Synthetic Pinnatoxins A and G Reversibly Block Mouse Skeletal Neuromuscular Transmission In Vivo and In Vitro

 , , ,
, , ,
Abstract
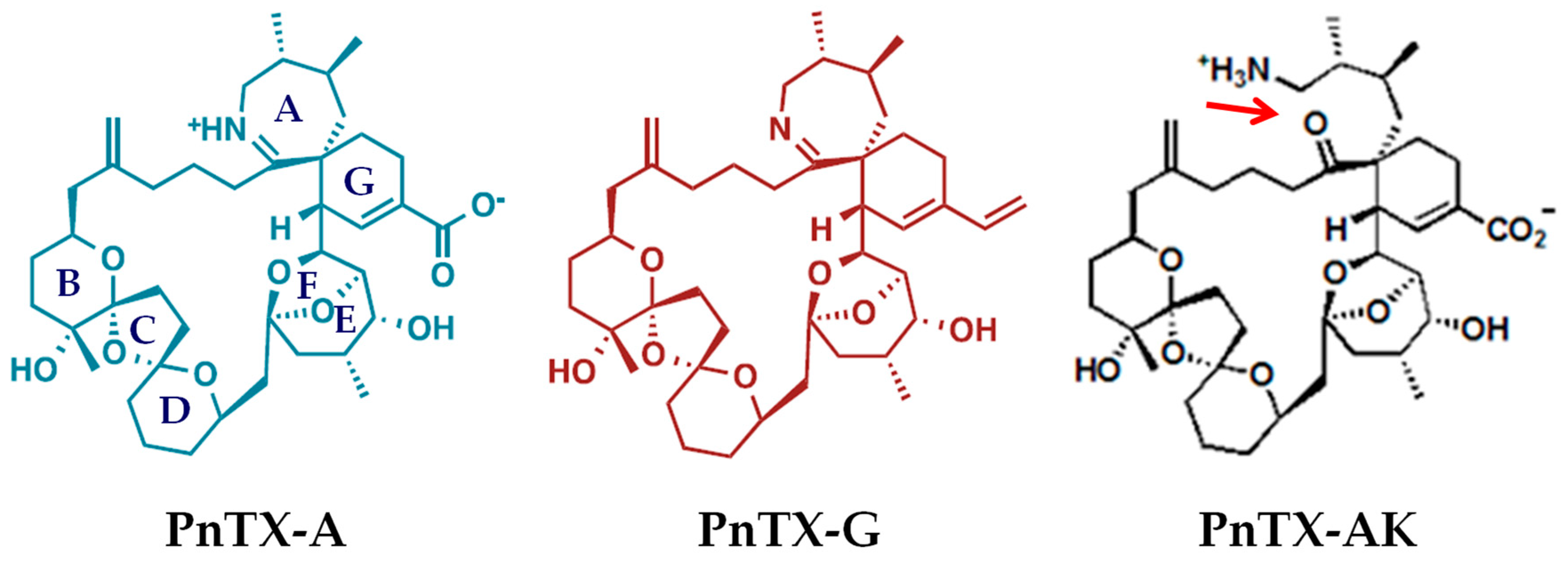
:1. Introduction
2. Results
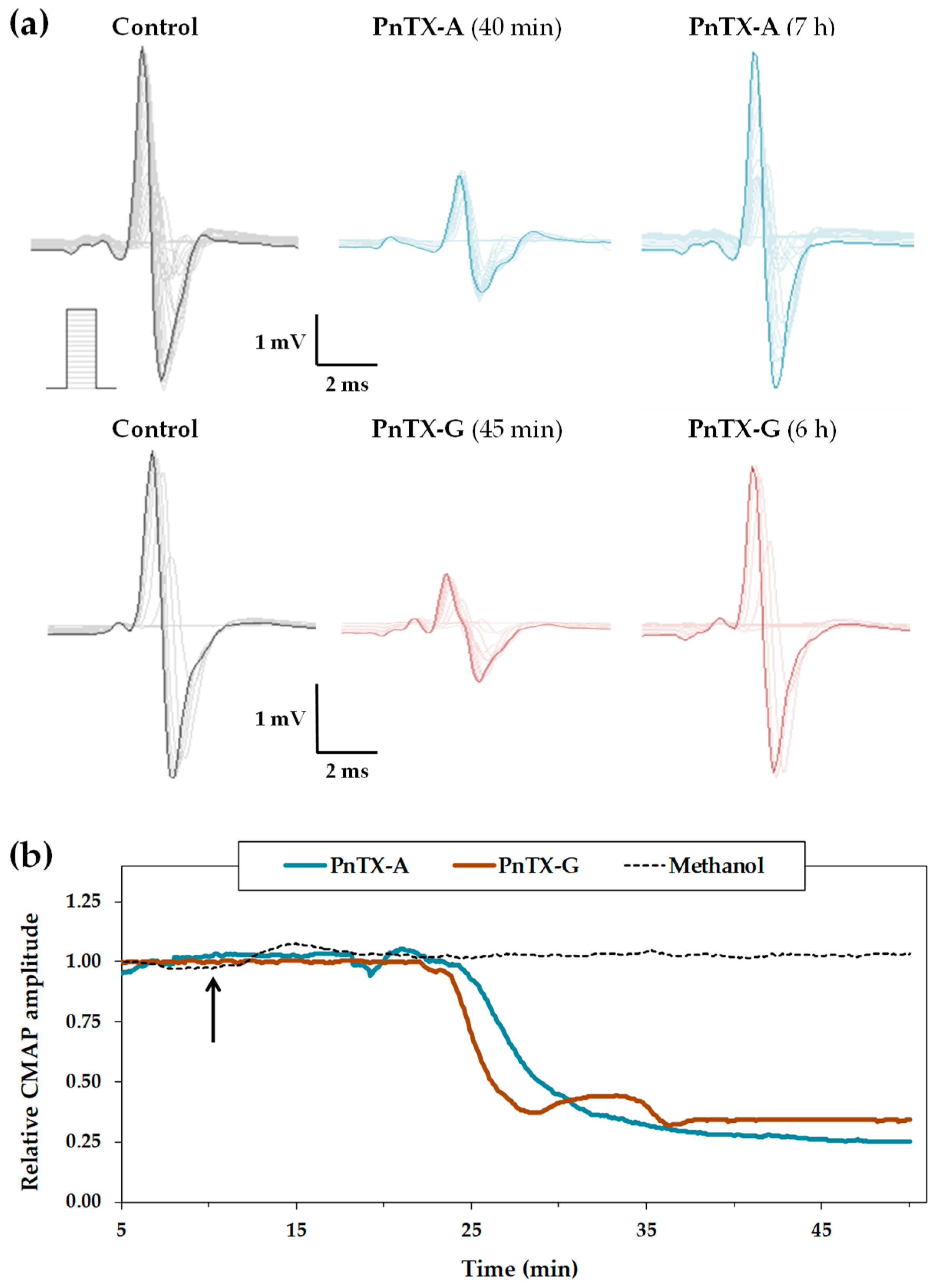
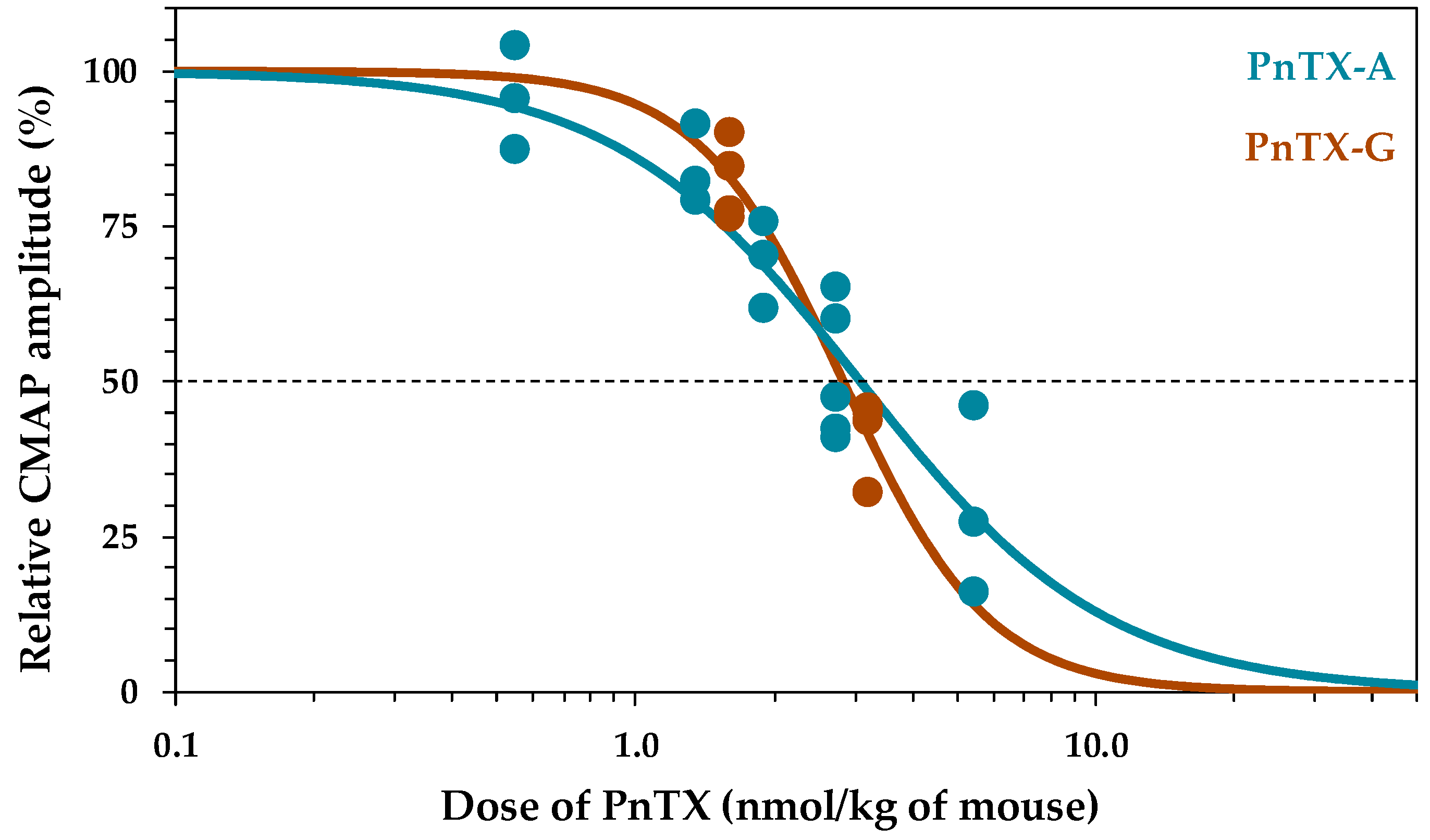
2.1. Effects of PnTX-A and G on the Excitability Properties of Mouse Neuromuscular System In Vivo
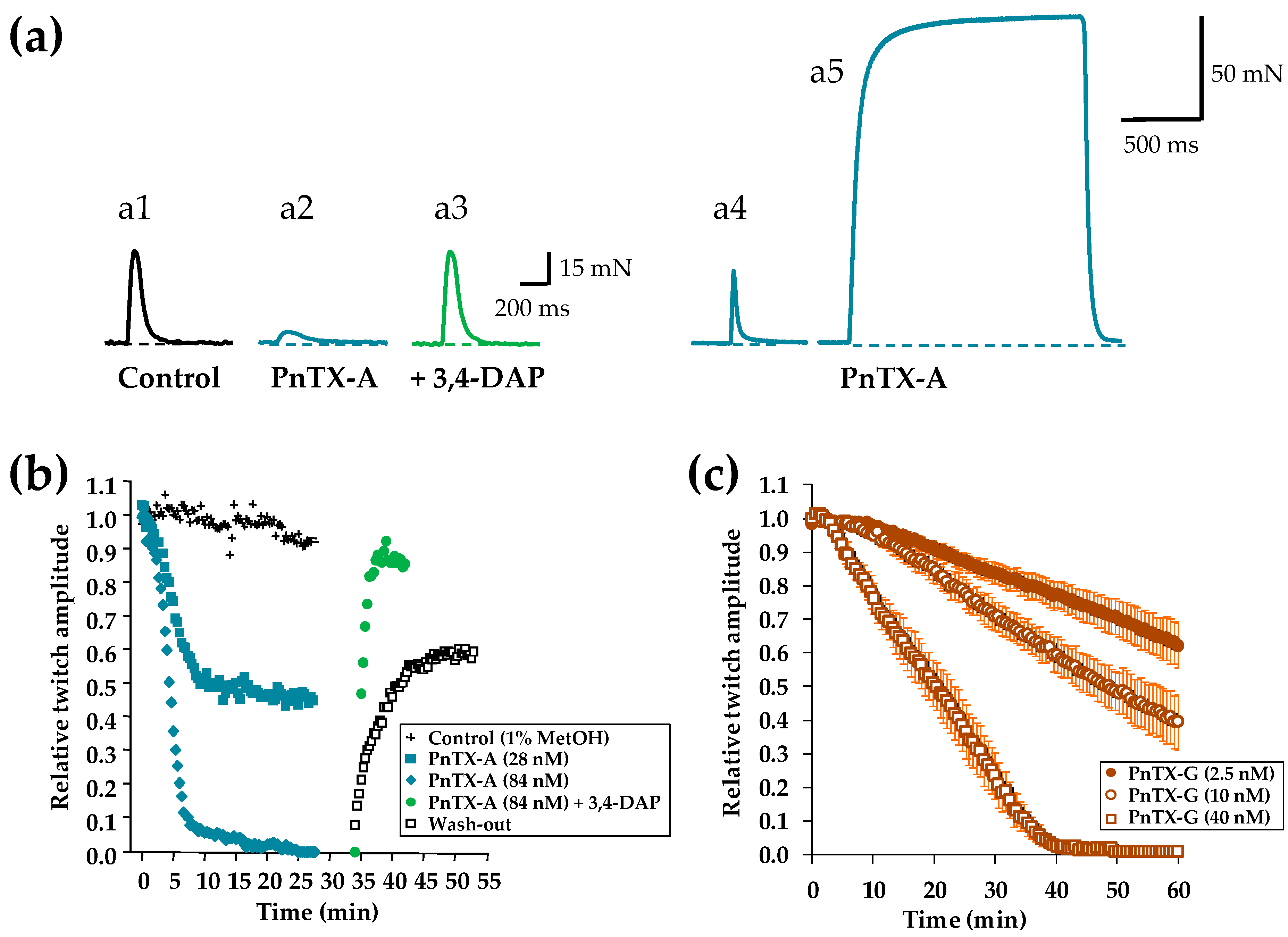
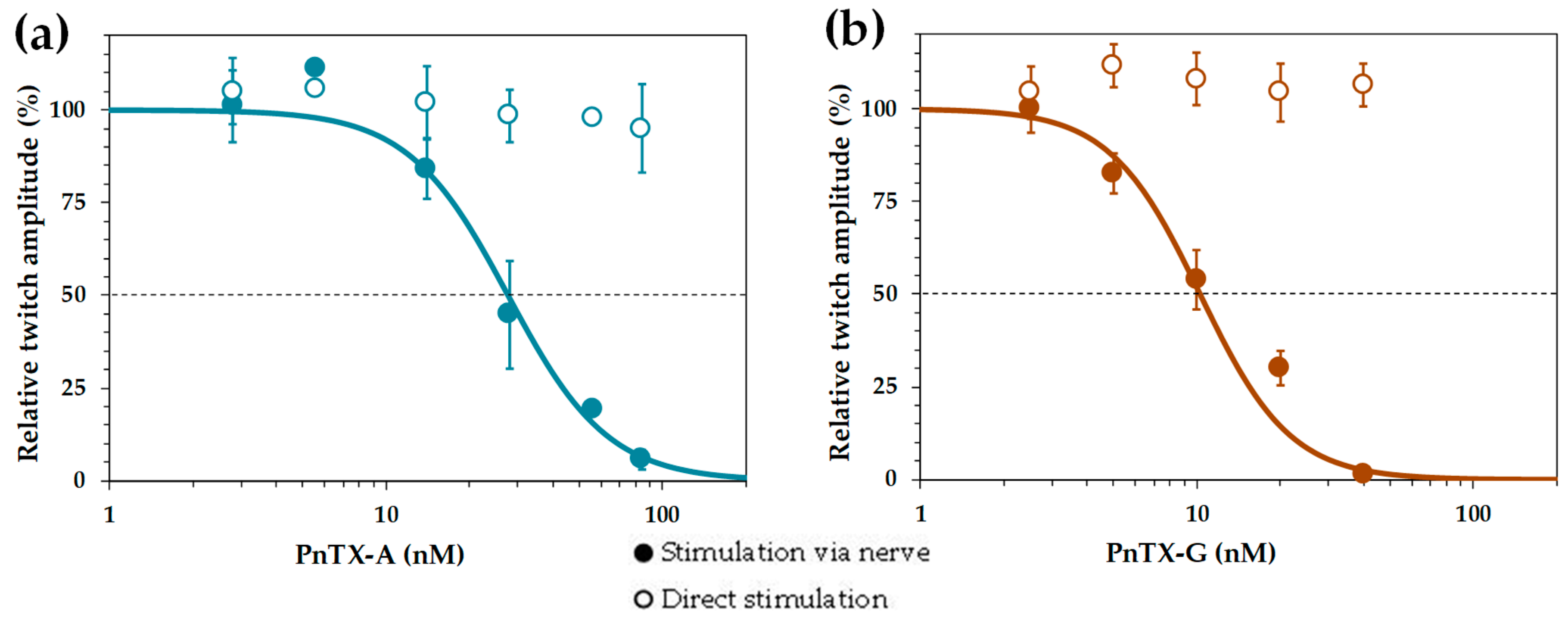
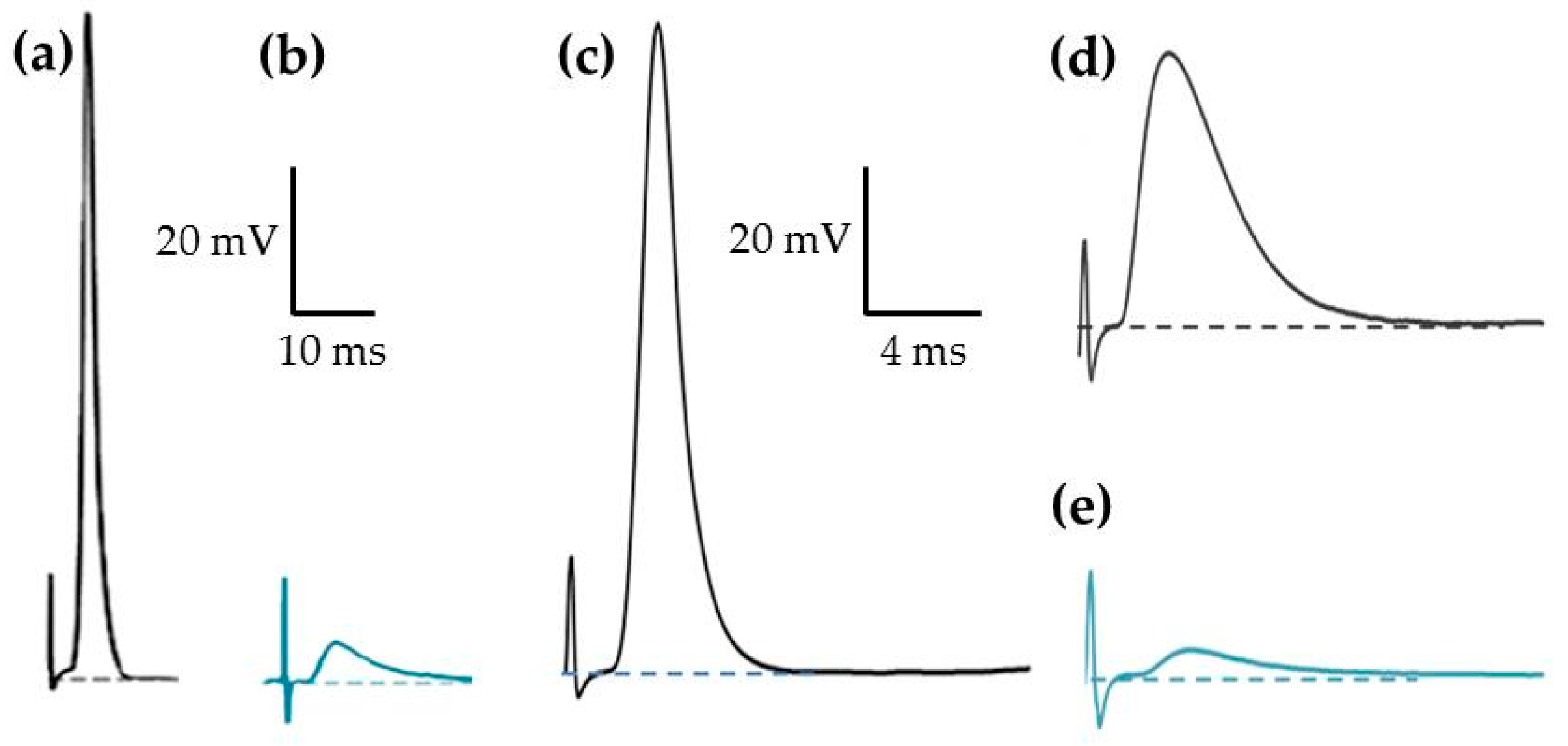
2.2. Effects of PnTX-A and G on Isometric Twitch Tension In Vitro
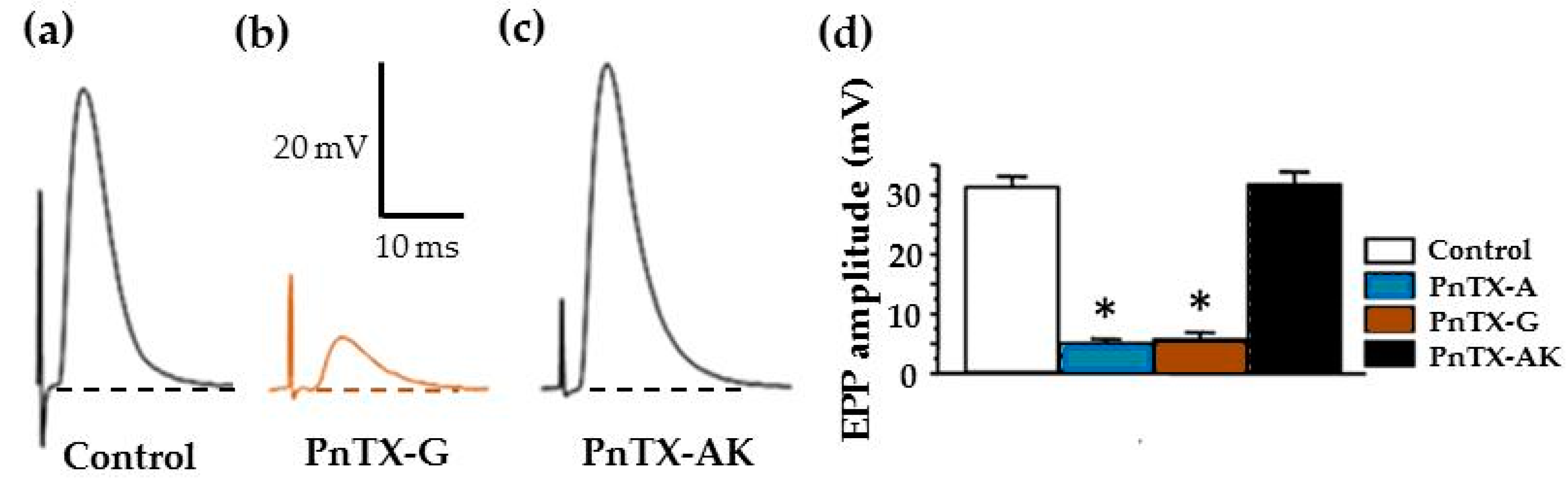
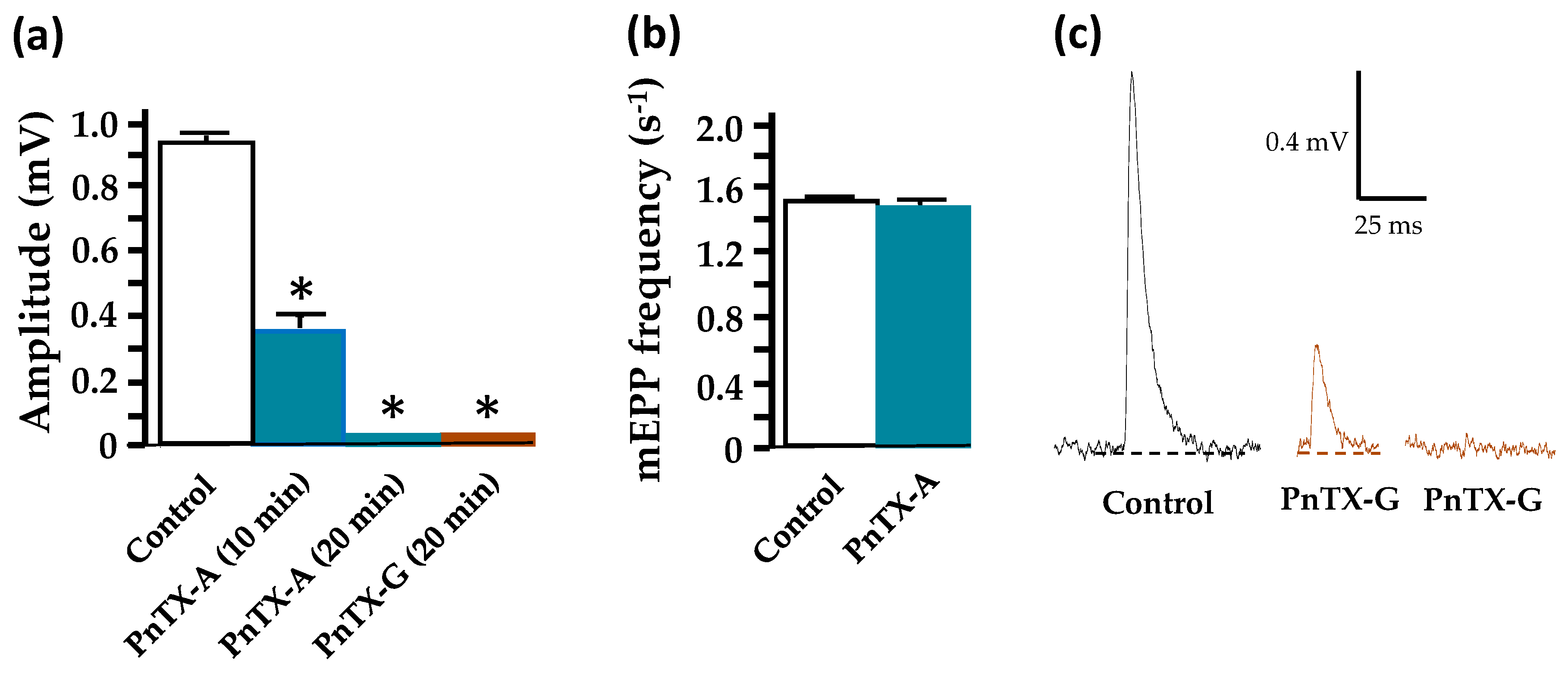
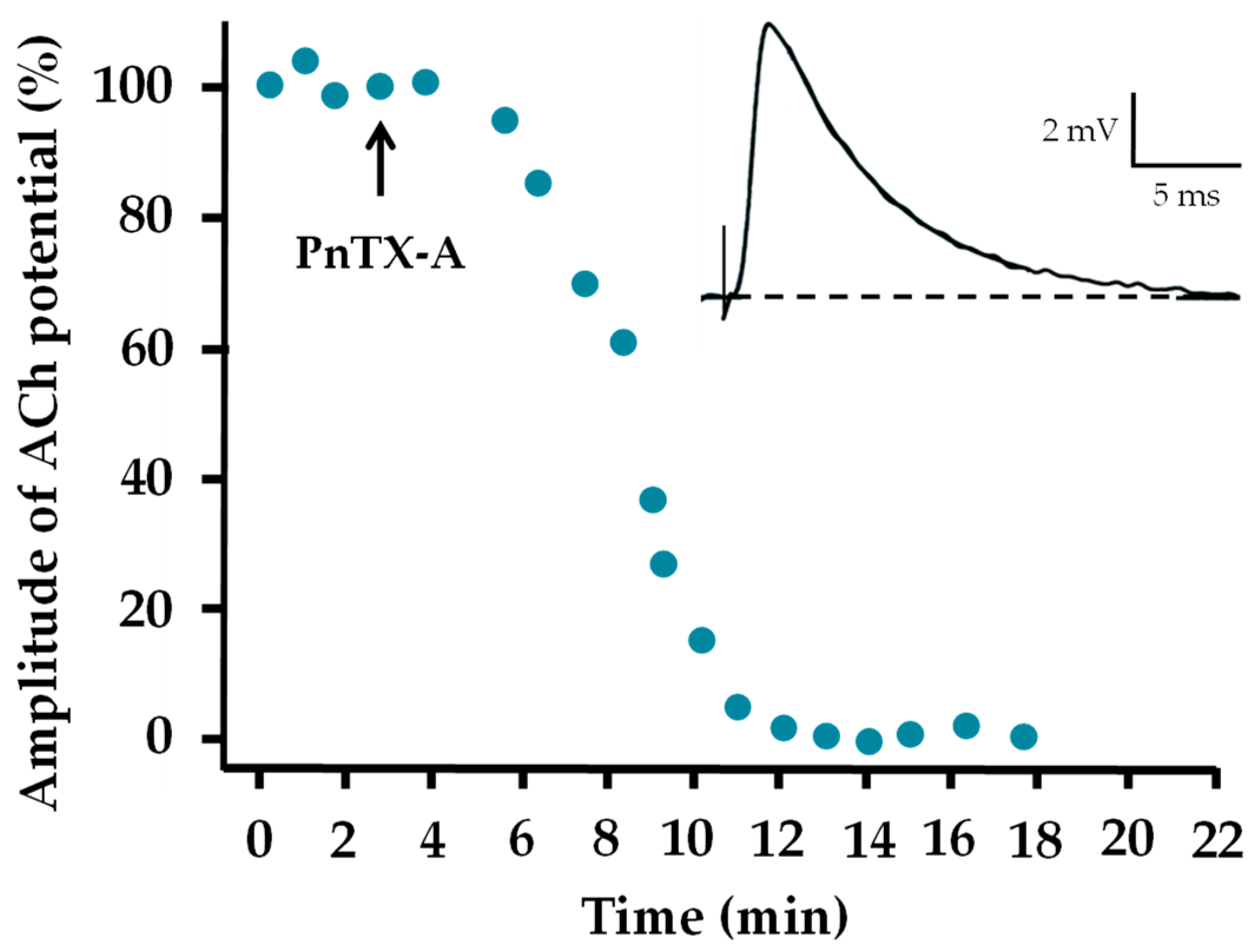
2.3. Effects of PnTX-A and G on Neuromuscular Transmission In Vitro
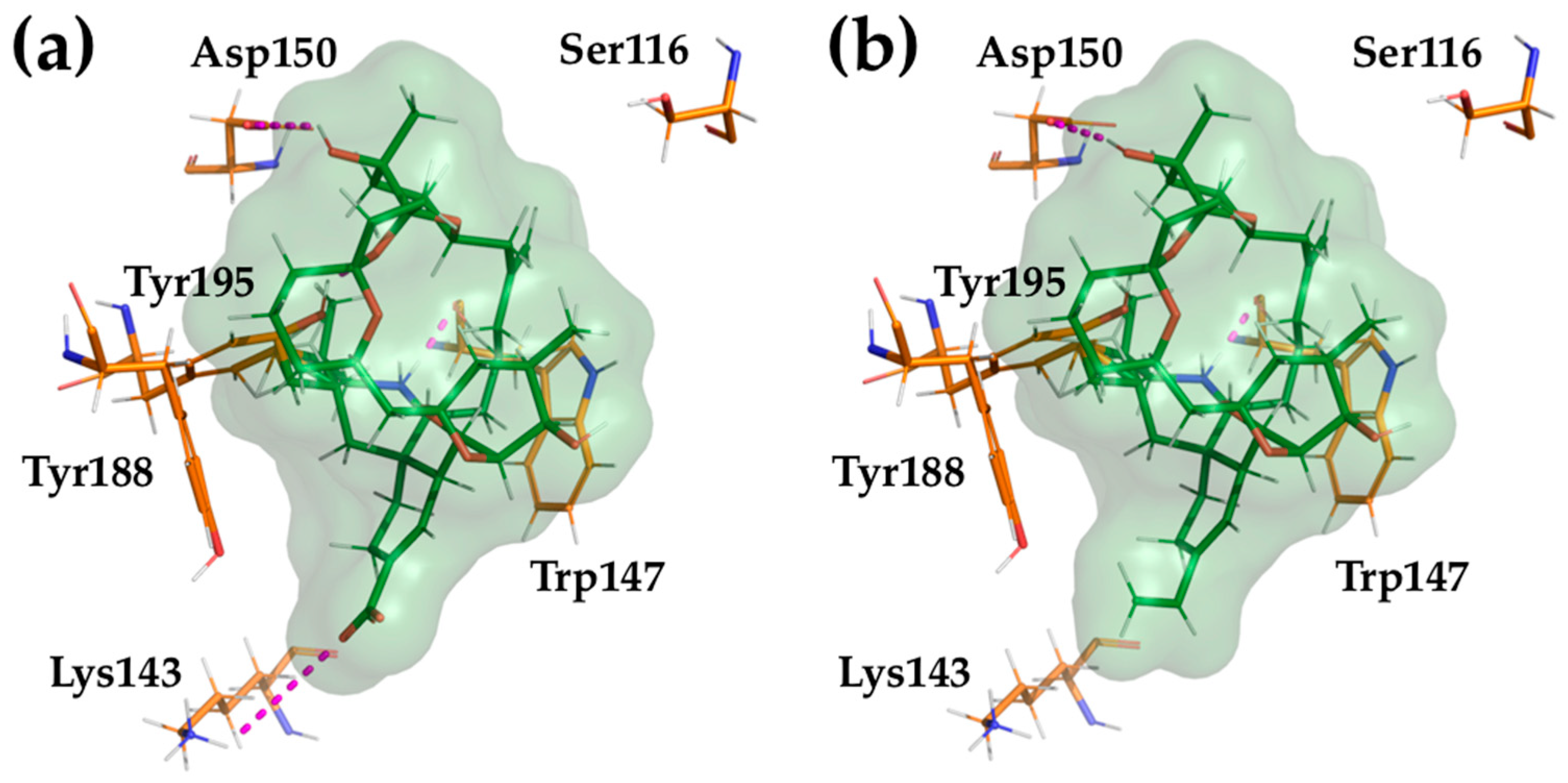
2.4. Computational Modeling of PnTX-A and G Interactions with the Nicotinic Acetylcholine Receptor
3. Discussion
4. Materials and Methods
4.1. Toxins and Chemicals
4.2. Animals
4.3. Recordings from the Neuromuscular System of Anesthetized Mice In Vivo
4.4. Recordings from Isolated Mouse Nerve-muscle Preparations In Vitro
4.5. Data and Statistical Analyses
4.6. Molecular Modeling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zheng, S.Z.; Huang, F.L.; Chen, S.C.; Tan, X.F.; Peng, J.; Xie, R.W. The isolation and bioactivities of pinnatoxin. Chin. J. Mar. Drugs 1990, 9, 33–35. (In Chinese) [Google Scholar]
- Uemura, D.; Chou, T.; Haino, T.; Nagatsu, A.; Fukuzawa, S.; Zheng, S.-Z.; Chen, H.-S. Pinnatoxin A: A toxic amphoteric macrocycle from the Okinawan bivalve Pinna muricata. J. Am. Chem. Soc. 1995, 117, 1155–1156. [Google Scholar] [CrossRef]
- Takada, N.; Umemura, N.; Suenaga, K.; Chou, T.; Nagatsu, A.; Haino, T.; Yamada, K.; Uemura, D. Pinnatoxins B and C, the most toxic components in the pinnatoxin series from the Okinawan bivalve Pinna muricata. Tetrahedron Lett. 2001, 42, 3491–3494. [Google Scholar] [CrossRef]
- Chou, T.; Haino, T.; Kuramoto, M.; Uemura, D. Isolation and structure of pinnatoxin D, a new shellfish poison from the Okinawan bivalve Pinna muricata. Tetrahedron Lett. 1996, 37, 4027–4030. [Google Scholar] [CrossRef]
- McNabb, P.; Rhodes, L.; Selwood, A. Results of analyses for brevetoxins and pinnatoxins in Rangaunu Harbour oysters, 1993–2008. Cawthron Rep. 2008, 1453, 18. [Google Scholar]
- Selwood, A.I.; Miles, C.O.; Wilkins, A.L.; van Ginkel, R.; Munday, R.; Rise, F.; McNabb, P. Isolation, structural determination and acute toxicity of pinnatoxins E, F and G. J. Agric. Food Chem. 2010, 58, 6532–6542. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, L.; Smith, K.; Selwood, A.; McNabb, P.; van Ginkel, R.; Holland, P.; Munday, R. Production of pinnatoxins by a peridinoid dinoflagellate isolated from Northland, New Zealand. Harmful Algae 2010, 9, 384–389. [Google Scholar] [CrossRef]
- Rhodes, L.; Smith, K.; Selwood, A.; McNabb, P.; Molenaar, S.; Munday, R.; Wilkinson, C.; Hallegraeff, G. Production of pinnatoxins E, F and G by scrippsielloid dinoflagellates isolated from Franklin Harbour, South Australia. N. Z. J. Mar. Freshw. Res. 2011, 45, 703–709. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, L.; Smith, K.; Selwood, A.; McNabb, P.; Munday, R.; Suda, S.; Molenaar, S.; Hallegraeff, G. Dinoflagellate Vulcanodinium rugosum identified as the causative organism of pinnatoxins in Australia, New Zealand and Japan. Phycologia 2011, 50, 624–628. [Google Scholar] [CrossRef]
- Smith, K.F.; Rhodes, L.L.; Suda, S.; Selwood, A.I. A dinoflagellate producer of pinnatoxin G, isolated from sub-tropical Japanese waters. Harmful Algae 2011, 10, 702–705. [Google Scholar] [CrossRef]
- Nézan, E.; Chomérat, N. Vulcanodinium rugosum gen. et sp. nov. (Dinophyceae), un nouveau dinoflagellé marin de la côte méditerranéenne française. Cryptogam. Algol. 2011, 32, 3–18. [Google Scholar] [CrossRef]
- Hess, P. First report of pinnatoxin in mussels and a novel dinoflagellate, Vulcanodinium rugosum, from France. In Proceedings of the 8th International Conference on Molluscan Shellfish Safety, Charlottetown, PE, Canada, 12–17 June 2011. [Google Scholar]
- Hess, P.; Abadie, E.; Hervé, F.; Berteaux, T.; Séchet, V.; Aráoz, R.; Molgó, J.; Zakarian, A.; Sibat, M.; Rundberget, T.; et al. Pinnatoxin G is responsible for atypical toxicity in mussels (Mytilus galloprovincialis) and clams (Venerupis decussata) from Ingril, a French Mediterranean lagoon. Toxicon 2013, 75, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Miles, C.O.; Rundberget, T.; Sandvik, M.; Aasen, J.A.B.; Selwood, A.I. The Presence of Pinnatoxins in Norwegian Mussels; National Veterinary Institute’s Report Series 7b-2010; National Veterinary Institute: Oslo, Norway, 2010. [Google Scholar]
- Rundberget, T.; Aasen, J.A.; Selwood, A.I.; Miles, C.O. Pinnatoxins and spirolides in Norwegian blue mussels and seawater. Toxicon 2011, 58, 700–711. [Google Scholar] [CrossRef] [PubMed]
- García-Altares, M.; Casanova, A.; Bane, V.; Diogène, J.; Furey, A.; de la Iglesia, P. Confirmation of pinnatoxins and spirolides in shellfish and passive samplers from Catalonia (Spain) by liquid chromatography coupled with triple quadrupole and high-resolution hybrid tandem mass spectrometry. Mar. Drugs 2014, 12, 3706–3732. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.; Bane, V.; García-Altares, M.; van Pelt, F.N.; Furey, A.; O’Halloran, J. Assessment of emerging biotoxins (pinnatoxin G and spirolides) at Europe’s first marine reserve: Lough Hyne. Toxicon 2015, 108, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Rambla-Alegre, M.; Miles, C.O.; de la Iglesia, P.; Fernandez-Tejedor, M.; Jacobs, S.; Sioen, I.; Verbeke, W.; Samdal, I.A.; Sandvik, M.; Barbosa, V.; et al. Occurrence of cyclic imines in European commercial seafood and consumers risk assessment. Environ. Res. 2018, 161, 392–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarron, P.; Rourke, W.A.; Hardstaff, W.; Pooley, B.; Quilliam, M.A. Identification of pinnatoxins and discovery of their fatty acid ester metabolites in mussels (Mytilus edulis) from eastern Canada. J. Agric. Food Chem. 2012, 60, 1437–1446. [Google Scholar] [CrossRef]
- Garrett, M.J.; Puchulutegui, C.; Selwood, A.I.; Wolny, J.L. Identification of the harmful dinoflagellate Vulcanodinium rugosum recovered from a ballast tank of a globally traveled ship in Port Tampa Bay, Florida, USA. Harmful Algae 2014, 39, 202–209. [Google Scholar] [CrossRef]
- Zeng, N.; Gu, H.; Smith, K.F.; Rhodes, L.L.; Selwood, A.I.; Yang, W. The first report of Vulcanodinium rugosum (Dinophyceae) from the South China Sea with a focus on the life cycle. N. Z. J. Mar. Freshw. Res. 2012, 46, 511–521. [Google Scholar] [CrossRef]
- Al Muftah, A.; Selwood, A.I.; Foss, A.J.; Al-Jabri, H.M.; Potts, M.; Yilmaz, M. Algal toxins and producers in the marine waters of Qatar, Arabian Gulf. Toxicon 2016, 122, 54–66. [Google Scholar] [CrossRef]
- Selwood, A.I.; Wilkins, A.L.; Munday, R.; Gu, H.; Smith, K.F.; Rhodes, L.L.; Rise, F. Pinnatoxin H: A new pinnatoxin analogue from a South China Sea Vulcanodinium rugosum isolate. Tetrahedron Lett. 2014, 55, 5508–5510. [Google Scholar] [CrossRef]
- Molgó, J.; Aráoz, R.; Benoit, E.; Iorga, B. Cyclic imine toxins: Chemistry, origin, metabolism, pharmacology, toxicology and detection. In Seafood and Freshwater Toxins: Pharmacology, Physiology and Detection; Botana, L.M., Ed.; CRC Press: Boca Raton, FL, USA, 2014; pp. 951–989. [Google Scholar]
- Stivala, C.E.; Benoit, E.; Aráoz, R.; Servent, D.; Novikov, A.; Molgó, J.; Zakarian, A. Synthesis and biology of cyclic imine toxins, an emerging class of potent, globally distributed marine toxins. Nat. Prod. Rep. 2015, 32, 411–435. [Google Scholar] [CrossRef] [PubMed]
- Molgó, J.; Marchot, P.; Aráoz, R.; Benoit, E.; Iorga, B.I.; Zakarian, A.; Taylor, P.; Bourne, Y.; Servent, D. Cyclic imine toxins from dinoflagellates: A growing family of potent antagonists of the nicotinic acetylcholine receptors. J. Neurochem. 2017, 142 (Suppl. 2), 41–51. [Google Scholar] [CrossRef] [PubMed]
- Zurhelle, C.; Nieva, J.; Tillmann, U.; Harder, T.; Krock, B.; Tebben, J. Identification of novel gymnodimines and spirolides from the marine dinoflagellate Alexandrium ostenfeldii. Mar. Drugs 2018, 16, 446. [Google Scholar] [CrossRef]
- Fribley, A.M.; Xi, Y.; Makris, C.; Alves-de-Souza, C.; York, R.; Tomas, C.; Wright, J.L.C.; Strangman, W.K. Identification of portimine B, a new cell permeable spiroimine that induces apoptosis in oral squamous cell carcinoma. ACS Med. Chem. Lett. 2018, 10, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.J.; Stivala, C.E.; Iorga, B.I.; Molgó, J.; Zakarian, A. Stability of cyclic imine toxins: Interconversion of pinnatoxin amino ketone and pinnatoxin A in aqueous media. J. Org. Chem. 2012, 77, 10435–10440. [Google Scholar] [CrossRef] [PubMed]
- Munday, R.; Selwood, A.I.; Rhodes, L. Acute toxicity of pinnatoxins E, F and G to mice. Toxicon 2012, 60, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Aráoz, R.; Servent, D.; Molgó, J.; Iorga, B.I.; Fruchart-Gaillard, C.; Benoit, E.; Gu, Z.; Stivala, C.; Zakarian, A. Total synthesis of pinnatoxins A and G and revision of the mode of action of pinnatoxin A. J. Am. Chem. Soc. 2011, 133, 10499–10511. [Google Scholar] [CrossRef]
- Bourne, Y.; Sulzenbacher, G.; Radić, Z.; Aráoz, R.; Reynaud, M.; Benoit, E.; Zakarian, A.; Servent, D.; Molgó, J.; Taylor, P.; et al. Marine macrocyclic imines, pinnatoxins A and G: Structural determinants and functional properties to distinguish neuronal α7 from muscle α1(2)βγδ nAChRs. Structure 2015, 23, 1106–1115. [Google Scholar] [CrossRef]
- Hellyer, S.D.; Indurthi, D.; Balle, T.; Runder-Varga, V.; Selwood, A.I.; Tyndall, J.D.; Chebib, M.; Rhodes, L.; Kerr, D.S. Pinnatoxins E, F and G target multiple nicotinic receptor subtypes. J. Neurochem. 2015, 135, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Hellyer, S.D.; Selwood, A.I.; Rhodes, L.; Kerr, D.S. Marine algal pinnatoxins E and F cause neuromuscular block in an in vitro hemidiaphragm preparation. Toxicon 2011, 58, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Hellyer, S.D.; Selwood, A.I.; Rhodes, L.; Kerr, D.S. Neuromuscular blocking activity of pinnatoxins E, F and G. Neuromuscular blocking activity of pinnatoxins E, F and G. Toxicon 2013, 76, 214–220. [Google Scholar] [CrossRef]
- Marrouchi, R.; Rome, G.; Kharrat, R.; Molgó, J.; Benoit, E. Analysis of the action of gymnodimine-A and 13-desmethyl spirolide C on the mouse neuromuscular system in vivo. Toxicon 2013, 75, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Couesnon, A.; Aráoz, R.; Iorga, B.I.; Benoit, E.; Reynaud, M.; Servent, D.; Molgó, J. The dinoflagellate toxin 20-methyl spirolide-G potently blocks skeletal muscle and neuronal nicotinic acetylcholine receptors. Toxins 2016, 8, 249. [Google Scholar] [CrossRef] [PubMed]
- Kharrat, R.; Servent, D.; Girard, E.; Ouanounou, G.; Amar, M.; Marrouchi, R.; Benoit, E.; Molgó, J. The marine phycotoxin gymnodimine targets muscular and neuronal nicotinic acetylcholine receptor subtypes with high affinity. J. Neurochem. 2008, 107, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Aráoz, R.; Ouanounou, G.; Iorga, B.I.; Goudet, A.; Alili, D.; Amar, M.; Benoit, E.; Molgó, J.; Servent, D. The neurotoxic effect of 13,19-didesmethyl and 13-desmethyl Spirolide C phycotoxins is mainly mediated by nicotinic rather than muscarinic acetylcholine Receptors. Toxicol. Sci. 2015, 147, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; Gray, W.R.; Olivera, B.M.; Zeikus, R.D.; Kerr, L.; Yoshikami, D.; Moczydlowski, E. Conus geographus toxins that discriminate between neuronal and muscle sodium channels. J. Biol. Chem. 1985, 260, 9280–9288. [Google Scholar] [PubMed]
- Nguyen-Huu, T.; Molgó, J.; Servent, D.; Duvaldestin, P. Resistance to D-tubocurarine of the rat diaphragm as compared to a limb muscle: Influence of quantal transmitter release and nicotinic acetylcholine receptors. Anesthesiology 2009, 110, 1011–1015. [Google Scholar] [CrossRef]
- Molgó, J.; Lundh, H.; Thesleff, S. Potency of 3,4-diaminopyridine and 4-aminopyridine on mammalian neuromuscular transmission and the effect of pH changes. Eur. J. Pharmacol. 1980, 61, 25–34. [Google Scholar] [CrossRef]
- Molgó, J. Effects of aminopyridines on neuromuscular transmission. In: Aminopyridines and Similarly Acting Drugs-Effects on Nerves, Muscles and Synapses. In Advances in the Biosciences; Lechat, P., Thesleff, S., Bowman, W.C., Eds.; Pergamon Press: London, UK, 1982; pp. 95–116. [Google Scholar]
- Van der Kloot, W.; Molgó, J. Quantal acetylcholine release at the vertebrate neuromuscular junction. Physiol. Rev. 1994, 74, 899–991. [Google Scholar] [CrossRef]
- Gonçalves, T.C.; Boukaiba, R.; Molgó, J.; Amar, M.; Partiseti, M.; Servent, D.; Benoit, E. Direct evidence for high affinity blockade of NaV1.6 channel subtype by huwentoxin-IV spider peptide, using multiscale functional approaches. Neuropharmacolgy 2018, 133, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Cerles, O.; Benoit, E.; Chereau, C.; Chouzenoux, S.; Morin, F.; Guillaumot, M.A.; Coriat, R.; Kavian, N.; Loussier, T.; Santulli, P.; et al. Niclosamide inhibits oxaliplatin neurotoxicity while improving colorectal cancer therapeutic response. Mol. Cancer Ther. 2017, 16, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, M.C.; Bostock, H. Effects of membrane polarization and ischaemia on the excitability properties of human motor axons. Brain 2000, 123, 2542–2551. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, A.V.; Lin, C.S.Y.; Park, S.B.; Kiernan, M.C. Assessment of nerve excitability in toxic and metabolic neuropathies. J. Peripher. Nerv. Syst. 2008, 13, 7–26. [Google Scholar] [CrossRef] [PubMed]
- Ouanounou, G.; Baux, G.; Bal, T. A novel synaptic plasticity rule explains homeostasis of neuromuscular transmission. Elife 2016, 5, e12190. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, E.M.; Martin, A.R. Non-linear summation of end-plate potentials in the frog and mouse. J. Physiol. 1981, 311, 307–324. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. Methods Mol. Biol. 2017, 1654, 39–54. [Google Scholar] [CrossRef]
- Bourne, Y.; Radić, Z.; Aráoz, R.; Talley, T.T.; Benoit, E.; Servent, D.; Taylor, P.; Molgó, J.; Marchot, P. Structural determinants in phycotoxins and AChBP conferring high affinity binding and nicotinic AChR antagonism. Proc. Natl. Acad. Sci. USA 2010, 107, 6076–6081. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID50 | PnTX-A | PnTX-G | GYM-A | 13-SPX-C | 20-meSPX-G |
|---|---|---|---|---|---|
| Mean | 3.080 | 2.830 | 3.474 | 0.009 | 0.002 |
| S.D. | 0.203 | 0.090 | 0.166 | 0.001 | 0.000 |
| n (mice) | 18 | 8 | 15 | 19 | 26 |
| Reference | Present study | Present study | [36] | [36] | [37] |
| Test 1 | Excitability Parameter 2 | Control | PnTX-A | P |
|---|---|---|---|---|
| T1 | Peak response (mV) | 3.299 ± 1.140 | 1.847 ± 1.370 | 0.050 * |
| Latency (ms) | 3.359 ± 0.065 | 3.349 ± 0.106 | 0.893 | |
| Stimulus (mA) for 50% max response | 0.481 ± 1.060 | 0.479 ± 1.130 | 0.927 | |
| Stimulus-response slope | 3.004 ± 1.180 | 2.650 ± 1.310 | 0.743 | |
| T2 | Resting slope | 0.638 ± 0.077 | 0.916 ± 0.257 | 0.182 |
| Minimum slope | 0.230 ± 0.013 | 0.218 ± 0.022 | 0.703 | |
| Hyperpolarizing slope | 0.536 ± 0.071 | 0.490 ± 0.074 | 0.757 | |
| T3 | Strength-duration time constant (ms) | 0.377 ± 0.035 | 0.316 ± 0.036 | 0.274 |
| Rheobase (mA) | 0.333 ± 1.070 | 0.327 ± 1.130 | 0.861 | |
| T4 | TEd (10–20 ms) | 47.680 ± 2.570 | 45.250 ± 4.930 | 0.700 |
| TEd (peak) | 48.640 ± 2.320 | 45.240 ± 4.110 | 0.555 | |
| TEd (40–60 ms) | 39.440 ± 3.000 | 38.080 ± 1.980 | 0.826 | |
| TEd (90–100 ms) | 36.000 ± 3.010 | 35.230 ± 1.810 | 0.878 | |
| TEd (accommodation) | 12.590 ± 1.230 | 10.170 ± 2.890 | 0.441 | |
| TEd (undershoot) | −11.820 ± 1.120 | −5.188 ± 0.512 | 0.019 * | |
| TEh (10–20 ms) | −97.600 ± 4.780 | −86.230 ± 2.660 | 0.318 | |
| TEh (20–40 ms) | −132.000 ± 8.430 | −123.900 ± 7.160 | 0.686 | |
| TEh (90–100 ms) | −159.20 ± 12.60 | −150.300 ± 4.910 | 0.753 | |
| TEh (overshoot) | 13.350 ± 1.460 | 7.238 ± 1.450 | 0.079 | |
| T5 | Relative refractory period (ms) | 2.349 ± 1.090 | 2.532 ± 1.100 | 0.632 |
| Superexcitability (%) | −7.596 ± 1.490 | −9.049 ± 1.970 | 0.600 | |
| Subexcitability (%) | 3.844 ± 0.708 | 3.316 ± 1.160 | 0.697 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benoit, E.; Couesnon, A.; Lindovsky, J.; Iorga, B.I.; Aráoz, R.; Servent, D.; Zakarian, A.; Molgó, J. Synthetic Pinnatoxins A and G Reversibly Block Mouse Skeletal Neuromuscular Transmission In Vivo and In Vitro. Mar. Drugs 2019, 17, 306. https://doi.org/10.3390/md17050306
Benoit E, Couesnon A, Lindovsky J, Iorga BI, Aráoz R, Servent D, Zakarian A, Molgó J. Synthetic Pinnatoxins A and G Reversibly Block Mouse Skeletal Neuromuscular Transmission In Vivo and In Vitro. Marine Drugs. 2019; 17(5):306. https://doi.org/10.3390/md17050306
Chicago/Turabian StyleBenoit, Evelyne, Aurélie Couesnon, Jiri Lindovsky, Bogdan I. Iorga, Rómulo Aráoz, Denis Servent, Armen Zakarian, and Jordi Molgó. 2019. "Synthetic Pinnatoxins A and G Reversibly Block Mouse Skeletal Neuromuscular Transmission In Vivo and In Vitro" Marine Drugs 17, no. 5: 306. https://doi.org/10.3390/md17050306