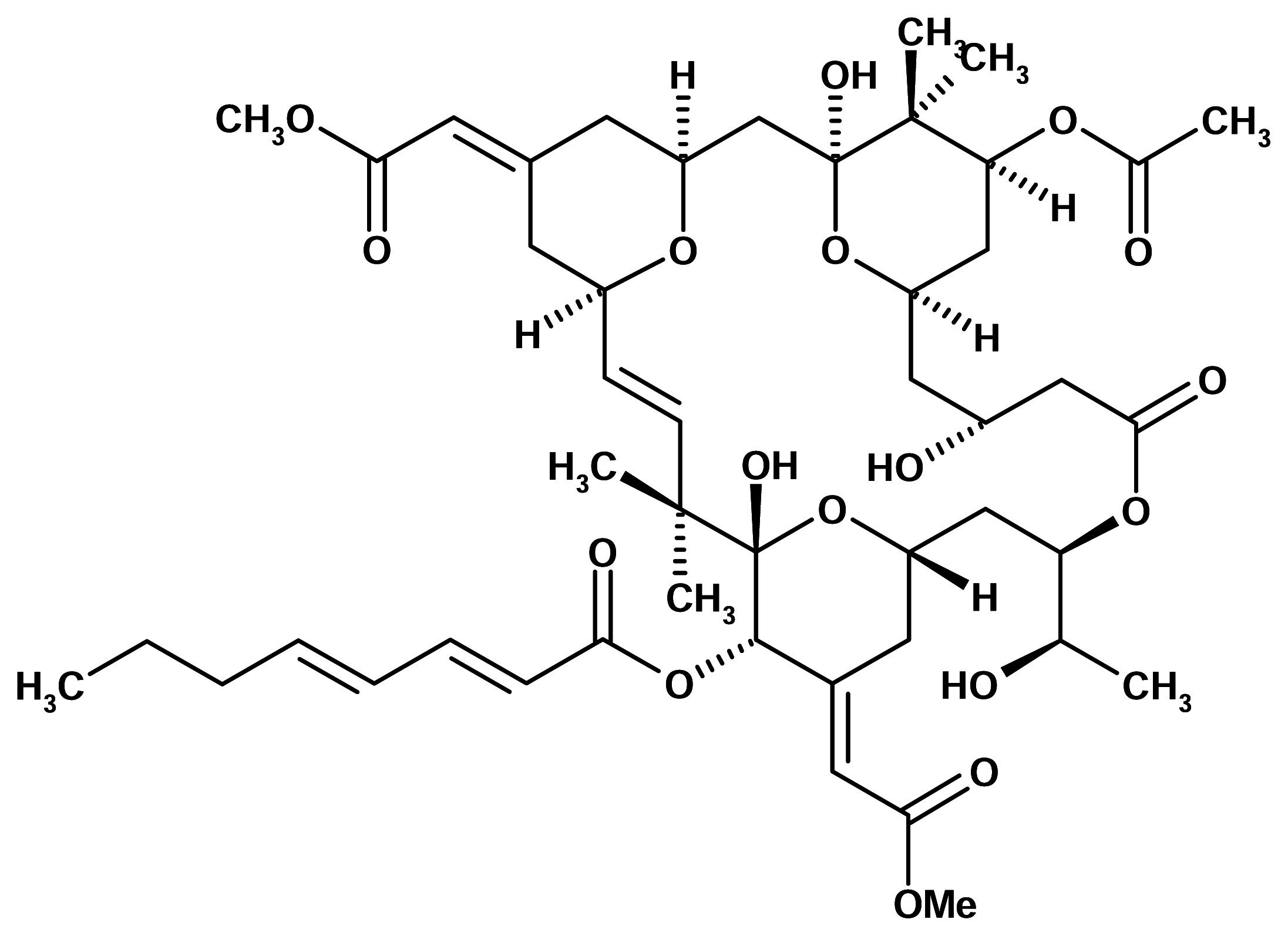
Bryostatin 1 has been licensed to Bristol-Mayers Squibb. It has just completed Phase I clinical trials in the United States and is now being tested in Phase II human clinical trials by the NCI under an agreement with Bristol-Mayers. However, bryostatin 1 is not effective in cancer treatment by itself but it seems to enhance the activity of such chemotherapies as taxol and cisplatin. Bryostatin 1 may be used in tandem with cancer treatments that respond to taxol, such as breast, ovarian and lung cancers.
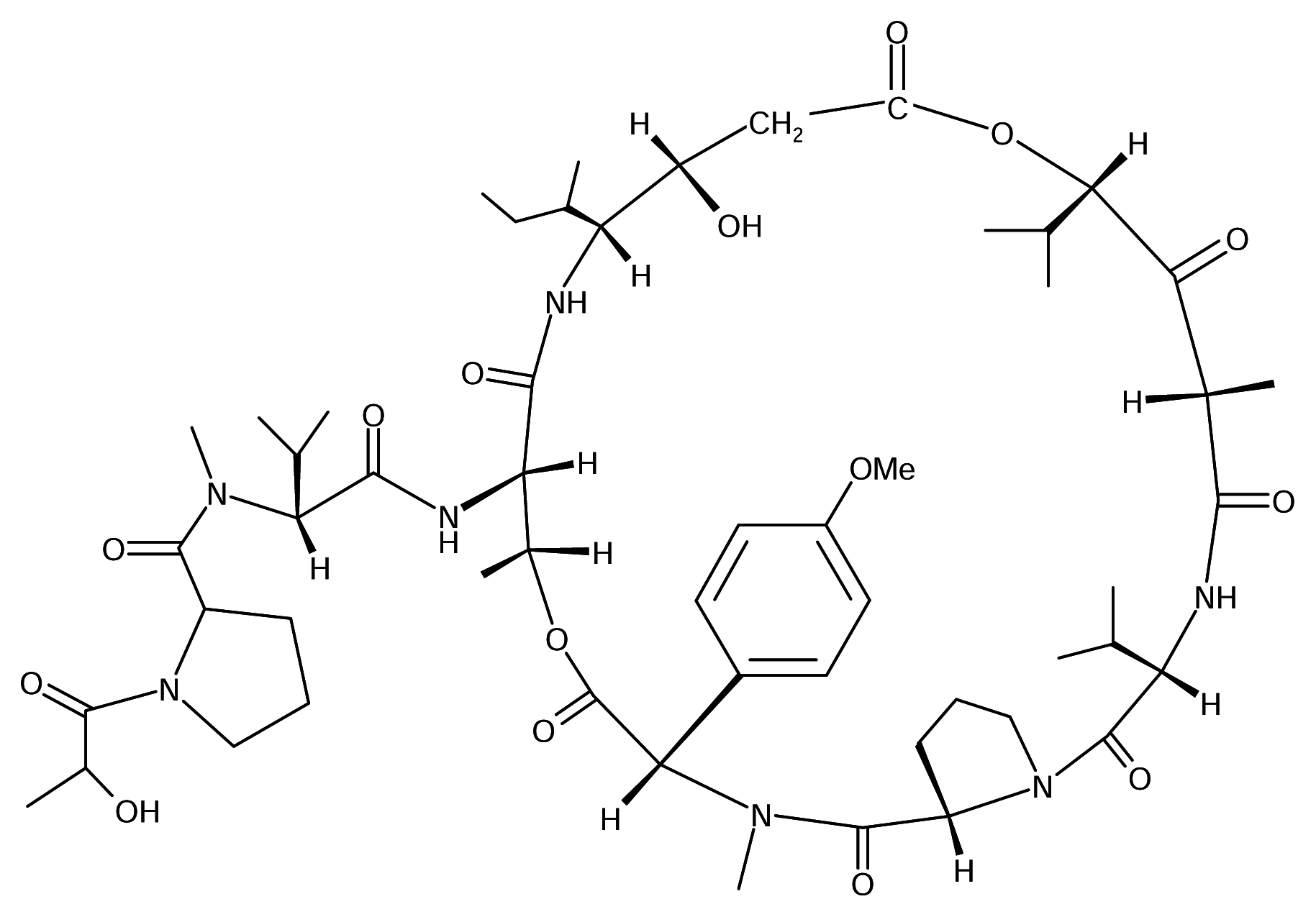
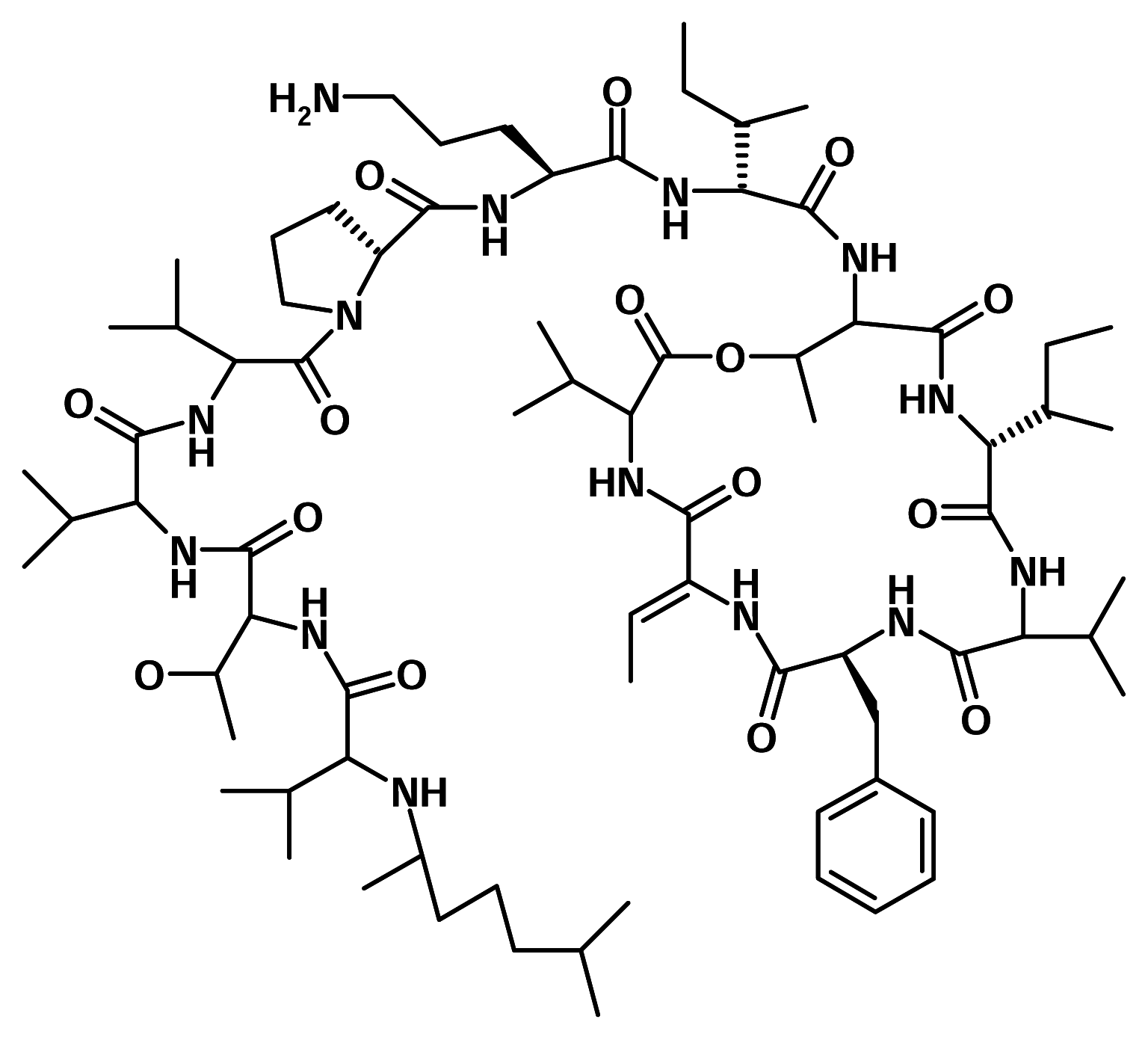
Didemnin B
Didemnin B is one of a number of related depsipeptides isolated from the Caribbean tunicate Trididemnum solidum (Didemnidae). It was later found to display antineoplastic, antiviral and subsequently immunosuppresive activities [6]. Mechanistically, didemnin B acts at the GTP-binding protein elongation factor [5]. This compound, though, is too toxic to be useful as antiviral or immunosuppressive agent, it has been in Phase I clinical trials as an anticancer agent. Phase II clinical trials are underway.
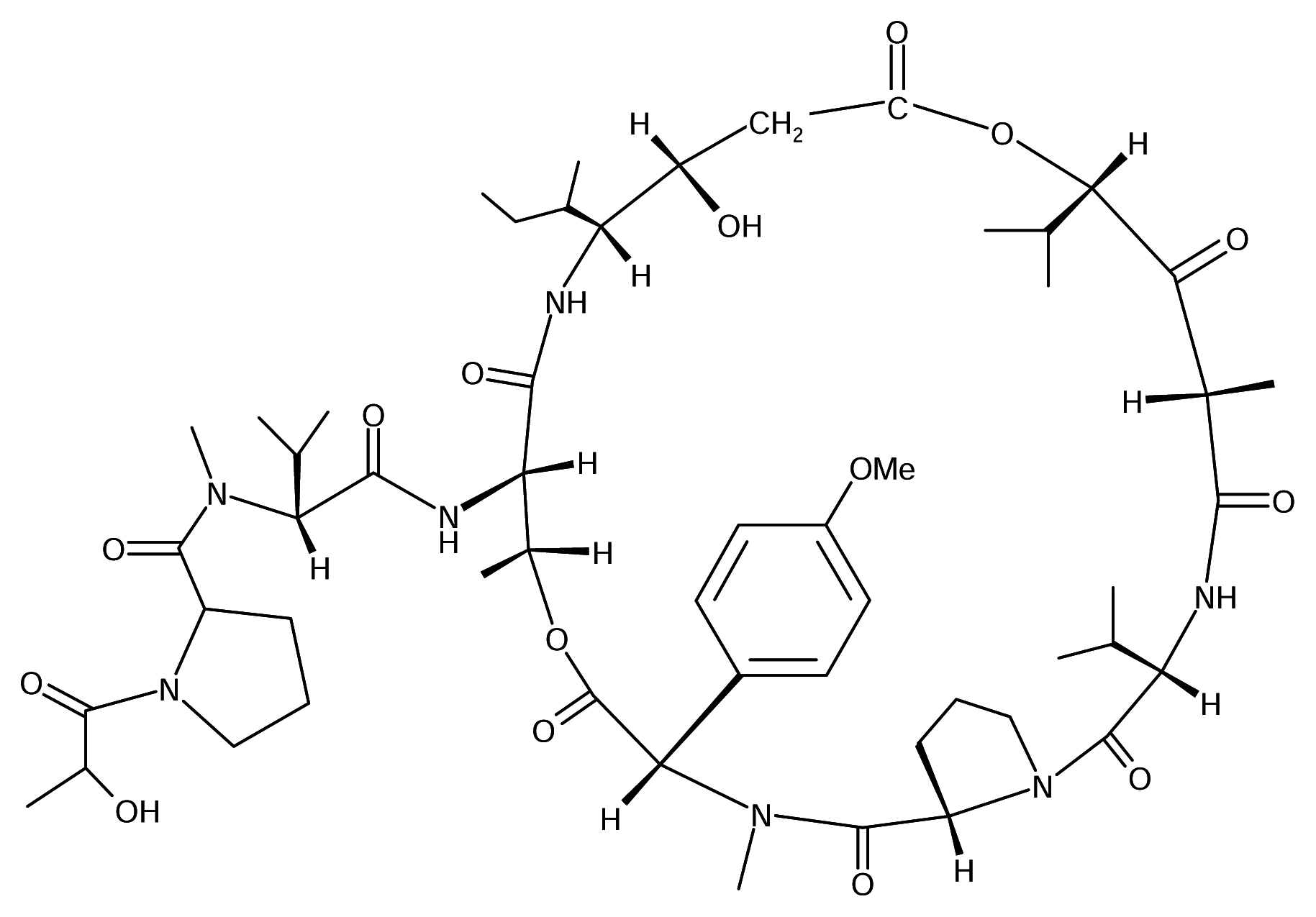
A close relative of didemnin B - dehydrodidemnin B, isolated from a Mediterranean tunicate Aplidium albicans, is currently in Phase II studies in the United States and Europe, to determine its anticancer properties. PharmaMar SA of Spain owns the rights to the compound, which has been shown to be six times more effective than didemnin B in animal tests.
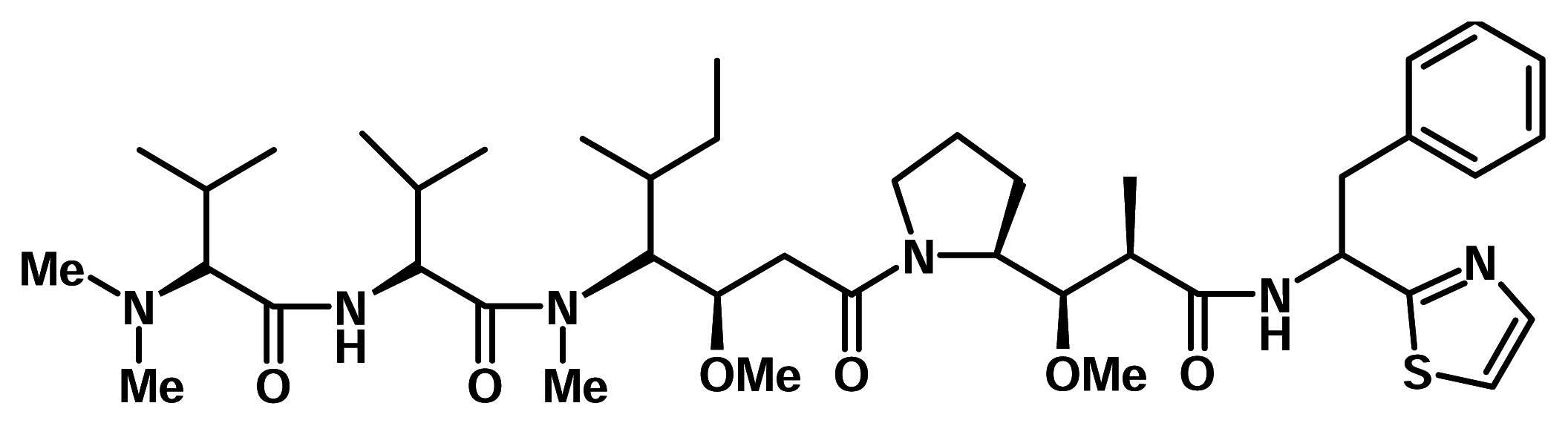
Dolastatin 10
Dolastatin 10 is a linear peptide isolated from the sea hare Dollabella auricularia from the Indian Ocean and is a well known antitumour agent with ED50 = 0.046 ng/ml against P 388 cells. It displayed unprecedented potency in experimental antineoplastic and tubulin assembly systems. Dolastatin 10 is in Phase I clinical trials as anticancer agent for use in the treatment of breast and liver cancers, solid tumours and leukemia [7].

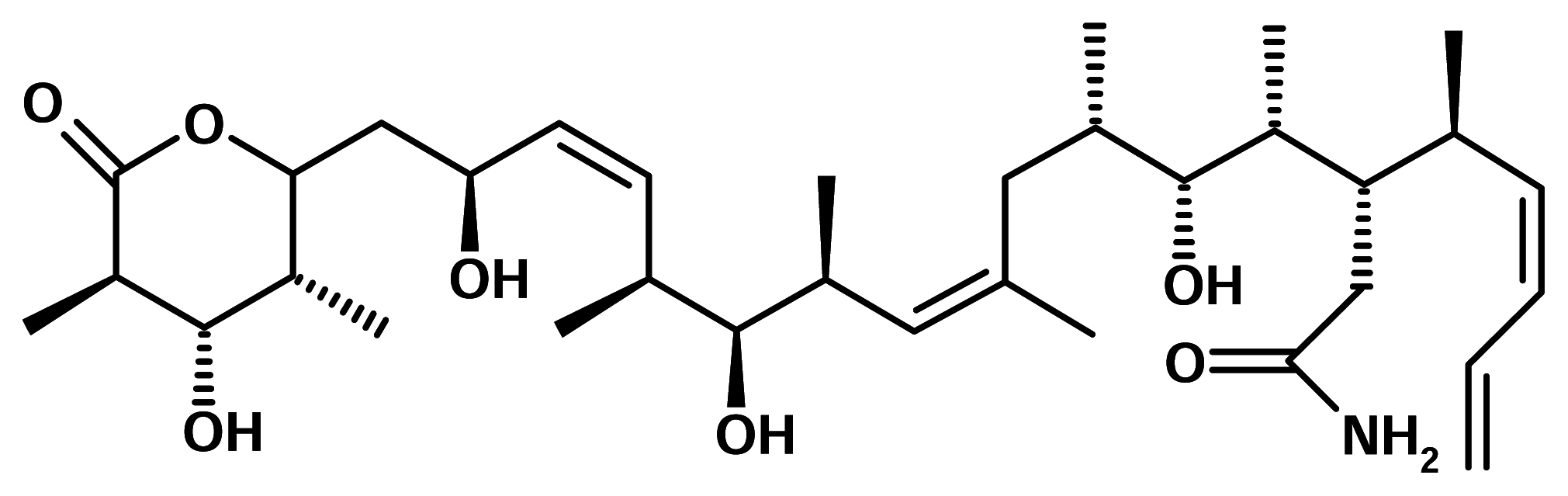
Discodermolide
Discodermolide is a polyhydroxylated lactone, isolated from the deep-sea sponge Discodermia ssp. Descodermolide is an immunosuppresive and cytotoxic agent [8]. The study of its mechanism has revealed that discodermolide was able to stabilize microtubules. In 1998 Novartis Pharma AG licensed this compound for development as a candidate agent for treatment of cancers.

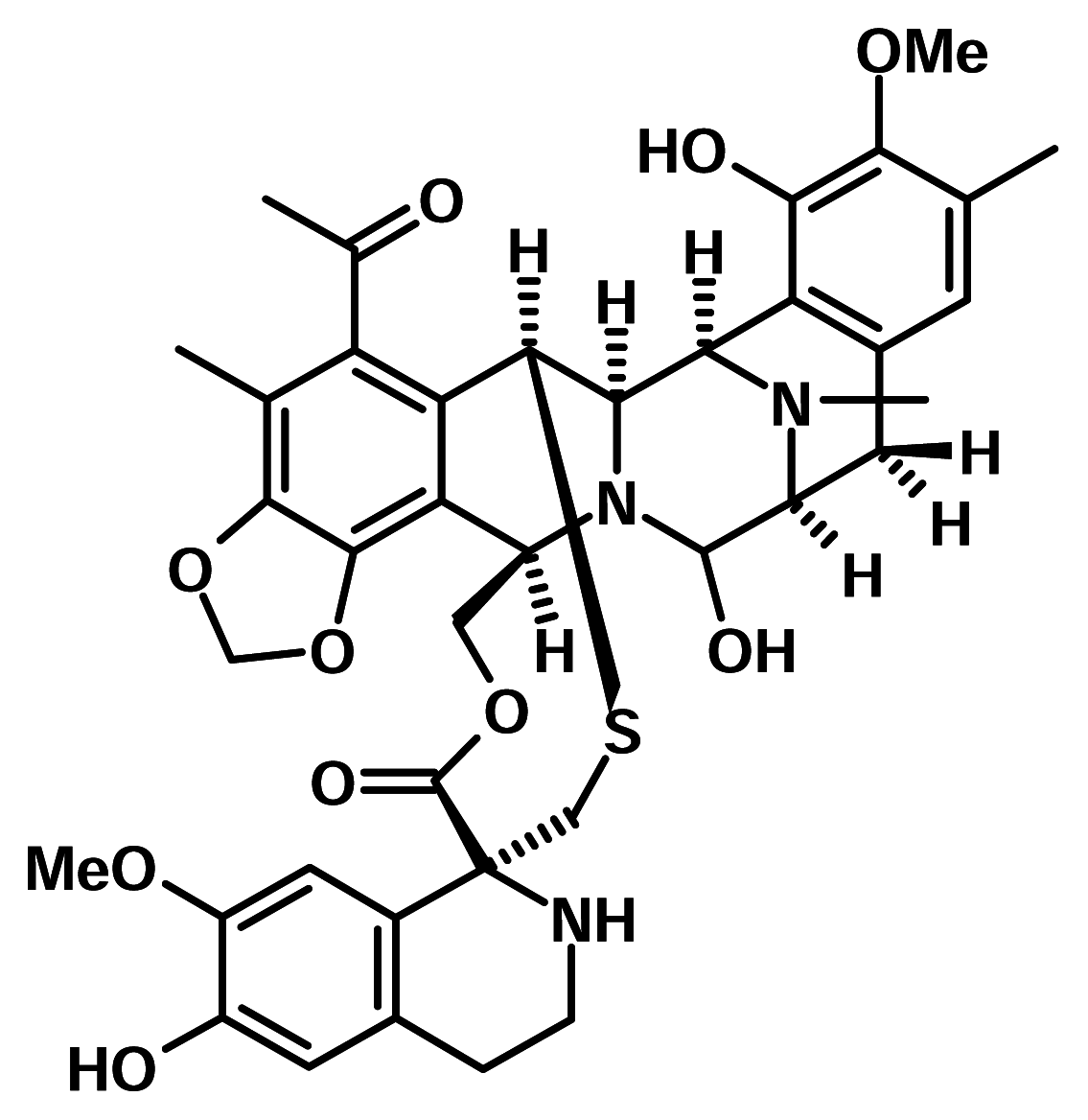
Ecteinascidin-743 (ET-743)
Ecteinascidin-743 or ET-743 is a tetrahydroisoquinoline alkaloid derived from the colonial tunicate Ecteinascidia turbinata, a sea squirt that lives in clusters in the Caribbean and Mediterranean seas. Early on, the compound demonstrated very potent activity against a broad spectrum of tumour types in animal models [9]. The initial sets of Phase I trials for this compound were completed in 1998, with the objective of finding the maximum tolerated dose and studying any possible toxicities. The studies identified a safe, tolerable dose and demonstrated the feasibility of applying it in multiple cycles. Early trial results have shown promising activity of ET-743 in the treatment of advanced soft tissue sarcoma (STS), osteosarcoma and metastatic breast cancers.
Development of ET-743 was followed with interest by the medical community, as anti-tumour activity was observed in all Phase I programs, which were conducted on patients with advanced-stage breast, colon, ovarian and lung cancers, melanoma, mesothelioma and several types of sarcoma. Phase II studies with ET-743 are being performed at 13 different centres in France, Belgium, the Netherlands, the United Kingdom and the United States, in cooperation with assisting groups such as the European Organisation for Research and Treatment of Cancer (EORTC). The European Commission’s Biomed II demonstration program provided partial funding for six of the European studies in 1999.

Recent research indicates that ET-743 may even be able to prevent tumours from becoming drug-resistant in the first place. It was found that ET-743 prevents the formation of P-glycoprotein, a protein associated with multidrug-resistant tumours [10]. P-glycoprotein is a membrane protein that transports toxins such as chemotherapy agents out of the cancer cells, preventing the chemotherapy drugs from destroying the tumour. Previous studies have indicated that when tumours are exposed to chemotherapy agents, they quickly boost the activity of the MDR1 gene, which is responsible for the formation of P-glycoprotein. By interfering with that process, ET-743 may keep the tumour cells vulnerable to chemotherapy. Even if ET-743 is not proven effective on its own, it may become a key ingredient in chemotherapy “cocktails” to prevent tumours from developing resistance to drugs currently used.
Kahalaide F
Kahalaide F is a depsipeptide discovered in Elysia rufescens, a marine mollusc found in Hawaii. Studies to determine the mechanism of action of Kahalaide F showed that, in certain experimental systems, it caused a disruption of lysosomal membranes and consequently the formation of large vacuoles. This mechanism is unique among anticancer agents and may cause increasing acidification of the intra-cellular space, a stimulatory event that initiates a pathway for apoptosis [11]. In addition, Kahalaide F leads to an inhibition of erb 2 transmembrane tyrosine kinase activity and inhibits TGF–a gene expression.
The pattern of Kahalaide F cytotoxicity was distinct from any of the other standard chemotherapeutic agents. Kahalaide F showed in vitro and in vivo selectivity for prostate-derived cell lines and tumours. It is further selective for hormone-independent prostate tumour cells, which generally represent the more aggressive and harder to treat type of prostate cancer. Phase I clinical trials in patients with androgen-independent prostate cancer have begun.

It is interesting to note that the majority of the compounds under clinical trials described so far are either cytotoxic or immunosuppresive agents. However, as the advance in mechanism-based bioassays continues, other pharmacologically active marine natural products have been discovered. These include:
Ziconitide (Conotoxin MVIIV)
Ziconitide is a 25 aminoacid peptide from the venom of a predatory snail Conus magnus. It acts by binding to and inhibiting presynaptic calcium channels, thereby preventing neurotransmitter release [12]. It is licensed by Elan Pharmaceuticals under the name Prialt.
Prialt blocks nerve impulses in a key region of the spinal cord, where pain fibers from the body connect with the nerve cells that send pain to the brain. This is why Prialt, which is 50 times more potent than morphine, is so exquisitely precise and does not cause the adverse effects of opiates. It stops pain messages from getting through while allowing the rest of the nervous system to function normally.
However, it would be inaccurate to assume that all of the biologically active compounds being tested are toxic. As the secondary metabolites from marine organisms show diverse structural types, it is expected that they should have biological activities across the board. These compounds are:
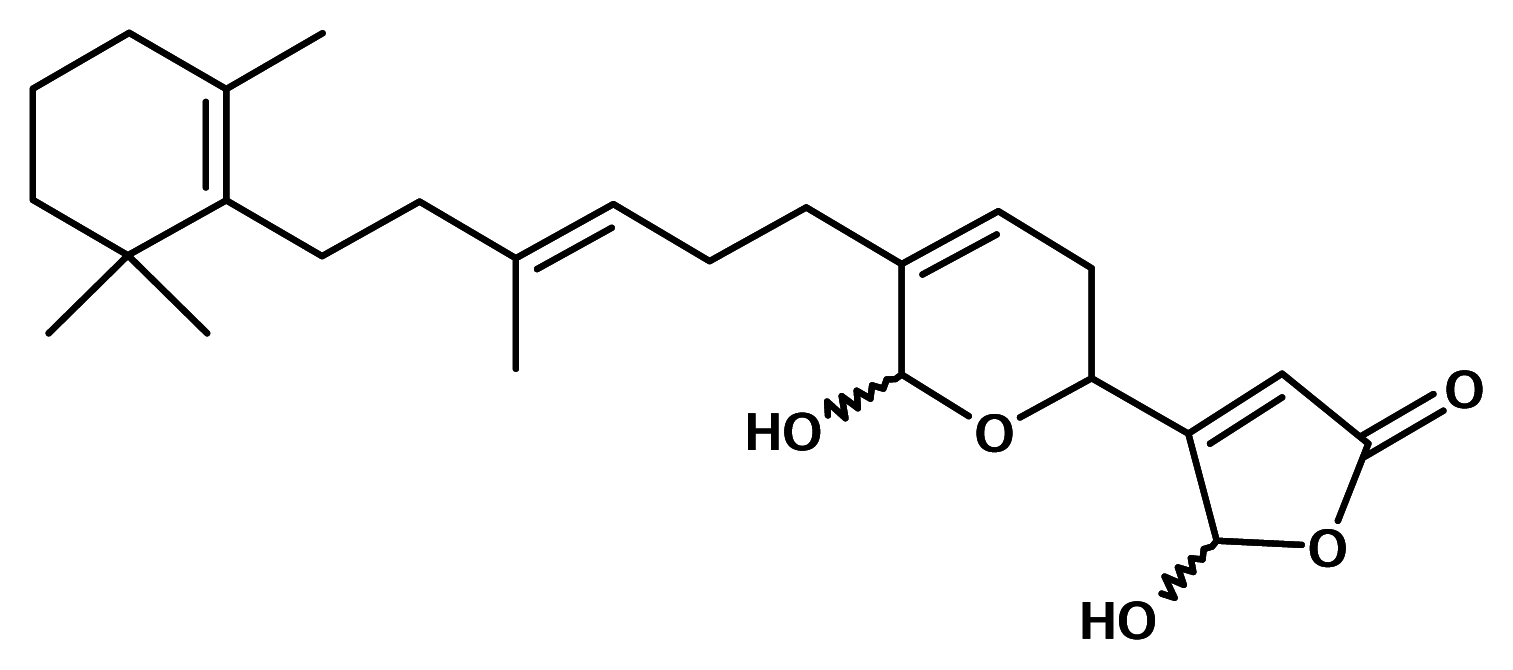
Manoalide
Manoalide is a sesquiterpenoid isolated from the Indo-Pacific sponge Luffariella variabilis. It is a potent analgesic and anti-inflammatory agent. Manoalide is by far the best characterised PLA2 inhibitor from natural sources. At low concentrations manoalide inhibited calcium channels with no effect on phosphoinositide metabolism. Manoalide was licensed by Allergan Pharmaceuticals who took the compound through Phase I clinical trials for the treatment of psoriasis, then launched a medicinal chemistry program with it. Though no pharmaceutical based on manoalide has yet reached the drugstores, manoalide itself is commercially available as a standard probe for PLA2 inhibition [13].
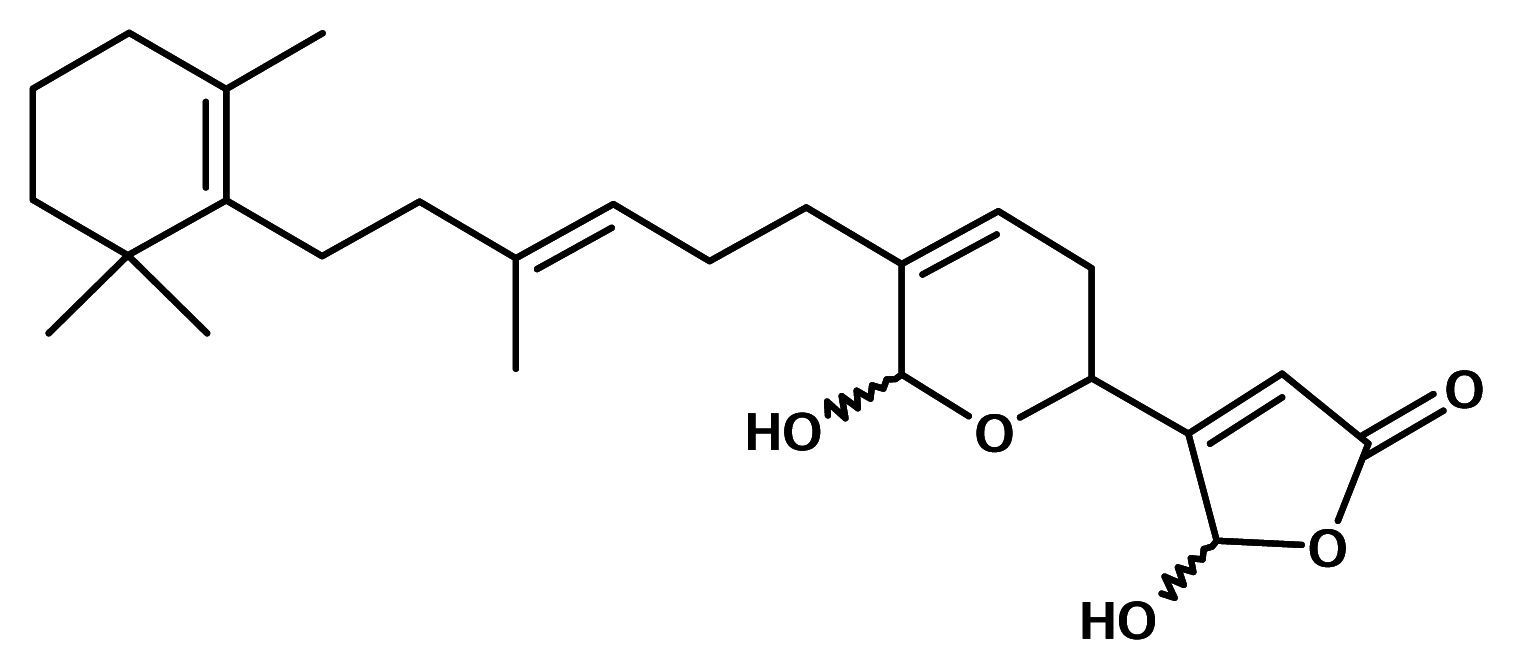
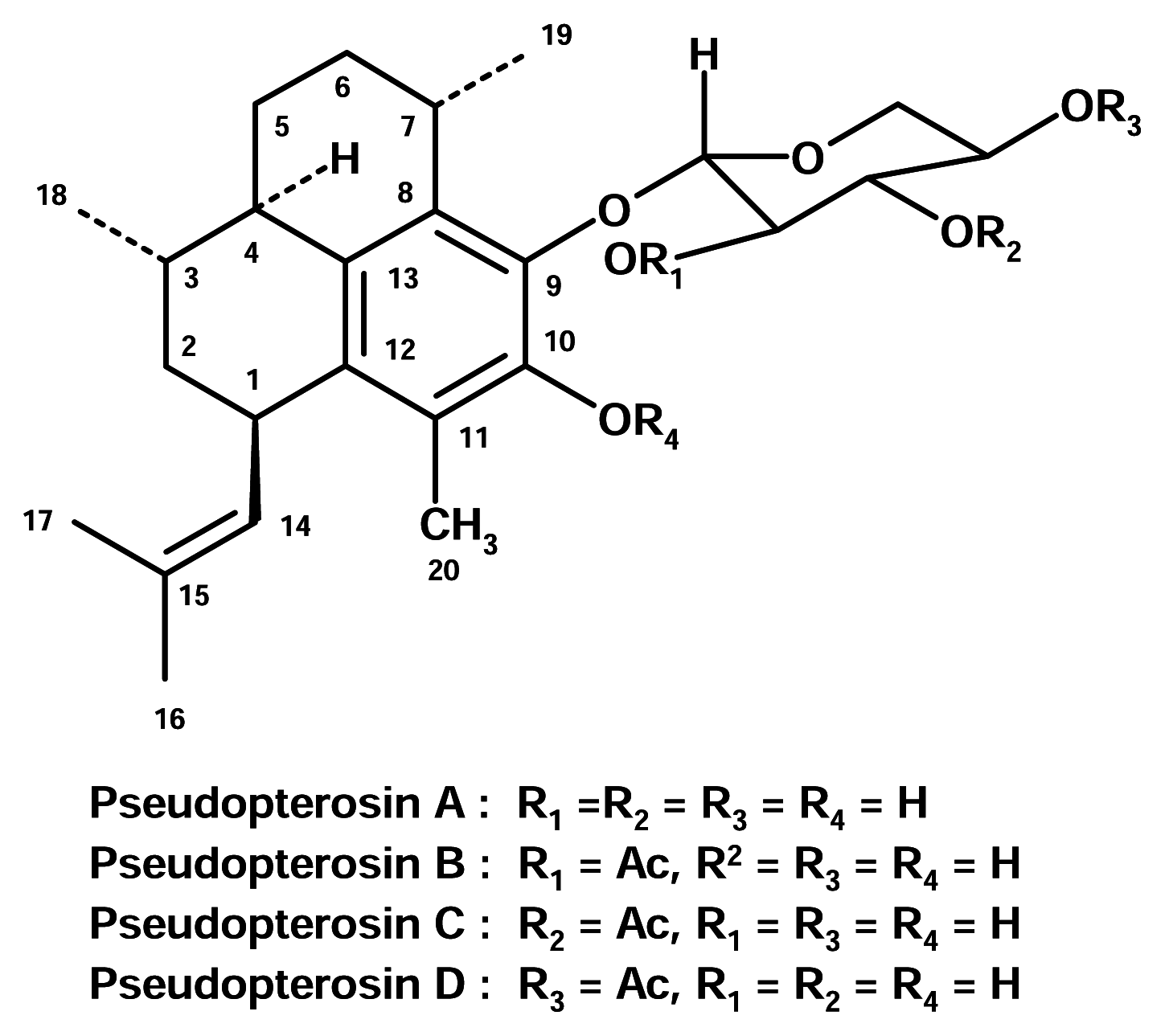
Pseudopterosins
The pseudopterosins are tricyclic diterpene glycosides isolated from the Caribbean sea whip (gorgonian) Pseudopterogorgia elisabethae (Gorgoniidae). They are potent anti-inflammatory and analgesic agents and appear to inhibit eicosanoid biosynthesis by inhibition of both PLA2 and 5-lipoxygenase. Interestingly, the pseudopterosins are found to inhibit only PMN-PLA2, and not PLA2 from other sources. It is also thought that the cell type selectivity of the pseudopterosins may well be a function of the glycoside moiety and a novel example of drug targeting [13].

The pseudopterosins have been licensed to a small pharmaceutical firm, OsteoArthritis Sciences Inc., for medical use as potential anti-inflammatory drugs. The company has completed preclinical tests of one of pseudopterosins, a potent tropical anti-inflammatory compound, and filed an Investigational New Drug (IND) application with the U.S. food and Drug Administration. Clinical trials on human subjects for irritant contact dermatitis are anticipated.
However, the pseudopterosins extract has found its way to the marketplace. It is used as an additive to prevent irritation caused by exposure to the sun or the chemicals in the Estée Lauder cosmetic skin care product, Resilience®[14].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Compound | Organism | Origin |
|---|---|---|---|
| Cancer | Aplidine | Tunicate | Mediterranean |
| Bryostatin1 | Bryozoan | Gulf of California | |
| Didemnin B | Tunicate | Caribbean | |
| Dolastatin 10 | Sea hare | Indian Ocean | |
| Ecteinascidin-743 | Tunicate | Caribbean | |
| Halichondrin B | Sponge | Okinawa | |
| Kahalaide F | Mollusk | Hawaii | |
| Mycaperoxide B | Sponge | Thailand | |
| HIV | Cyclodidemniserinol trisulfate | Tunicate | Palau |
| Lamellarin a 20 sulfate | Tunicate | Australia | |
| Nematode infection | Dithiocyanates | Sponge | Australia |
| Asthma | Contignasterol | Sponge | Papua, New Guinea |
| Pain | Conotoxins | Snail | Australia |
Acknowledgements
We wish to thank Fundação para a Ciência e Tecnologia (FCT) of Portugal, POCTI (QCA III) and FEDER for support.
References
- Carté, B. K. Biomedical Potential of Marine Natural Products. Biosciences 1996, 271–286. [Google Scholar]
- Halvorson, H. O. Aquaculture, Marine Sciences and Oceanography: A Confluence Connection. New Engl. J. Higher Ed. Econ. Dev 1998, 13, 28–42. [Google Scholar]
- Faulkner, D. J. Chemical Riches from the Ocean. Chem. Brit 1995, 680–684. [Google Scholar]
- Scheuer, P. J. Marine Metabolites as Drug Leads-Retrospect and Prospect. In Biochemical Aspects of Marine Pharmacology; Lazarovici, P., Spira, M. E., Zlotkin, Eliahu, Eds.; Alaken, Inc: Fort Collins, Colorado, 1996; pp. 1–12. [Google Scholar]
- de Vries, D. J.; Beart, P. M. Fishing for Drugs from the Sea: Status and Strategies. TiPS 1995, 16, 275–279. [Google Scholar]
- Rinehart, K. L.; Kishore, V.; Bible, K. C.; Sakai, R.; Sullins, D. W.; Li, K.M. Didemnins and Tunichlorin: Novel Natural Products from the Marine Tunicate Trididemnum solidum. J. Nat. Prod 1988, 51, 1–21. [Google Scholar]
- Yamada, K; Okija, M.; Kigoshi, H.; Suenaga, K. Cytotoxic Substances from Opisthobranch Molluscs. In Drugs From The Sea; Fusetani, N., Ed.; Karger AG; Basel, 2000; pp. 59–73. [Google Scholar]
- Gunasekera, S. P.; Gunasekera, M.; Longley, R. E.; Schulte, G.K. J. Org. Chem 1990, 55, 4912–4915.
- Rinehart, K. L. Antitumor Compounds from Tunicates. Med. Res. Rev 2000, 20, 1–27. [Google Scholar]
- Zewail-Foote, M.; Hurley, L.H. Ecteinascidin 743: A Minor Groove Alkylator that Binds DNA Toward the Major Groove. J. Med. Chem 1999, 42, 2493–2497. [Google Scholar]
- Hamann, M. T.; Scheuer, P. J. Kahalide F, A Bioactive Depsipeptide from the Sacoglossan Mollusk Elisia refescens and the Green Alga Bryopsis sp. J. Am. Chem. Soc 1993, 115, 5825–5826. [Google Scholar]
- Oliveira, B. M.; Gray, W. R.; Zeikus, R.; McIntosh, J. M.; Varga, J.; Revier, J.; de Santos, W.; Cruz, L. J. Peptide Neurotoxins from Fish-Hunting Cone Snails. Science 1985, 230, 1338–1343. [Google Scholar]
- Potts, B. C. M.; Faulkner, D. J. Phospholipase A2 Inhibitors from Marine Organisms. J. Nat. Prod 1992, 55, 1701–1717. [Google Scholar]
- Rouhi, A. M. Supply Issues Complicate Trek of Chemicals from the Sea to Market. C&EN 1995, 42–44. [Google Scholar]
© 2004 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Kijjoa, A.; Sawangwong, P. Drugs and Cosmetics from the Sea. Mar. Drugs 2004, 2, 73-82. https://doi.org/10.3390/md202073
AMA Style
Kijjoa A, Sawangwong P. Drugs and Cosmetics from the Sea. Marine Drugs. 2004; 2(2):73-82. https://doi.org/10.3390/md202073
Chicago/Turabian StyleKijjoa, Anake, and Pichan Sawangwong. 2004. "Drugs and Cosmetics from the Sea" Marine Drugs 2, no. 2: 73-82. https://doi.org/10.3390/md202073