1. Introduction
Dextranases (α-1,6-
d-glucan-6-glucanohydrolase; EC 3.2.1.11) hydrolyze dextran to oligosaccharides at the α-1,6 glucosidic bond, and are widely used in medical, dental, and sugar industries. In clinical applications, specific clinical dextran produced by dextranase can be used as a blood substitute in emergencies [
1,
2,
3]. In the sugar industry, dextranase has been used to resolve the poor clarification and throughput that dextran can cause in sugarcane juice [
4,
5,
6,
7]. It is worth noting that this enzyme can be used with commercial dextran to directly synthetize isomaltose and isomaltooligosaccharides which exhibit prebiotic effects [
8,
9,
10]. A previous report proposed that dextranase may be capable of treating dental plaques [
2,
3,
11]. For this reason, the use of dextranase to treat dental caries has attracted a great deal of attention, particularly with respect to the degradation of dextran in dental plaques. Bacterial cells form biofilms as a protective barrier from external conditions, serving as a mechanism for improving survival and dispersion [
12,
13].
Streptococcus mutans is the main cause of dental decay in human teeth and key modulator of the development of cariogenic biofilms [
14,
15]. Accumulation of this cariogenic bacterium within the biofilm may lead to the onset of periodontal inflammation. Thus, dislodging the biofilm is the main therapy for periodontal inflammation.
An alkaline dextranase may be suitable for the treatment of dental caries because alkaline tooth-rinse products are expected to be more amenable to enamel than acidic products [
16]. In addition, dextranase works efficiently at temperatures of about 37 °C and may also contribute to the degradation of human dental plaques. However, the most common source of dextranase, fungi, produce many acidic and megathermal dextranases, which catalyze at pH values ranging from pH 5.0 to 6.5 and temperatures above 50 °C, and are unstable under alkaline conditions. Dextranases are seldom capable of catalysis under both alkaline and moderate-temperature conditions [
1]; however, enzymes from marine microorganisms may be an exception [
17,
18,
19].
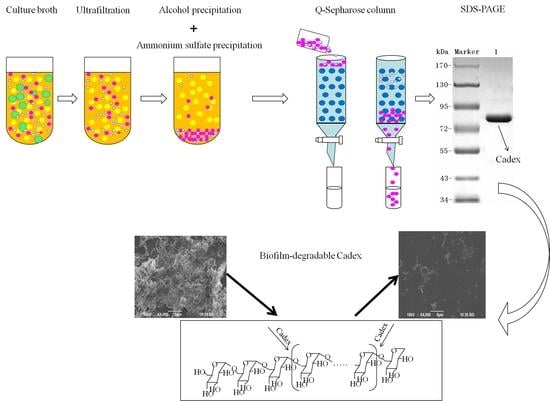
In this study, a dextranase produced by a marine bacterium
Catenovulum sp. DP03 (CGMCC No. 7386) was sequenced and selected for extracellular experiments [
20]. This study demonstrated a method for purifying, characterizing and hydrolyzing products of dextranase from the marine strain DP03 and analyzed its effects on
S. mutans biofilm. The results provide insights into additional applications for this enzyme.
3. Discussion
A psychrotolerant dextranase-producing bacterium named
Catenovulum sp. DP03 was previously studied [
20]. However, to the best of our knowledge, this is the first report of the purification and characterization of dextranase from
Catenovulum. Purification of crude dextranase by ammonium sulfate fractionation and Sepharose 6B chromatography, which resulted in a 6.69-fold increase in specific activity and an 11.27% recovery, was previously reported [
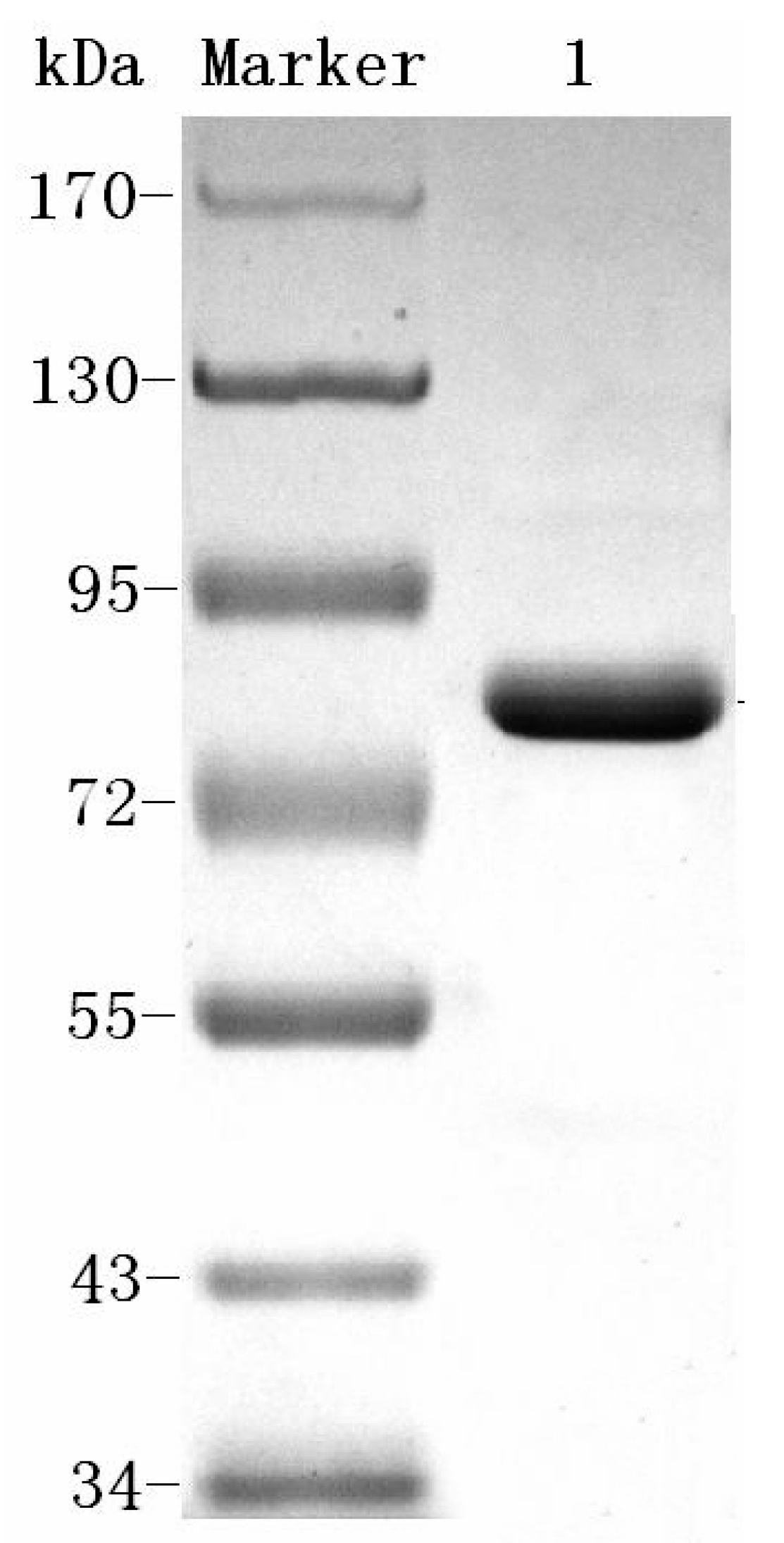
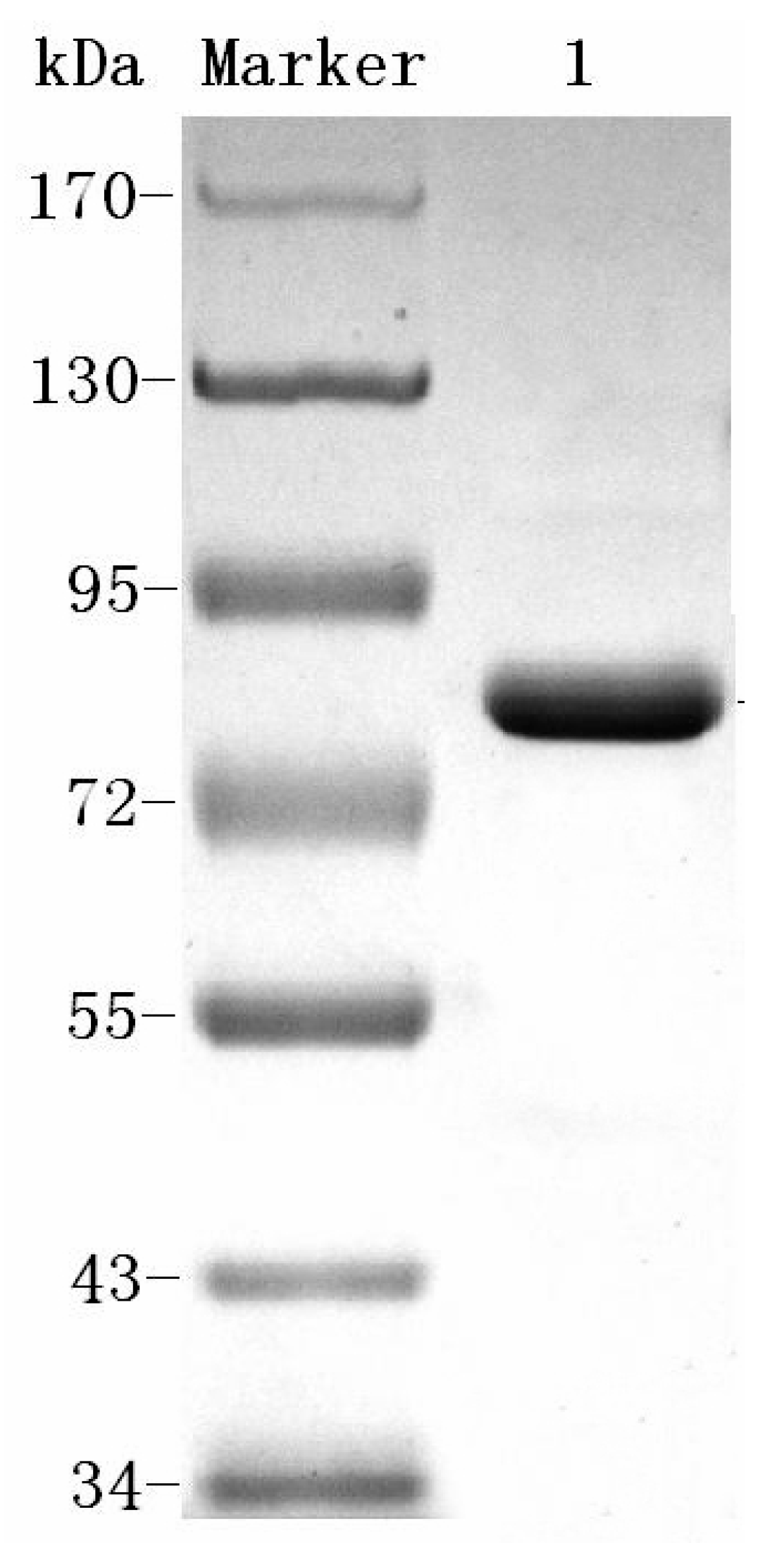
23]. This system of the aforementioned procedure may be used to produce homogenetic dextranase. The process can easily be scaled up and is cost-effective. The molecular weight of Cadex was about 75 kDa, which resembled that of dextranase from
Sporotrix schencki (79 kDa) [
27]. Bacteria producing dextranases generally have molecular weights ranging from 60 to 114 kDa [
28,
29,
30]. The smallest dextranase (23 kDa) is from
Lipomyces starkeyi [
24], and the largest (175 kDa) is from
Streptococcus sobrinus [
31].
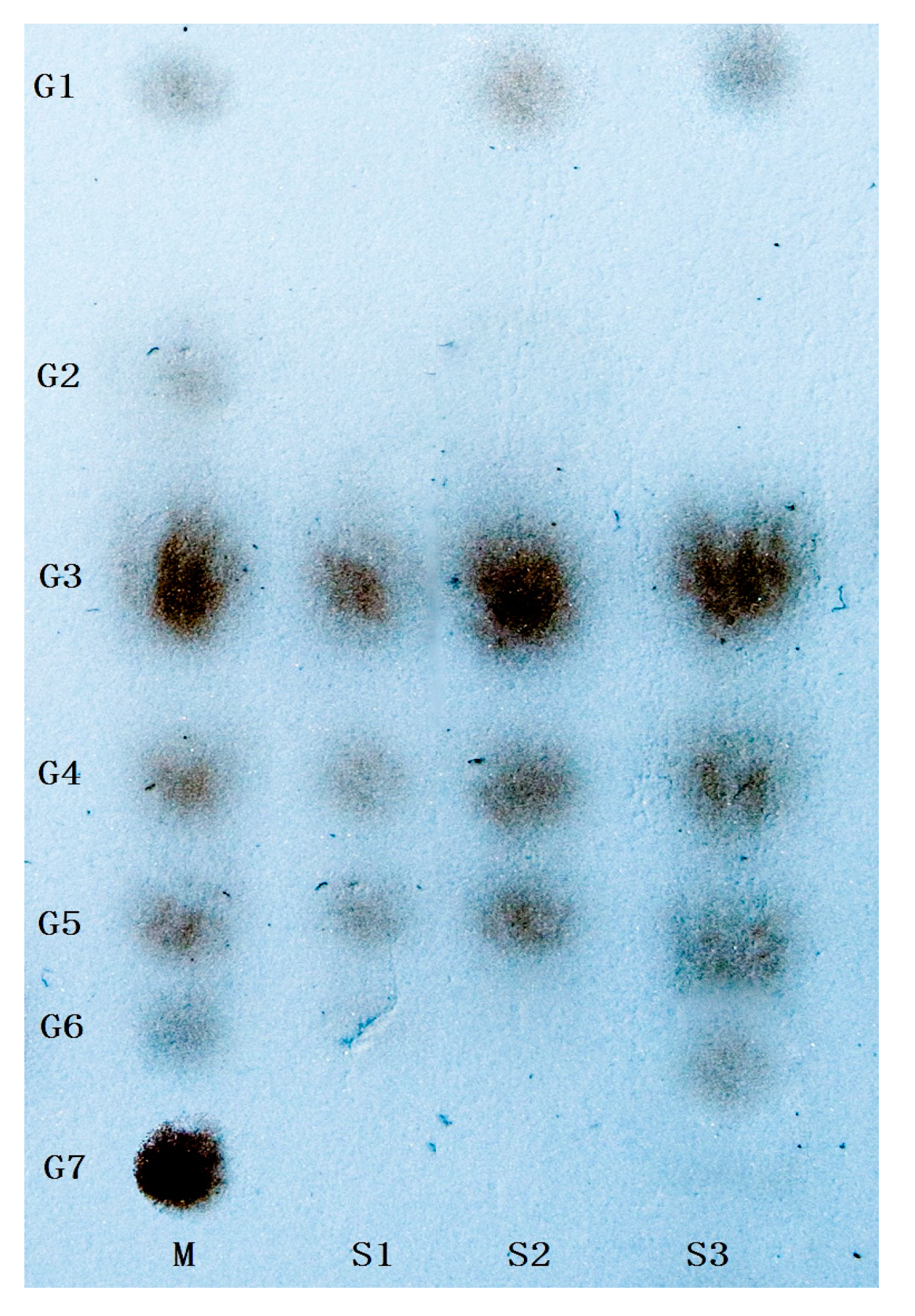
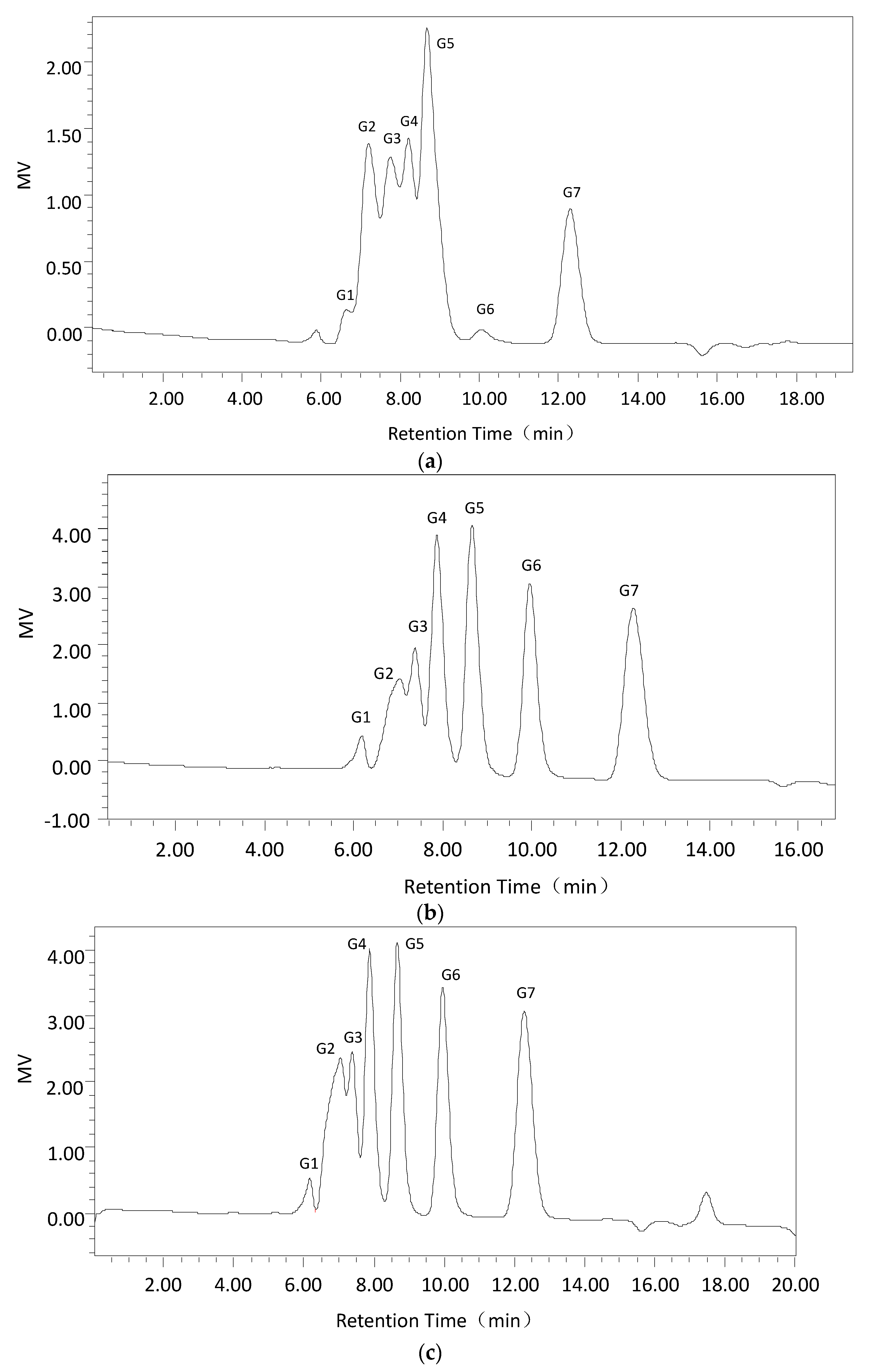
Endo-type Cadex showed high specificity towards dextrans containing α-1,6 glucosidic bonds. Moreover, the main hydrolysis products of Cadex were isomaltooligomers [
30,
32,
33]. Dextranase from
Chaetomium [
34],
Aspergillus [
35],
Penicillium [
36], and
Fusarium [
37] synthesize comparatively low amounts of glucose and higher amounts of isomaltooligosaccharides. Isomaltooligosaccharides can promote the growth and proliferation of
Bifidobacteria and
Lactobacillus [
1,
38]. Numerous isomaltooligosaccharides are prebiotics, which are produced endodextranases and have garnered much commercial interest [
33].
The optimum pH for Cadex activity tends to be alkaline, and recently reported alkalophilic cases were
Streptomyces sp. NK458 and
Bacillus subtilis NRC-B233b, which had maximum activities at pH 9.0 and pH 9.2 [
7,
39]. Evidence is accumulating that alkali generation plays a major role in pH homeostasis which may modulate the initiation and progression of dental caries [
40]. Therefore, alkalophilic Cadex may be suitable for the development of novel marine agents for the treatment of this condition [
16]. Cadex had catalytic efficiency at 0 °C, similar characteristics to other cold-adapted enzymes: for example, a cold-adapted ι-carrageenase showed 36.5% relative activity at 10 °C [
41] and a cold-adapted β-glucosidase retained more than 60% of its activity at temperatures ranging from 15 °C to 35 °C [
42]. Cold-adapted enzymes have optimal catalyst temperatures near 30 °C and remain efficient at 0 °C. Cadex can be classified as a cold-adapted enzyme according to the system developed by Margesin and Schinner [
43]. The excellent pH stability of Cadex distinguishes it from other dextranases, which are generally unstable across a broad pH range [
1,
7,
22,
27]. It would be easier to hydrolyze dextran than dextranases in acid/alkaline catalysis conditions. We speculate that Cadex may be suitable for widespread use. We have classified Cadex as a cold-adapted dextranase, which may explain its lower thermo-stability than terrestrial dextranases [
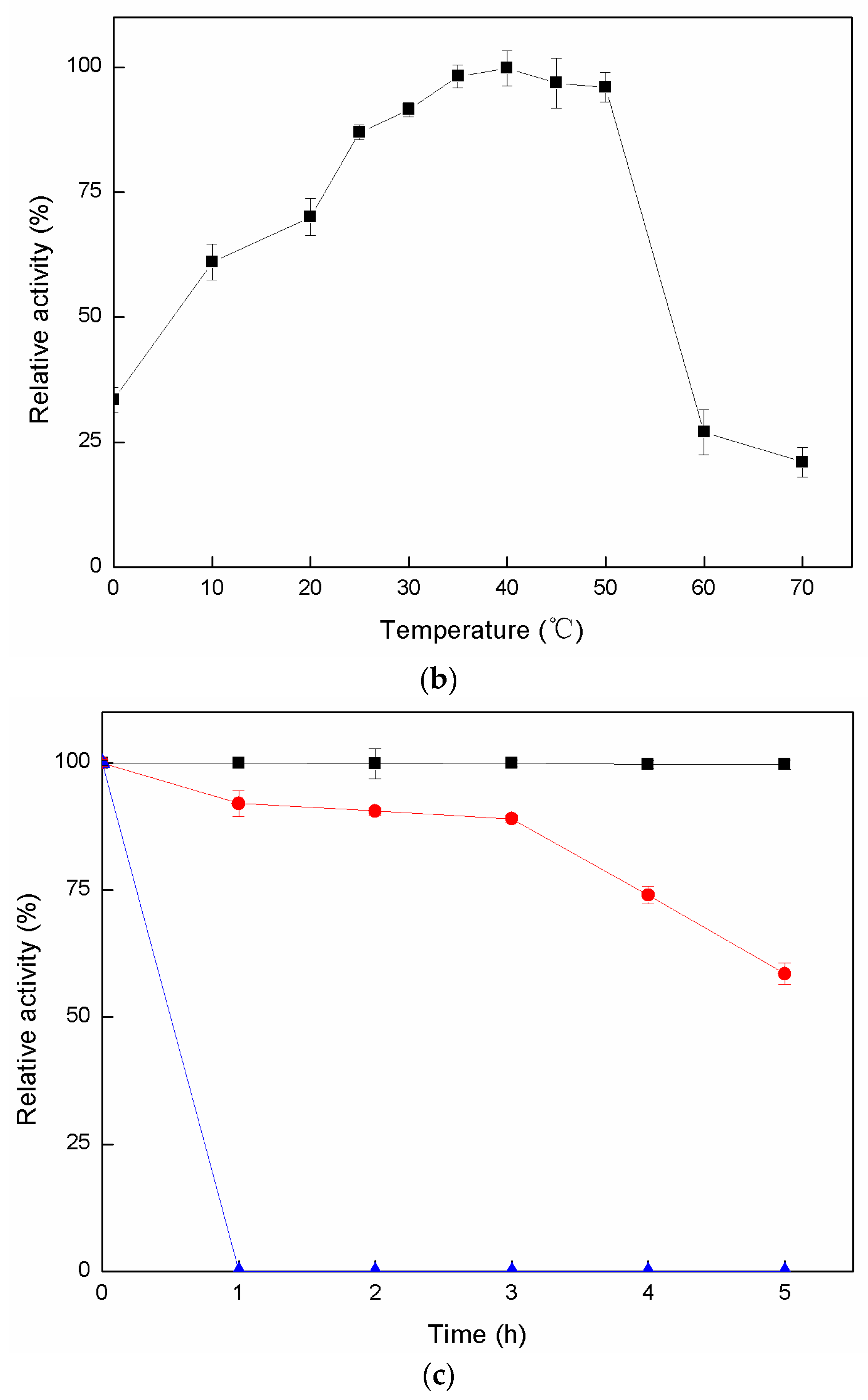
1]. Nevertheless, in our early studies, crude Cadex showed greater thermostability than purified dextranase, as it was stable at 45 °C, and its half-life was 10 h (data not shown).
The present study proposes that purified
Catenovulum dextranase, namely Cadex, is an alkalophilic and cold-adapted dextranase that is considered to be a novel marine dextranase of dealing with biofilms. The failure of biofilm formation is attributable to the failure of extracellular polysaccharides to form efficiently, possibly due to cleavage of the α-1,6 glucosidic linkages in the biofilm that occurs in the presence of Cadex. The oral
Streptococcus biofilm is formed by α-(1,3)-glucan and α-(1,6)-glucan, in which the α-1,6 glucosidic linkages are degradable by dextranase while the α-1,3 glucosidic linkages can be cleaved by mutanase [
44].
Penicillium dextranase is often used as the standard dextranase in studies of this enzyme such as studies showing dextran removal during sugar manufacturing [
7]. Cadex was a favorable biofilm inhibitor that surpassed the inhibitory ability of
Penicillium dextranase at the same concentration. In addition, common teeth rinsing products, such as carboxybenzene, ethanol, sodium fluoride, and xylitol, had no negative effects on Cadex activity. Marine organisms are regarded as a prolific resource of novel bioactive metabolites, including a vast array of macrolides, cyclic peptides, pigments, polyketides, terpenes, steroids and alkaloids [
45]. At the same time, marine enzymes are important bioactive metabolites which characterized by high salt tolerance, hyperthermostability, and low ideal temperature tolerance. These beneficial properties make Cadex an attractive candidate for development as a novel reagent for dental plaque treatment [
46,
47].
4. Materials and Methods
4.1. Chemicals
Q-Sepharose FF, dextran (T20, T40, T70, and T500), and PhastGel IEF 3-9 were obtained from GE Healthcare (Uppsala, Sweden). A prestained protein PAGE ruler was obtained from Fermentas (Waltham, MA, USA). An oligosaccharide kit, an IEF protein mix 3.6–9.3, bovine serum albumin, crystal violet, (CV), and Coomassie brilliant blue R250 and G250 were purchased from Sigma-Aldrich (St. Louis, MO, USA). All other reagents were purchased from Sinopharm Chemical Reagent Corporation (Shanghai, China) and were of the highest analytical grade.
4.2. Crude Dextranase Production
Extracellular dextranase production was performed in medium containing 5 g/L yeast extract, 5 g/L peptone, 10 g/L dextran T20, and 5 g/L NaCl. The pH was adjusted to about 8.0 before autoclaving. Then, the production medium was inoculated with Catenovulum sp. DP03. After fermenting at 30 °C for 28 h, a cell-free culture broth was obtained by centrifugation for 20 min at 12,000× g and 4 °C.
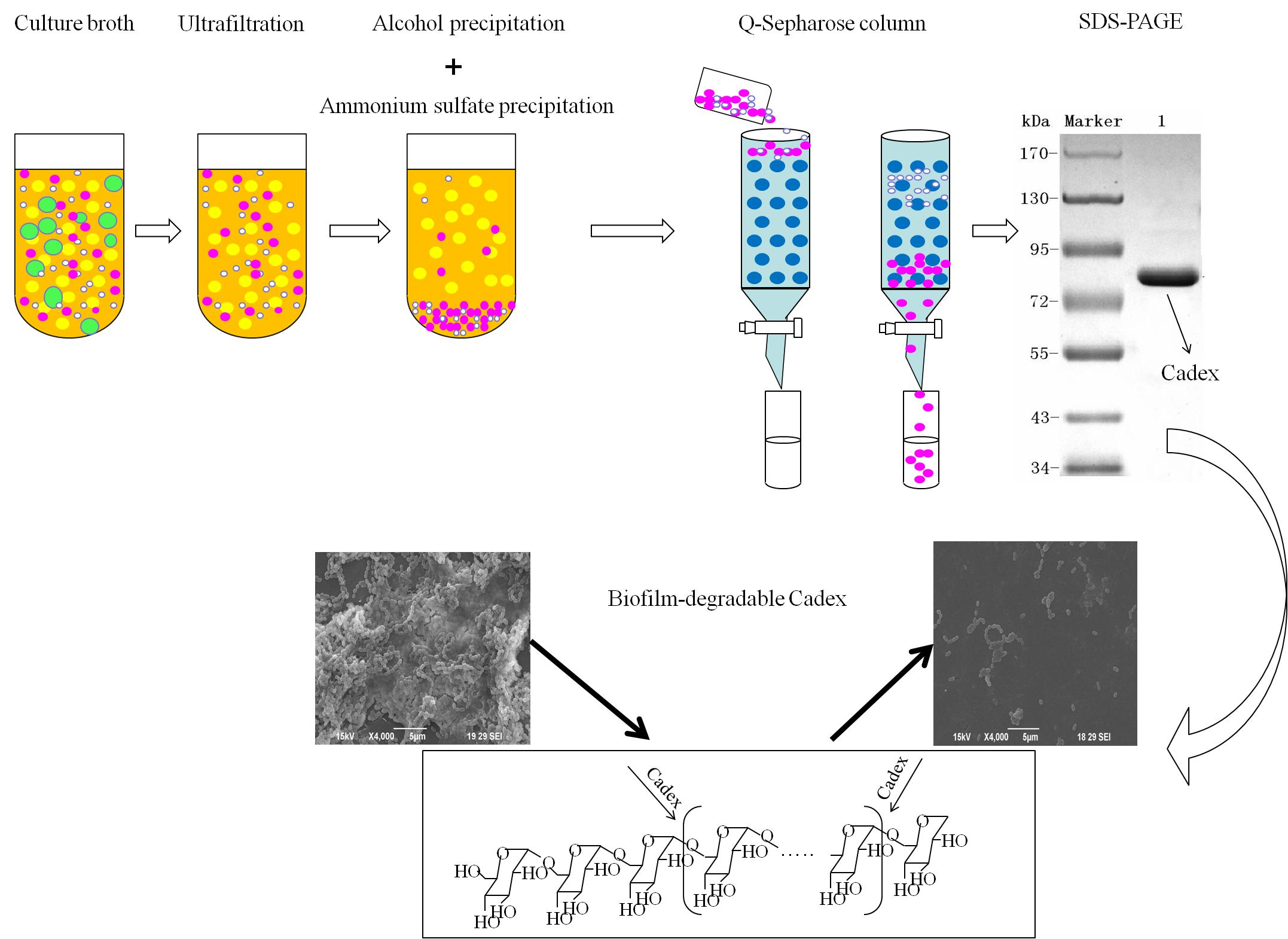
4.3. Purification of Dextranase
First, excess water and other matter were removed from the crude dextranase product by ultrafiltration (Watson Marlow, Cornwall, UK) using a 30,000 NMWC hollow fiber cartridge (GE Healthcare) at room temperature. Deionized water was added three times. The crude enzyme was finally concentrated to one tenth of the original volume of the culture broth. Second, pre-cooled ethanol (−40 °C) was added to the crude enzyme solution slowly and agitated gently for 10 min. An ethanol: enzyme ratio of 0.6:1.2 (v:v) was found to be optimal. After centrifugation for 15 min at 12,000× g and 4 °C, the precipitate was dissolved in 10 mM Tris-HCl buffer (pH 7.5). Third, ammonium sulfate precipitation was performed using a magnetic stirrer. The supernatant from ethanol precipitation was placed in a beaker within an ice tray. Then 25% saturated ammonium sulfate was added to the solution, which then was allowed to stand at 4 °C for 1 h, after which it was centrifuged at 12,000× g at 4 °C for 20 min. The supernatant was collected, and 60% saturated ammonium sulfate was added. The mixture was allowed to stand for 1 h, and precipitated protein was collected by centrifugation 20 min at 12,000× g and 4 °C. Then the pellet was dissolved and dialyzed using 10 mM Tris-HCl buffer (pH 7.5). Finally, the enzyme sample was loaded onto a Q-Sepharose column (1.6 cm × 10 cm; GE Healthcare). Chromatographic analysis was conducted by a fast protein liquid chromatography (FPLC; Bio-Rad, Hercules, CA, USA) at room temperature. Proteins were eluted with several NaCl gradients in 10 mM Tris-HCl (pH 7.5) at a flow rate of 0.8 mL/min. Protein content and enzyme activity for each fraction were monitored. The enzyme-containing fractions were concentrated using ultracel-10k centrifugal filters (Millipore, Burlington, MA, USA).
4.4. SDS-PAGE and Isoelectric Focusing
SDS-PAGE analysis of dextranase was performed according to Laemmli [
48] with minor modifications. After electrophoresis, the gel was stained with Coomassie brilliant blue R-250. Isoelectric focusing (IEF) was performed using a Pharmacia PhastGel System (GE Healthcare) on PhastGel IEF 3–9 according to the manufacturer’s instructions. An IEF protein marker (mix 3.6–9.3) was used as the pI standard.
4.5. Enzyme Assay and Protein Measurement
Enzyme activity was measured using dextran T70 as the substrate (3%,
m/
v) in 0.1 M Tris-HCl buffer (pH 8.0) using the 3,5-dinitrosalicylic acid method. Maltose served as the standard. One unit of dextranase activity was defined as the amount of enzyme capable of hydrolyzing dextran to 1 μM of reducing sugar in1 min [
49]. Protein concentration was measured by the Bradford protein assay [
50] using bovine serum albumin as the standard.
4.6. Enzyme Properties
4.6.1. Effects of pH on Activity and Stability of Cadex
Dextranase activity was measured in buffers of different pH values and containing 3% dextran T70. To determine the effects of pH on dextranase stability, the enzyme solution was incubated at 25 °C for 1 h, and residual activity was measured at the optimum pH. Solutions (50 mM) with different pH values were used as follows: citrate buffer (pH 4.0–6.0), sodium phosphate buffer (pH 6.0–7.5), Tris-HCl (pH 7.5–9.0), and NaHCO3-Na2CO3 (pH 9.0–11.0).
4.6.2. Effects of Temperature on Activity and Stability of Cadex
The temperature for the enzymatic reaction was optimized by experimentation at different temperatures (0–70 °C). To assess thermal stability, the enzyme solution was pre-heated at temperatures of 30 °C, 40 °C, and 50 °C for 0–5 h. Residual enzyme activity was measured at each interval.
4.6.3. Effects of Metal Ions and Chemicals on Cadex Activity
The effects of different solutions containing chloride metal ion salts (final concentrations of 1 mM and 5 mM) and chemicals on purified Cadex activities were determined. The relative enzyme activity in the presence of metal ions and chemicals was calculated based on activity in the absence of reagent.
4.6.4. Substrate Specificity
To determine the substrate specificity of Cadex, dextranase activity in the enzymatic hydrolysis of carbohydrates with various glycosidic linkages was determined using the method described by Wu.et al. [
22]. Dextranase was incubated in 50 mM Tris-HCl (pH 8.0) with various carbohydrates at 40 °C for 20 min. Relative activity was expressed in percentage values of the highest activity, which was set as 100%.
4.6.5. Products of Cadex Hydrolysis
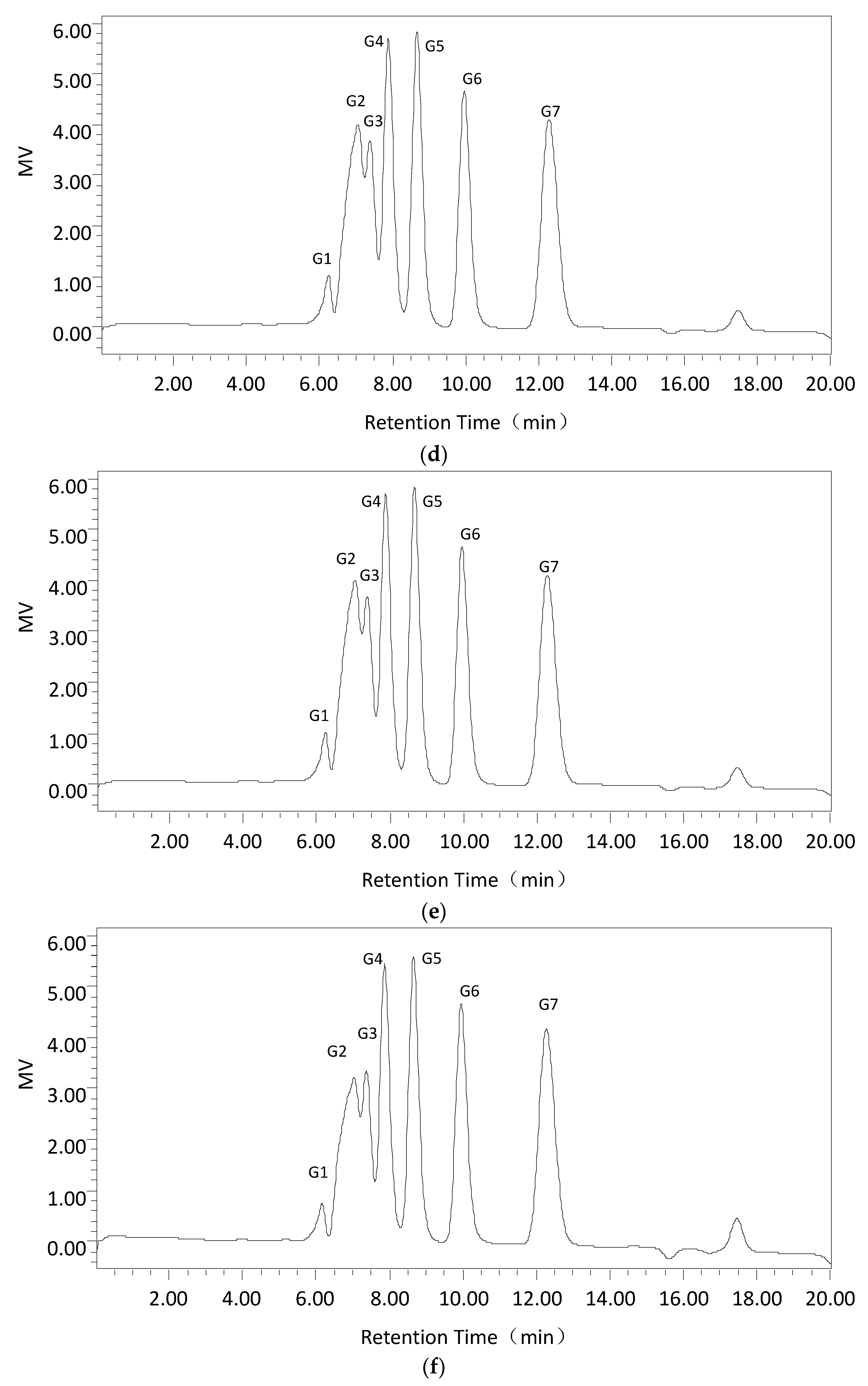
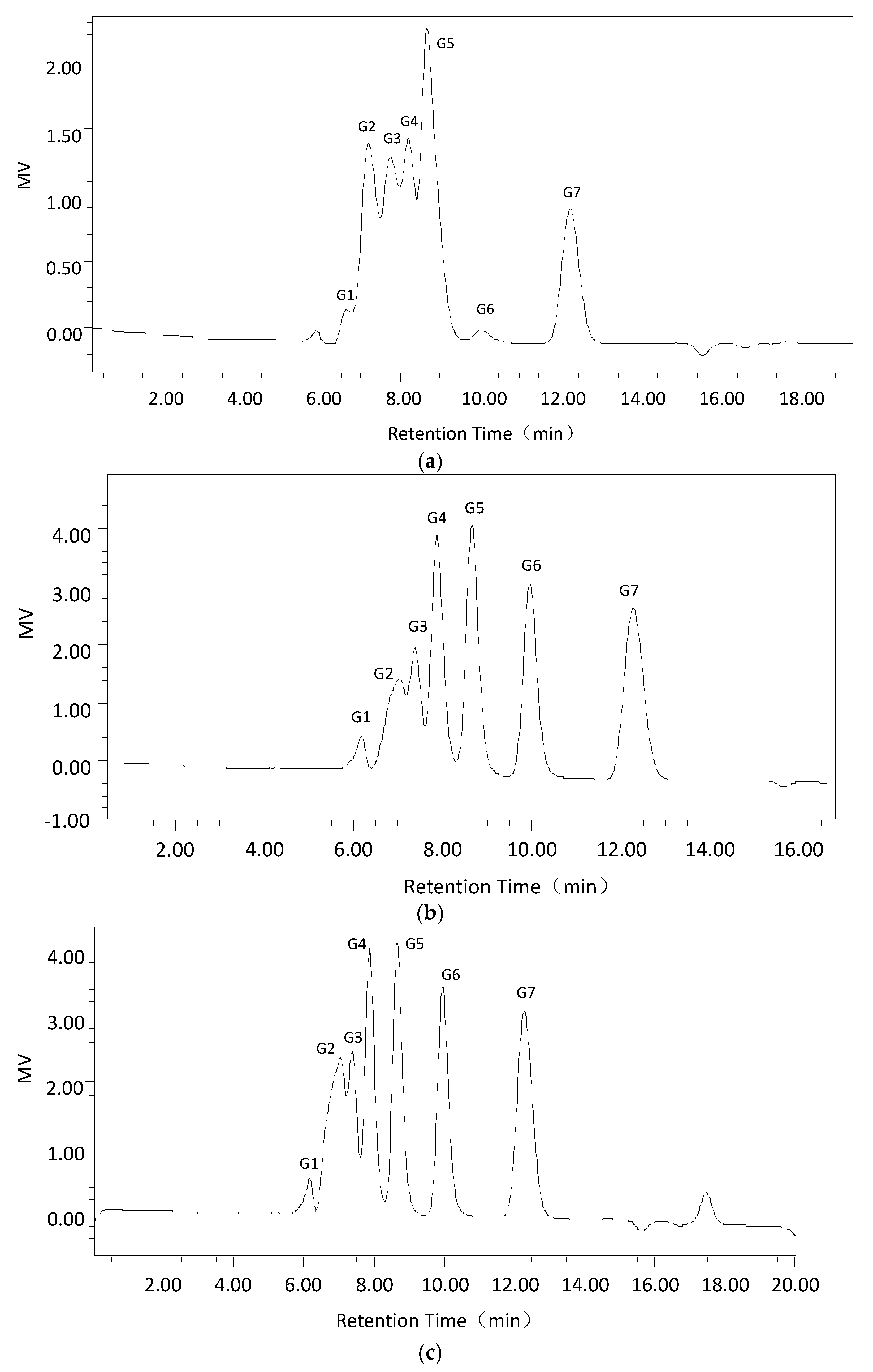
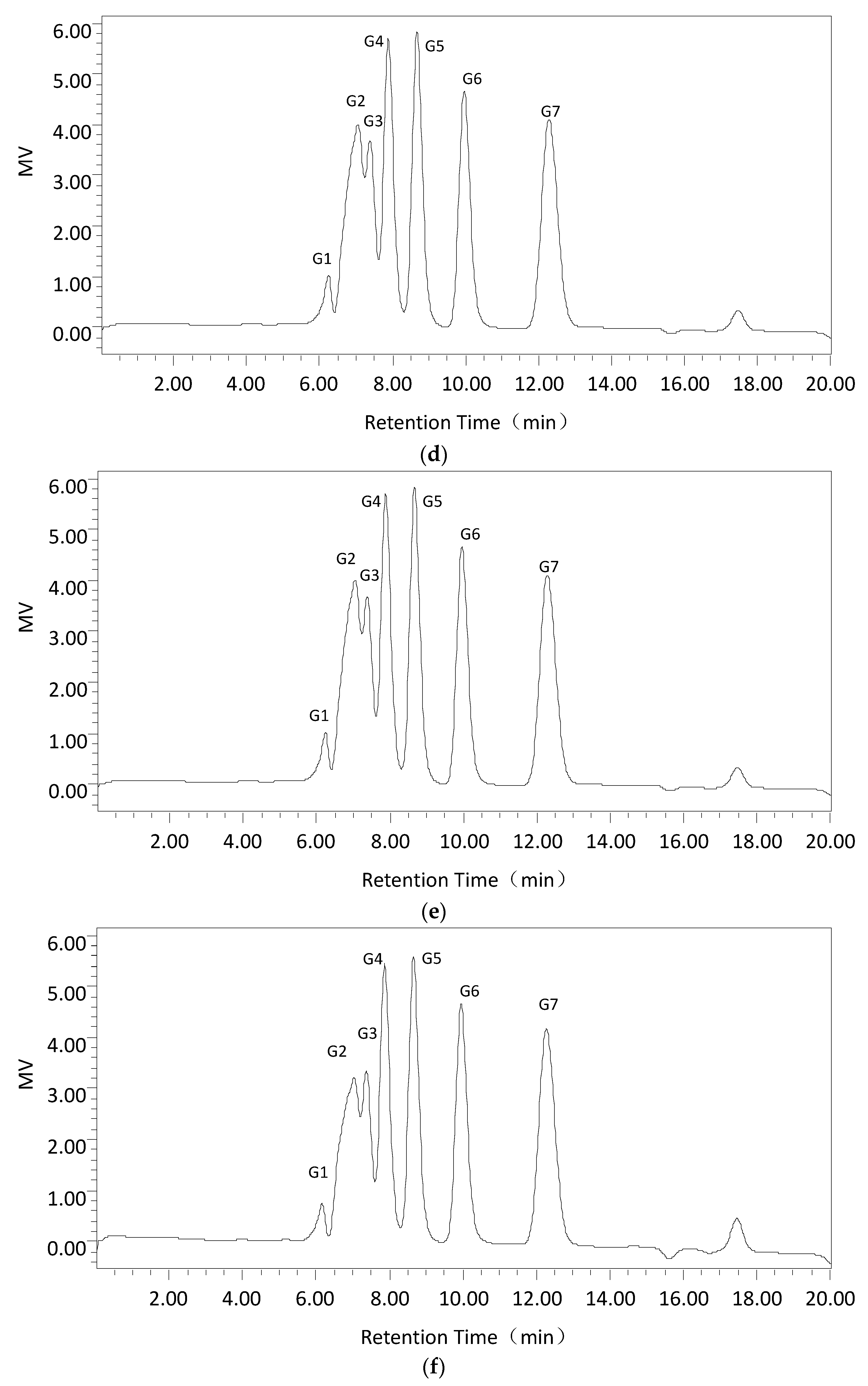
First, Cadex hydrolysis took place in dextran T70 solution, and the products were analyzed by TLC using a silica gel GF254 plate developed in a chloroform: acetic: acid: water ratio of = 5:7:1 (v:v:v). An oligosaccharide kit was used as the standard.
Second, using the optimum dextranase temperature and pH, 3% dextran T70 samples were digested for different periods (15 min, 30 min, 1 h, 3 h, and 5 h). The products were identified and analyzed with the Waters 600 and Waters Sugar-Pak1 (300 mm × 7.8 mm; Waters, Milford, MA, USA) HPLC with a differential refraction detector. The mobile phase was water at 0.4 mL/min, the column temperature was 85 °C and the injection volume was 20 μL. The standard sugars were glucose, maltose, maltotriose, isomaltotriose, isomaltotetraose, isomaltopentose, and isomaltohexose. For quantification, the peak areas were determined. Data acquisition and processing were conducted using Empower GPC software (Waters, Milford, MA, USA).
4.7. Effects of Cadex on Biofilm
4.7.1. Biofilm Mass Assay
The biofilm mass was assayed by CV staining according to the protocol of Cardoso et al. [
51] with some modifications. Briefly, biofilms were grown on a flat bottom sterile 96-well plate (Greiner, Frickhausen, Germany) in which the cultured medium was removed. To each well, 0.2 mL of 0.2 M phosphate buffer was added three times to clean the unattached biofilms, which were left to air dry and fixed for 60 min. Then 0.2 mL of 1% CV was added to each well for 5 min. Following the staining step, the CV solution was removed and the biofilms were cleaned and dried, after which 0.2 mL of 95% ethyl alcohol was added to re-solubilize the dyed biofilms. The CV solutions were obtained and transferred to a new 96-well plate and the optical density of the content was measured using a microtiter plate spectrophotometer (Bio-Rad) at 595 nm.
4.7.2. Effects of Cadex on Biofilm Formation
Base on the biofilm mass assay, MBIC of
Streptococcus mutans ATCC 25175 (American Type Culture Collection (ATCC), Manassas, VA, USA) was measured. The effects of Cadex and a homogenetic purity dextranase from
Penicillium (SA D8144; Sigma) on
S. mutans biofilm formation were investigated using SEM at MBIC
90.
S. mutans was pre-inoculated in BHI medium without sucrose at 37 °C for 15 h. Then, 1 mL of this precultured solution was inoculated into fresh BHI medium with 1% sucrose (20 mL in 100 mL Erlenmeyer-type flask). Sterile glass coverslips were placed in the BHI medium. The media were co-cultured with
S. mutans and incubated with Cadex at 37 °C for 3 h, 9 h, and 18 h. An identical assay with an equal volume of cell-free deionized water served as the blank control. All coverslips were collected for fixation, and were dehydrated and dried according to the procedure described by Tao et al. [
52]. The coverslips were sputter-coated with gold (JFC-1600, JEOL, Tokyo, Japan) and viewed by SEM (JSM-6390LA; JEOL).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}