
Palladium-Catalyzed Dehydrogenative Coupling: An Efficient Synthetic Strategy for the Construction of the Quinoline Core

Abstract
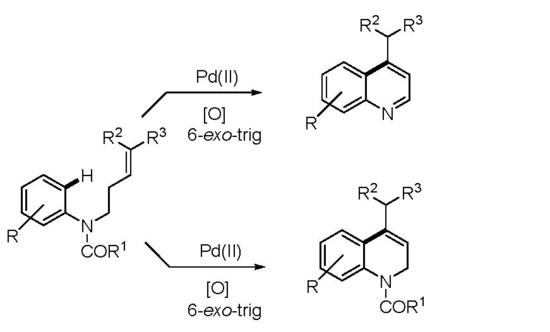
:
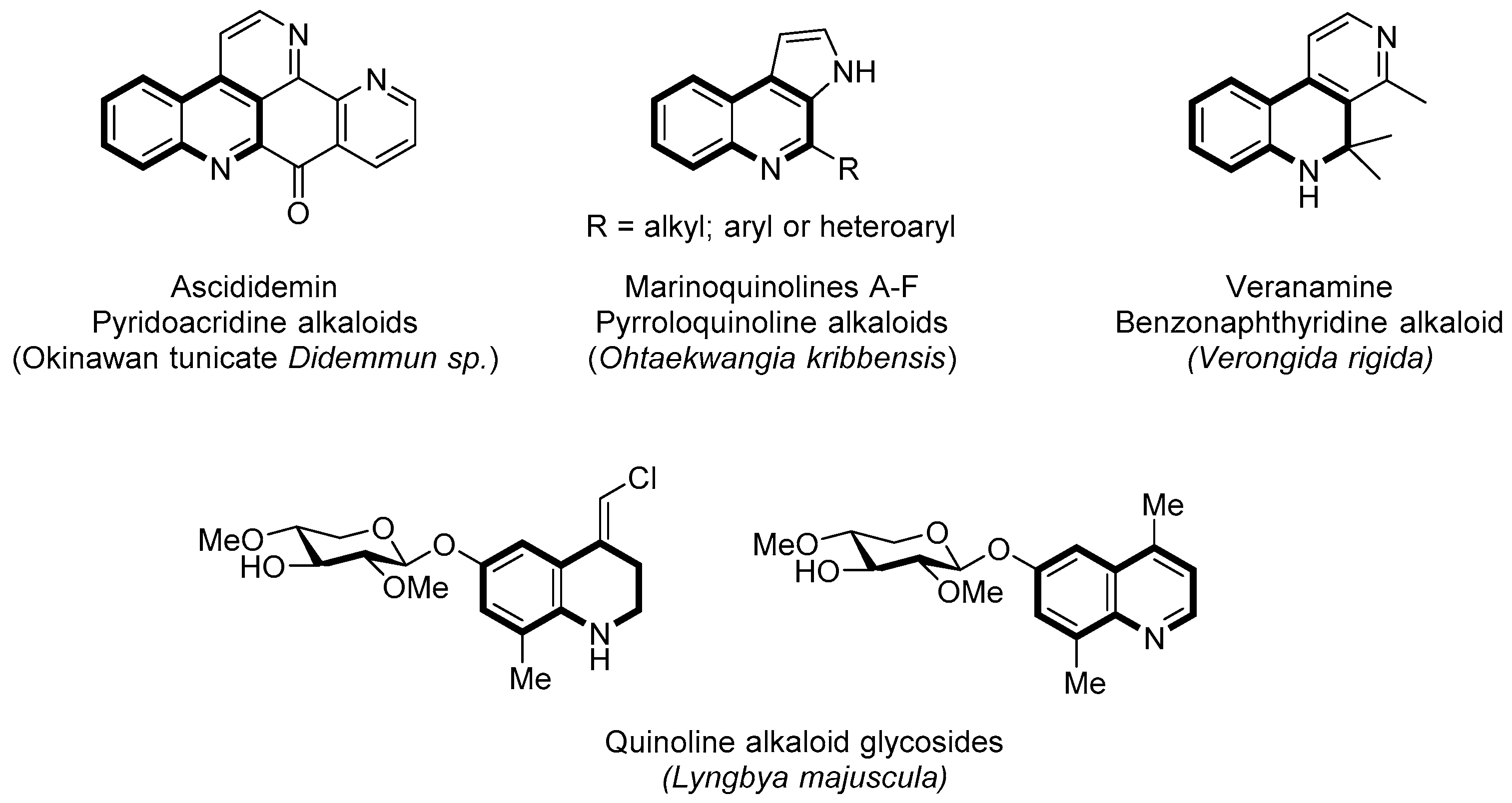
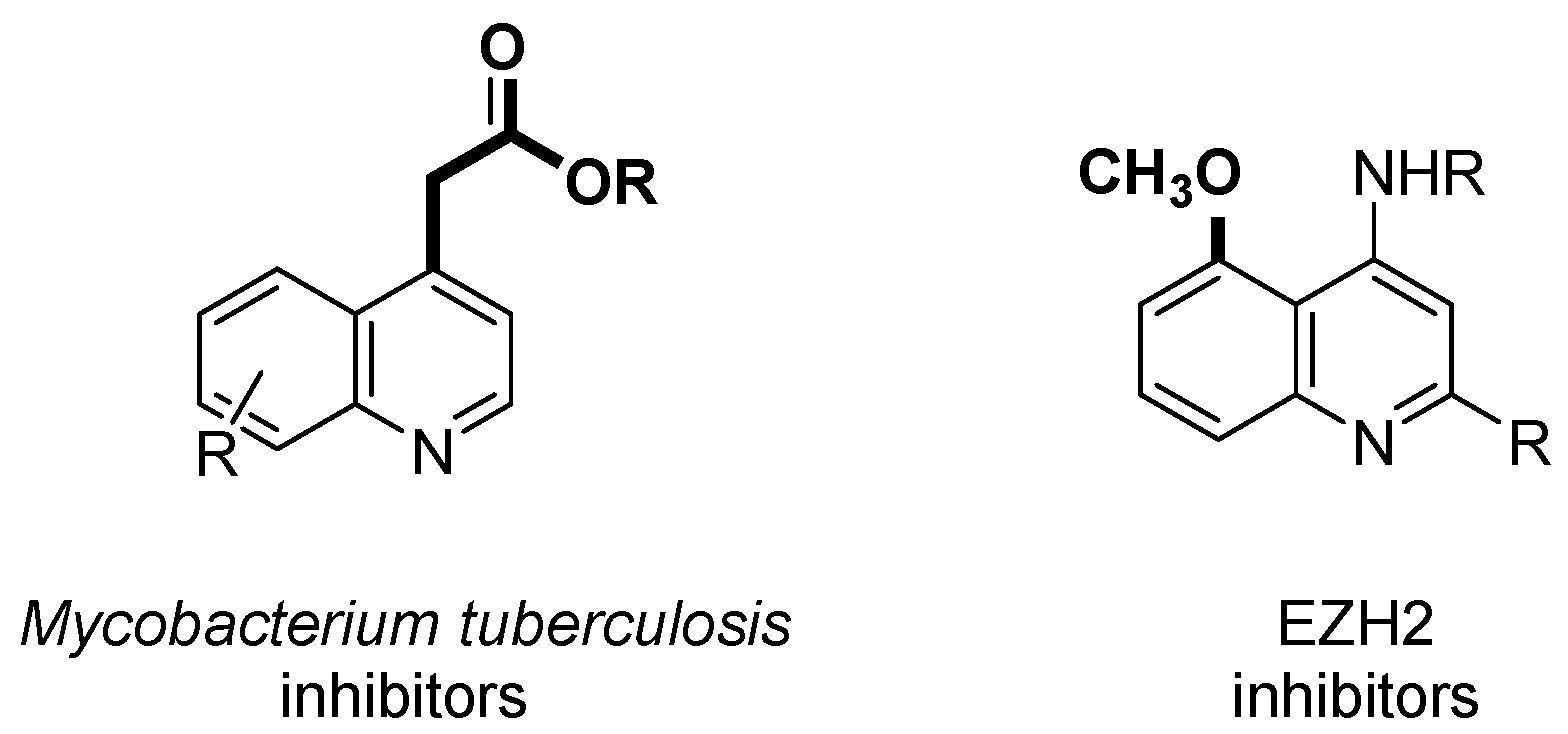
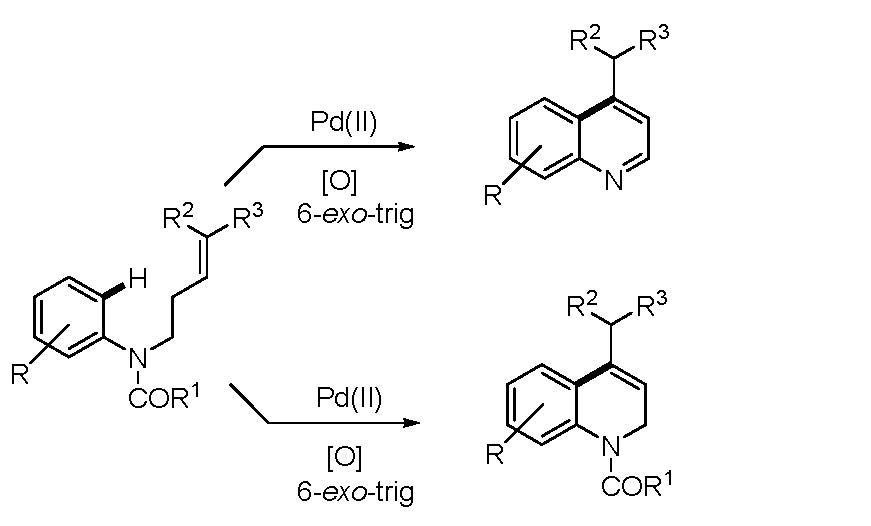
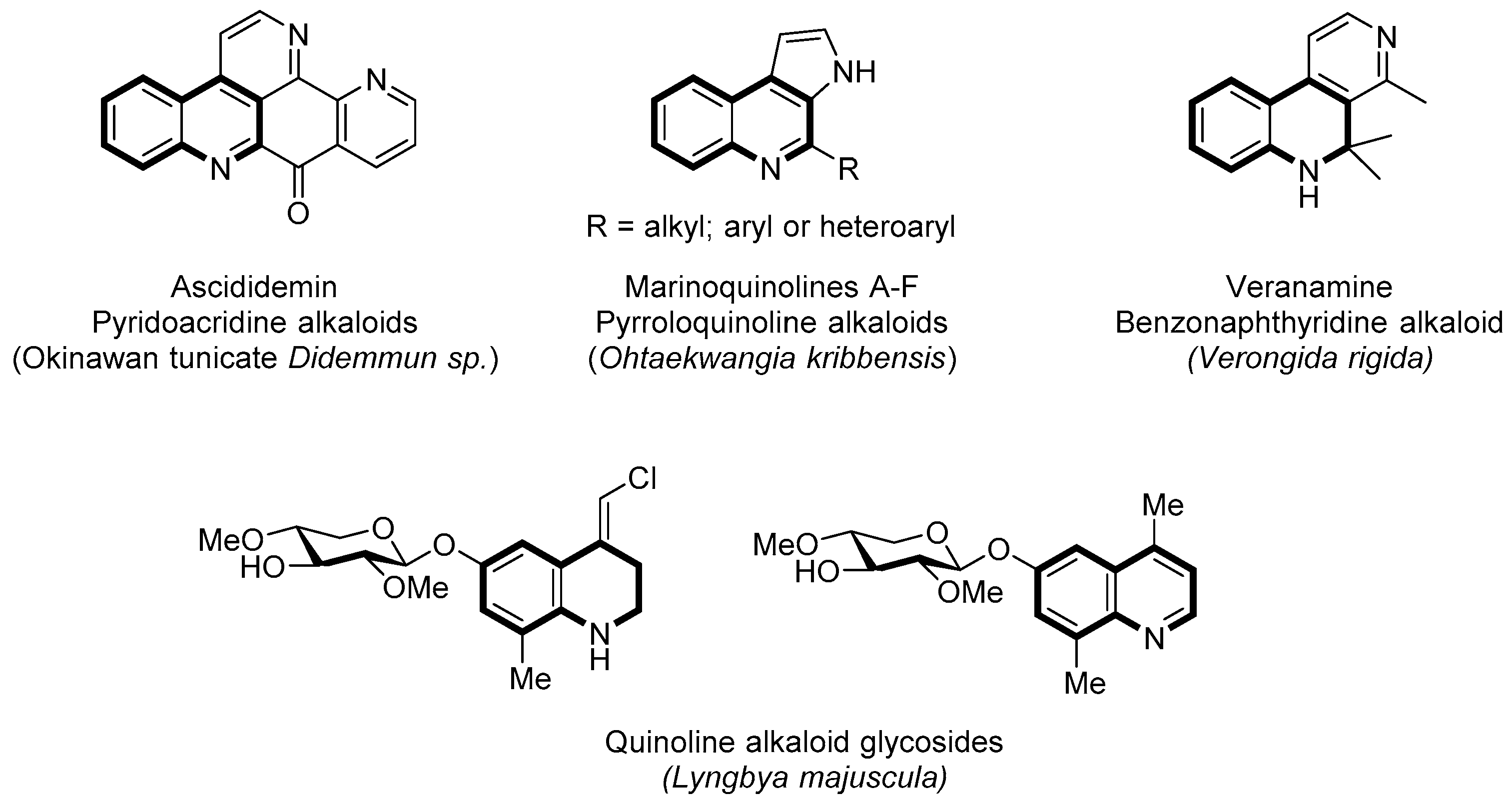
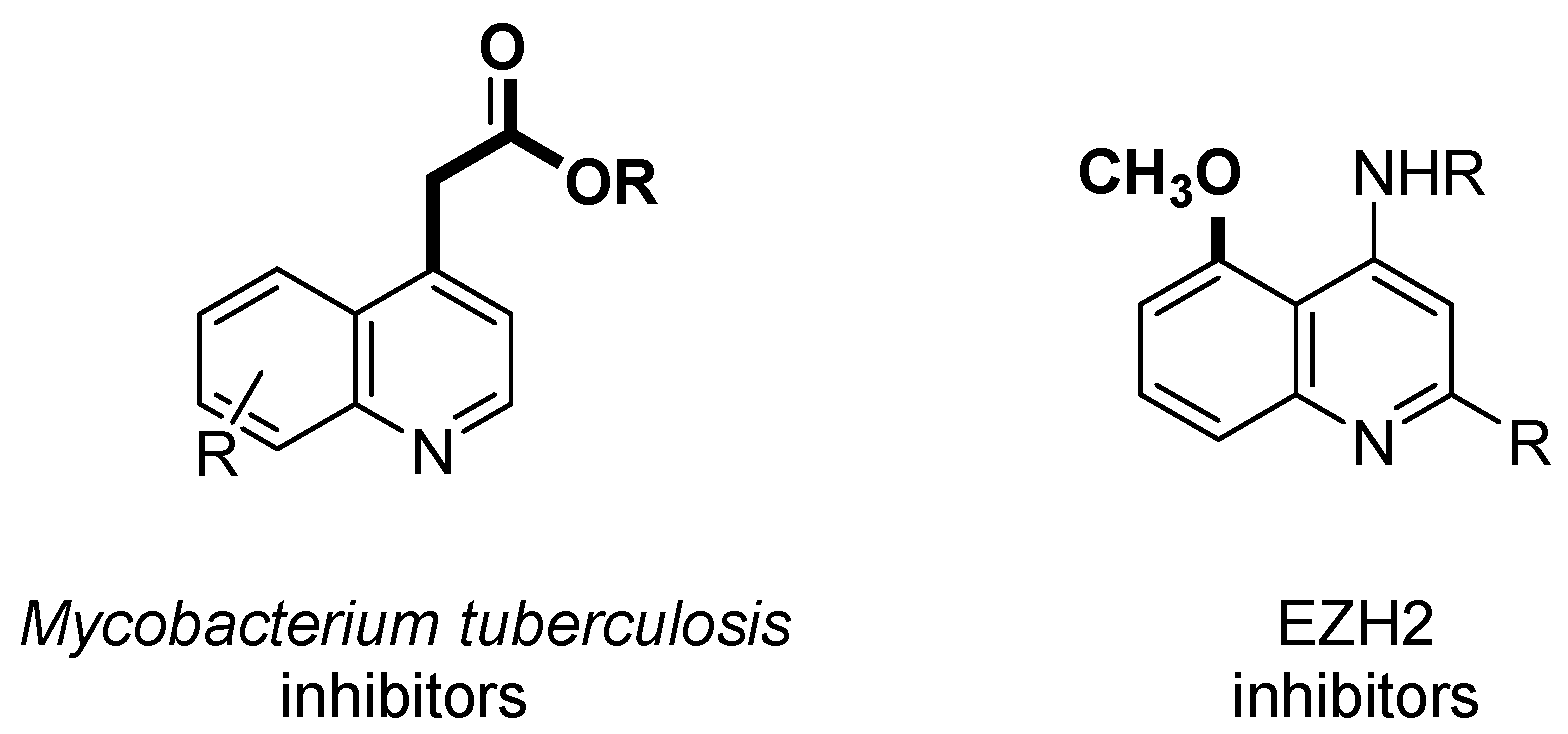
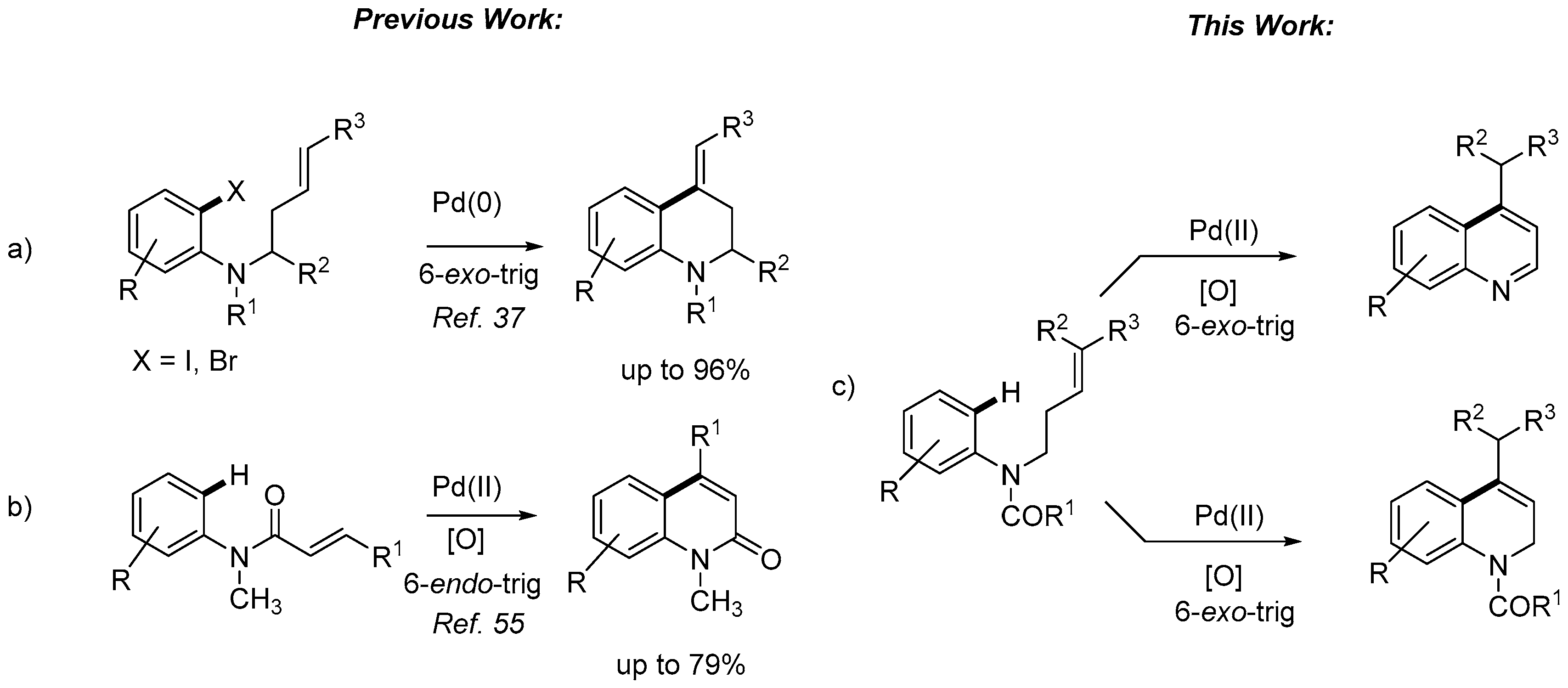
1. Introduction
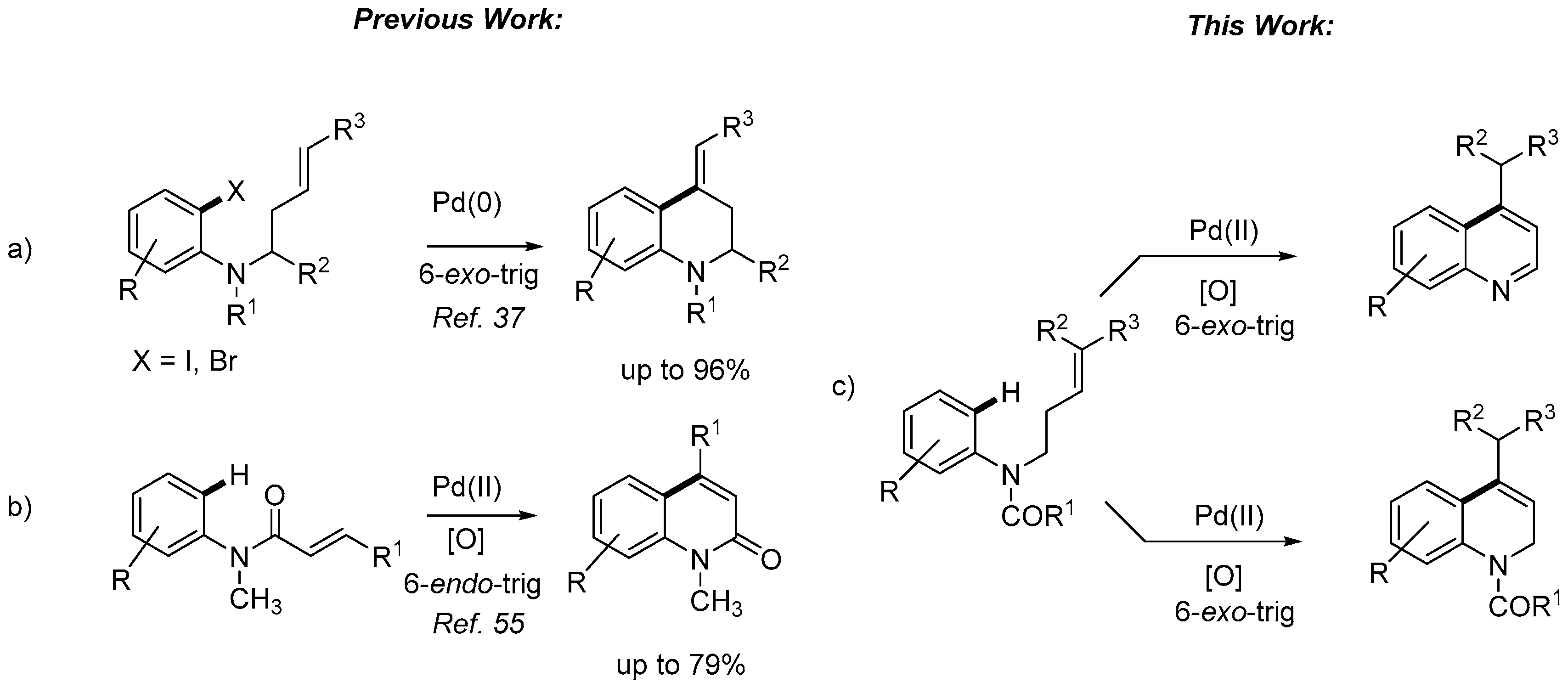
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Methods
3.2. Synthesis of 4-Substituted Quinolines 2
General Procedure for the Synthesis of 4-Substituted Quinolines 2b–f
3.3. Synthesis of 4-Substituted Dihydroquinolines 3 and 4
General Procedure
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Note
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.J. Sponging off nature for new drug leads. Biochem. Pharmacol. 2017, 139, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Hu, W.-P.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2007, 24, 31–86. [Google Scholar] [CrossRef] [PubMed]
- Michael, J.P. Quinoline, quinazoline and acridone alkaloids. Nat. Prod. Rep. 2008, 25, 166–187. [Google Scholar] [CrossRef] [PubMed]
- Molinski, T.F. Marine pyridoacridine alkaloids: Structure, synthesis, and biological chemistry. Chem. Rev. 1993, 93, 1825–1838. [Google Scholar] [CrossRef]
- Skyler, D.; Heathcock, C.H. The pyridoacridine family tree: A useful scheme for designing synthesis and predicting undiscovered natural products. J. Nat. Prod. 2002, 65, 1573–1581. [Google Scholar] [CrossRef] [PubMed]
- Delfourne, E.; Bastide, J. Marine pyridoacridine alkaloids and synthetic analogues as antitumor agents. Med. Res. Rev. 2003, 23, 234–252. [Google Scholar] [CrossRef] [PubMed]
- Imperatore, C.; Aiello, A.; D’Aniello, F.; Senese, M.; Menna, M.L. Alkaloids from marine invertebrates as important leads for anticancer drugs discovery and development. Molecules 2014, 19, 20391–20423. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Alfonso, E.; Avendaño, C.; Menéndez, J.C. Efficient synthesis of the pyrido[2,3,4-kl]acridin-4-one system common to several cytotoxic marine alkaloids. Tetrahedron Lett. 2003, 44, 6003–6005. [Google Scholar] [CrossRef]
- Melzer, B.; Plodek, A.; Bracher, F. Total synthesis of the marine pyridoacridine alkaloid Demethyldeoxyamphimedine. J. Org. Chem. 2014, 79, 7239–7242. [Google Scholar] [CrossRef] [PubMed]
- Okanya, P.W.; Mohr, K.I.; Gerth, K.; Jansen, R.; Müller, R. Marinoquinolines A-F, pyrroloquinolines from Ohtaekwangia kribbensis (Bacteroidetes). J. Nat. Prod. 2011, 74, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Hamann, M.T.; Kochanowska, A.J.; El-Alfy, A.; Matsumoto, R.R.; Boujos, A. Method Using Marine Sponge-Derived Compounds Having Antidepressant, Anxiolytic and Other Neurological Activity, and Compositions of Matter. U.S. Patent 20,090,093,513 A1, 9 April 2009. [Google Scholar]
- Liang, D.; Wang, Y.; Wang, Y.; Di, D. A simple synthesis of the debrominated analogue of veranamine. J. Chem. Res. 2015, 39, 105–107. [Google Scholar] [CrossRef]
- Orjala, J.; Gerwick, W.H. Two quinoline alkaloids from the Caribbean cyanobacterium Lyngbya majuscula. Phytochemistry 1997, 45, 1087–1090. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2005, 22, 15–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettit, G.R.; Hogan, F.; Toms, S. Antineoplastic agents. 592. Highly effective cancer cell growth inhibitory structural modifications of Dolastatin 10. J. Nat. Prod. 2011, 74, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Melody, N.; Chapuis, J.-C. Antineoplastic agents. 603. Quinstatins: Exceptional cancer cell growth inhibitors. J. Nat. Prod. 2017, 80, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Nijampatnam, B.; Dutta, S.; Velu, S.E. Cyanobacterial metabolite calothrixins: Recent advances in synthesis and biological evaluation. Mar. Drugs 2016, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Singh, P.P.; Jain, M.; Sachdeva, S.; Misra, V.; Kaul, C.L.; Kaur, S.; Vaitilingam, B.; Nayyar, A.; Bhaskar, P.P. Ring-Substituted Quinoline Analogs as Anti-Tuberculosis Agents. Indian Patent 2002DE00628, 11 March 2005. [Google Scholar]
- Khan, M.A.; Miller, K.; Rainsford, K.D.; Zhou, Y. Synthesis and antimicrobial activity of novel substituted ethyl 2-(quinolin-4-yl)-propanoates. Molecules 2013, 18, 3227–3240. [Google Scholar] [CrossRef] [PubMed]
- Xiang, P.; Jie, H.; Zhou, Y.; Yang, B.; Wang, H.-J.; Hu, J.; Hu, J.; Yang, S.-Y.; Zhao, Y.-L. 5-Methoxyquinoline derivatives as a new class of EZH2 inhibitors. Molecules 2015, 20, 7620–7636. [Google Scholar] [CrossRef] [PubMed]
- Kouznetsov, V.; Vargas Mendez, L.Y.; Melendez Gomez, C.M. Recent progress in the synthesis of quinolines. Curr. Org. Chem. 2005, 9, 141–161. [Google Scholar] [CrossRef]
- Barluenga, J.; Rodríguez, F.; Fañanás, F.J. Recent advances in the synthesis of indole and quinoline derivatives through cascade reactions. Chem. Asian J. 2009, 4, 1036–1048. [Google Scholar] [CrossRef] [PubMed]
- Alajarín, R.; Burgos, C. Six-membered heterocycles: Quinoline and isoquinoline. In Modern Heterocyclic Chemistry; Álvarez-Builla, J., Vaquero, J.J., Barluenga, J., Eds.; Wiley-VCH: Weinheim, Germany, 2011; Volume 3, pp. 1527–1629. [Google Scholar]
- Ramann, G.A.; Cowen, B.J. Recent advances in metal-free quinoline synthesis. Molecules 2016, 21, 986. [Google Scholar] [CrossRef] [PubMed]
- De Meijere, A.; Diederich, F. (Eds.) Metal Catalyzed Cross-Coupling Reactions, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Wu, X.-F.; Anbarasan, P.; Neumann, H.; Beller, M. From noble metal to Nobel Prize: Palladium-catalyzed coupling reactions as key methods in organic synthesis. Angew. Chem. Int. Ed. 2010, 49, 9047–9050. [Google Scholar] [CrossRef] [PubMed]
- Tymoshenko, D.; Jeges, G.; Gregg, B.T. Synthesis of heterocycles by palladium-catalyzed intramolecular heteroarylation. In Progress in Heterocyclic Chemistry; Gribble, G.W., Joule, J.A., Eds.; Elsevier: Oxford, UK, 2011; Volume 23, pp. 27–74. [Google Scholar]
- Majumdar, K.C.; Samanta, S.; Sinha, B. Recent developments in palladium-catalyzed formation of five- and six-membered fused heterocycles. Synthesis 2012, 44, 817–847. [Google Scholar] [CrossRef]
- Zeni, G.; Larock, R.C. Synthesis of heterocycles via palladium-catalyzed oxidative addition. Chem. Rev. 2006, 106, 4644–4680. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Gribble, G.W. (Eds.) Palladium in Heterocyclic Chemistry: A Guide for the Synthetic Chemist, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Muller, T.; Bräse, S. Formation of heterocycles. In The Mizoroki-Heck Reaction; Oestreich, M., Ed.; Wiley: Chichester, UK, 2009; pp. 215–258. [Google Scholar]
- Beletskaya, I.P.; Cheprakov, A.V. Modern Heck reactions. In RSC Catalysis Series: New Trends in Cross-Coupling: Theory and Applications; Colacot, T., Ed.; Royal Society of Chemistry: London, UK, 2015; Volume 21, pp. 355–478. [Google Scholar]
- Martínez-Estíbalez, U.; Sotomayor, N.; Lete, E. Pd-catalyzed arylation/ring-closing metathesis approach to azabicycles. Tetrahedron Lett. 2007, 48, 2919–2922. [Google Scholar] [CrossRef]
- Martínez-Estíbalez, U.; Sotomayor, N.; Lete, E. Intramolecular carbolithiation reactions for the synthesis of 2,4-disubstituted tetrahydroquinolines: Evaluation of TMEDA and (−)-sparteine as ligands in the stereoselectivity. Org. Lett. 2009, 11, 1237–1240. [Google Scholar] [CrossRef] [PubMed]
- García-Calvo, O.; Martínez-Estíbalez, U.; Lete, E.; Sotomayor, N. Synthesis of tetrahydroquinolines through intramolecular carbolithiation reactions. Heterocycles 2014, 88, 425–440. [Google Scholar] [CrossRef]
- Martínez-Estíbalez, U.; García-Calvo, O.; Ortiz-de-Elguea, V.; Sotomayor, N.; Lete, E. Intramolecular Mizoroki–Heck Reaction in the regioselective synthesis of 4-alkylidene-tetrahydroquinolines. Eur. J. Org. Chem. 2013, 2013, 3013–3022. [Google Scholar] [CrossRef]
- Ferreira, E.M.; Zhang, H.; Stoltz, B.M. Oxidative Heck-type reactions (Fujiwara-Moritani reactions). In The Mizoroki-Heck Reaction; Oestreich, M., Ed.; Wiley: Chichester, UK, 2009; pp. 345–382. [Google Scholar]
- Zhou, L.; Lu, W. Towards ideal synthesis. alkenylation of aryl C-H bonds by a Fujiwara-Moritani reaction. Chem. Eur. J. 2014, 20, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Fujiwara, Y. Dehydrogenative Heck-type reactions: The Fujiwara-Moritani reaction. In RSC Green Chemistry Series: From C-H to C-C Bonds: Cross-Dehydrogenative-Coupling; Li, C.-J., Ed.; Royal Society of Chemistry: London, UK, 2015; Volume 26, pp. 33–54. [Google Scholar]
- Topczewski, J.J.; Sanford, M.S. Carbon-hydrogen (C-H) bond activation at PdIV: A frontier in C-H functionalization catalysis. Chem. Sci. 2015, 6, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Gensch, T.; Hopkinson, M.N.; Glorius, F.; Wencel-Delord, J. Mild metal-catalyzed C-H activation: Examples and concepts. Chem. Soc. Rev. 2016, 45, 2900–2936. [Google Scholar] [CrossRef] [PubMed]
- Neufeldt, S.R.; Sanford, M.S. Controlling site selectivity in palladium-catalyzed C-H bond functionalization. Acc. Chem. Res. 2012, 45, 936–946. [Google Scholar] [CrossRef] [PubMed]
- Rouquet, G.; Chatani, N. Catalytic functionalization of C(sp2)-H and C(sp3)-H bonds by using bidentate directing groups. Angew. Chem. Int. Ed. 2013, 52, 11726–11743. [Google Scholar] [CrossRef] [PubMed]
- Pichette Drapeau, M.; Gooßen, L.J. Carboxylic acids as directing groups for C-H bond functionalization. Chem. Eur. J. 2016, 22, 18654–18677. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Gandeepan, P.; Li, J.; Ackermann, L. Recent advances in positional-selective alkenylations: Removable guidance for twofold C-H activation. Org. Chem. Front. 2017, 4, 1435–1467. [Google Scholar] [CrossRef]
- Lyons, T.W.; Sanford, M.S. Palladium-catalyzed ligand-directed C-H functionalization reactions. Chem. Rev. 2010, 110, 1147–1169. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-H.; Engle, K.M.; Shi, B.-F.; Yu, J.-Q. Ligand-enabled reactivity and selectivity in a synthetically versatile aryl C-H olefination. Science 2010, 327, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Engle, K.M.; Yu, J.-Q. Developing Ligands for palladium(II)-catalyzed C–H functionalization: Intimate dialogue between ligand and substrate. J. Org. Chem. 2013, 78, 8927–8955. [Google Scholar] [CrossRef] [PubMed]
- Engle, K.M. The mechanism of palladium(II)-mediated C-H cleavage with mono-N-protected amino acid (MPAA) ligands: Origins of rate acceleration. Pure Appl. Chem. 2016, 88, 119–138. [Google Scholar] [CrossRef]
- Suna, E.; Shubin, K. Intramolecular coupling via C(sp2)-H activation. In Science of Synthesis. Cross Coupling and Heck-Type Reactions 3: Metal-Catalyzed Heck-Type Reactions and C-H Couplings via C-H Activation; Larhed, M., Ed.; Thieme: Stuttgart, Germany, 2013; Volume 3, pp. 643–724. [Google Scholar]
- Beck, E.M.; Gaunt, M.J. Pd-catalyzed C-H bond functionalization on the indole and pyrrole nucleus. Top. Curr. Chem. 2010, 292, 85–121. [Google Scholar] [CrossRef] [PubMed]
- Broggini, G.; Beccalli, E.M.; Fasana, A.; Gazzola, S. Palladium-catalyzed dual C-H or N-H functionalization of unfunctionalized indole derivatives with alkenes and arenes. Beilstein J. Org. Chem. 2012, 8, 1730–1746. [Google Scholar] [CrossRef] [PubMed]
- Schiffner, J.A.; Oestreich, M. All-carbon-substituted quaternary carbon atoms in oxindoles by an aerobic palladium(II)-catalyzed ring closure onto tri- and tetrasubstituted double bonds. Eur. J. Org. Chem. 2011, 2011, 1148–1154. [Google Scholar] [CrossRef]
- Ortiz-de-Elguea, V.; Sotomayor, N.; Lete, E. Two consecutive Palladium(II)-promoted C-H alkenylation reactions for the synthesis of 3-alkenylquinolones. Adv. Synth. Catal. 2015, 357, 463–473. [Google Scholar] [CrossRef]
- Grimster, N.P.; Gauntlett, C.; Godfrey, C.R.A.; Gaunt, M.J. Palladium-catalyzed intermolecular alkenylation of indoles by solvent-controlled regioselective C-H functionalization. Angew. Chem. Int. Ed. 2005, 44, 3125–3129. [Google Scholar] [CrossRef] [PubMed]
- García-Rubia, A.; Urones, B.; Gómez-Arrayás, R.; Carretero, J.C. Pd(II)-catalysed C-H functionalisation of indoles and pyrroles assisted by the removable N-(2-pyridyl)sulfonyl group: C2-alkenylation and dehydrogenative homocoupling. Chem. Eur. J. 2010, 16, 9676–9685. [Google Scholar] [CrossRef] [PubMed]
- Abbiati, G.; Beccalli, E.M.; Broggini, G.; Zoni, C. Regioselectivity on the palladium-catalyzed intramolecular cyclization of indole derivatives. J. Org. Chem. 2003, 68, 7625–7628. [Google Scholar] [CrossRef] [PubMed]
- García-Rubia, A.; Urones, B.; Gómez-Arrayás, R.; Carretero, J.C. PdII-catalyzed C-H olefination of N-(2-pyridyl)sulfonyl anilines and arylalkylamines. Angew. Chem. Int. Ed. 2011, 50, 10927–10931. [Google Scholar] [CrossRef] [PubMed]
- Kandukuri, S.R.; Schiffner, J.A.; Oestreich, M. Aerobic palladium(II)-catalyzed 5-endo-trig cyclization: An entry into the diastereoselective C-2 alkenylation of indoles with tri- and tetrasubstituted double bonds. Angew. Chem. Int. Ed. 2012, 51, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ferreira, E.M.; Stoltz, B.M. Direct oxidative Heck cyclizations: Intramolecular Fujiwara–Moritani arylations for the synthesis of functionalized benzofurans and dihydrobenzofurans. Angew. Chem. Int. Ed. 2004, 43, 6144–6148. [Google Scholar] [CrossRef] [PubMed]
- Kubota, A.; Emmert, M.H.; Sanford, M.S. Pyridine ligands as promoters in PdII/0-catalyzed C-H olefination reactions. Org. Lett. 2012, 14, 1760–1763. [Google Scholar] [CrossRef] [PubMed]
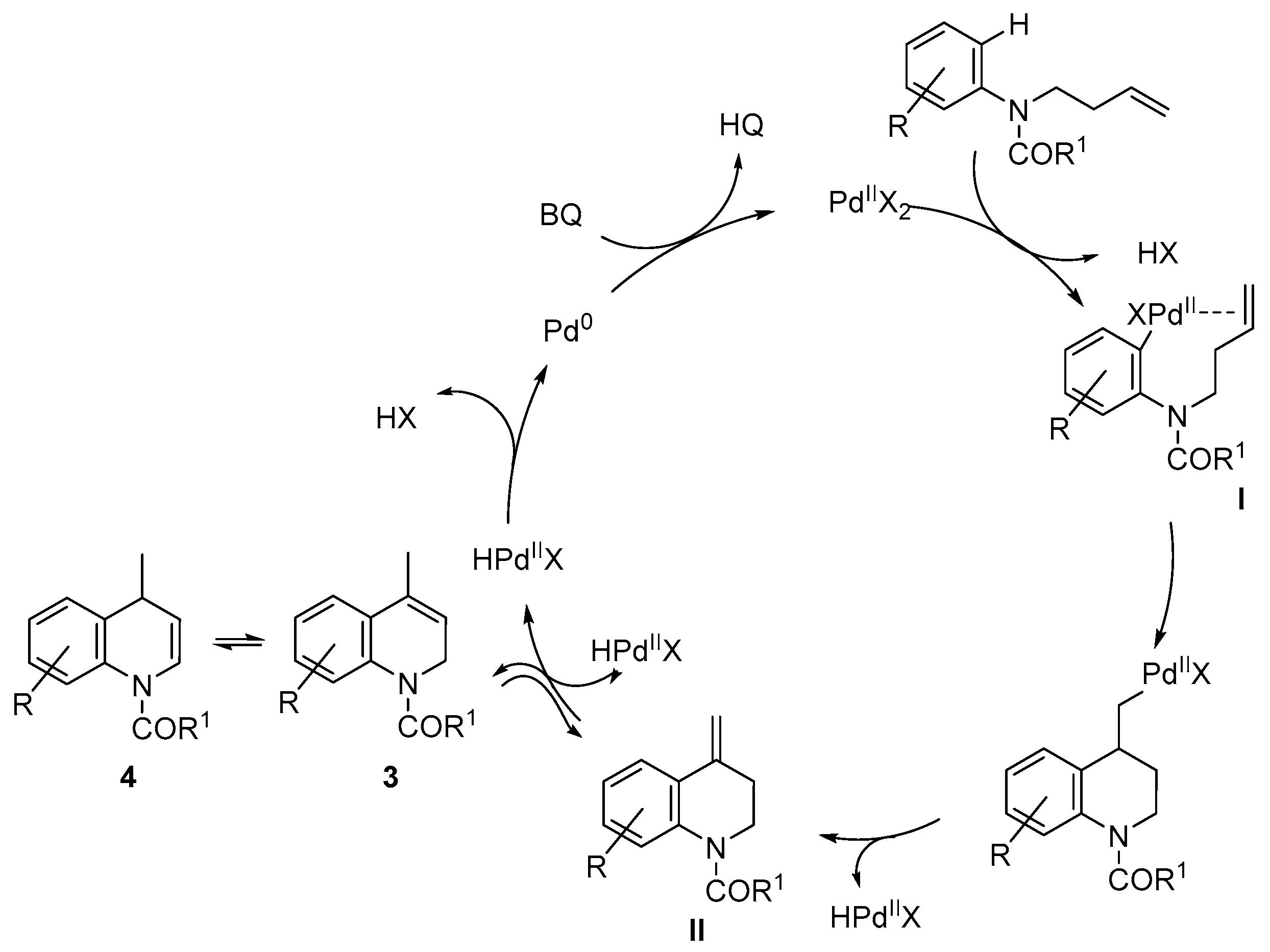
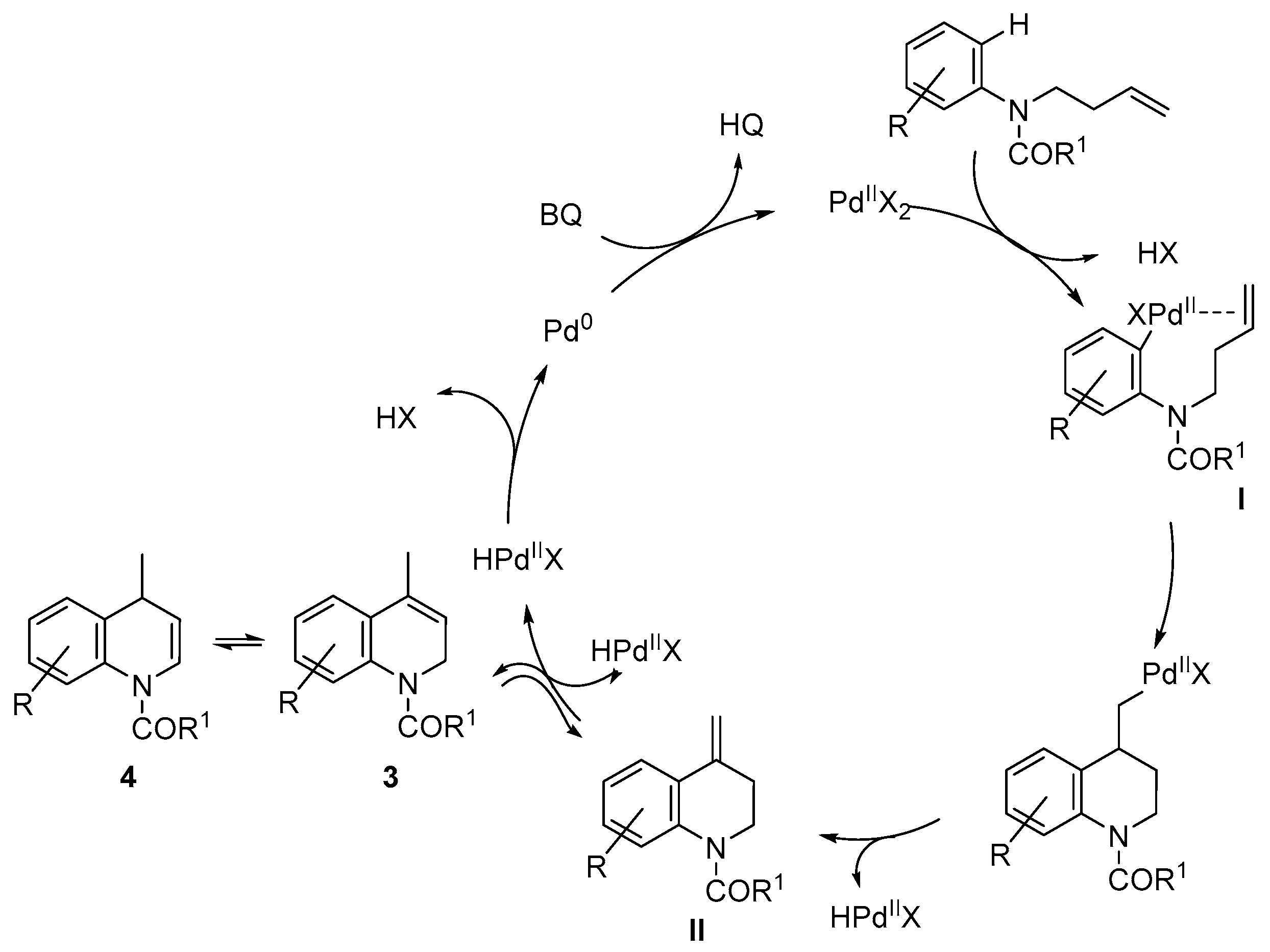
- Alternatively, to avoid the deprotection of the nitrogen, the corresponding N-(but-3-en-1-yl)-3,5-dimethoxy-N-methylaniline was prepared and submitted to cyclization conditions. However, only decomposition was observed under all conditions tested, using different palladium sources, oxidants and solvents, even in the absence of TsOH or AcOH.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
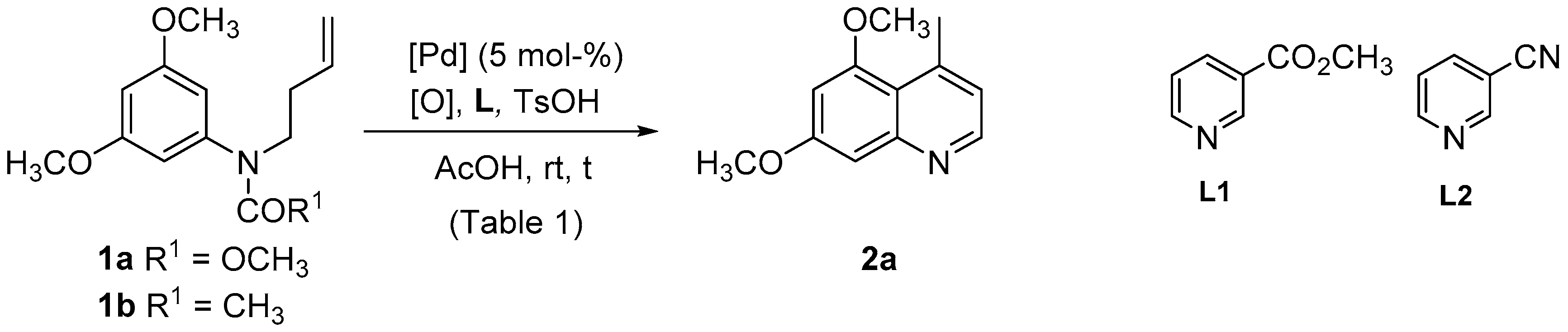
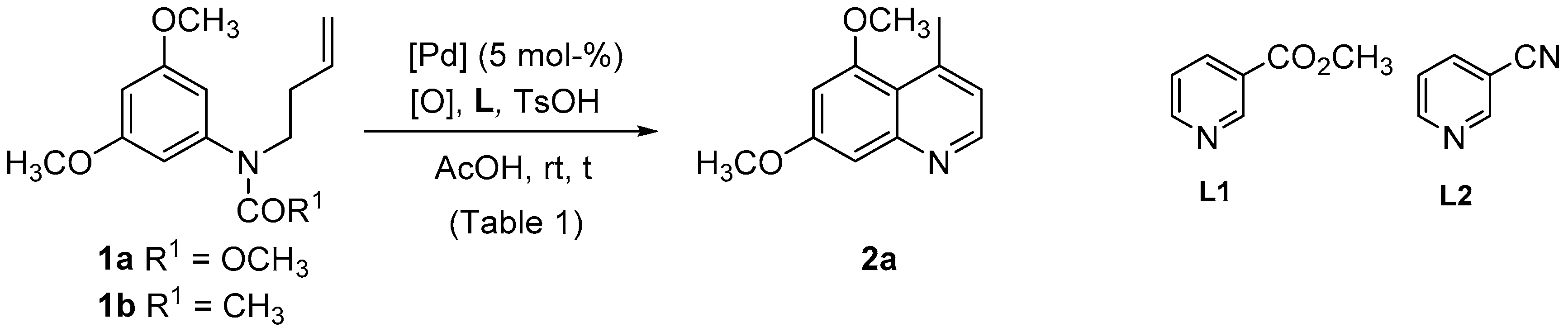
| Entry | Substrate | [Pd] | [O] | L (a) | t (h) | 2a Yield (%) |
|---|---|---|---|---|---|---|
| 1 | 1a | Pd(OAc)2 | PhCO3tBu (b) | - | 24 | 19 |
| 2 | 1a | Pd(OAc)2 | PhCO3tBu (b) | L1 | 24 | 11 |
| 3 | 1a | Pd(OAc)2 | PhCO3tBu (b) | L2 | 24 | 5 |
| 4 | 1b | Pd(OAc)2 | PhCO3tBu (b) | - | 5.5 | 12 |
| 5 | 1b | Pd(OAc)2 | PhCO3tBu (b) | L1 | 5.5 | 11 |
| 6 | 1b | Pd(OAc)2 | PhCO3tBu (b) | L2 | 5.5 | 9 |
| 7 | 1a | Pd(OAc)2 | Cu(OAc)2 (c) | - | 24 | 36 |
| 8 | 1a | Pd(OAc)2 | Cu(OAc)2 (c) | L1 | 24 | 35 |
| 9 | 1a | Pd(OAc)2 | Cu(OAc)2 (c) | L2 | 24 | 14 |
| 10 | 1b | Pd(OAc)2 | Cu(OAc)2 (c) | - | 5.5 | 20 |
| 11 | 1b | Pd(OAc)2 | Cu(OAc)2 (c) | L1 | 5.5 | nr |
| 12 | 1b | Pd(OAc)2 | Cu(OAc)2 (c) | L2 | 5.5 | nr |
| 13 | 1a | Pd(OAc)2 | p-BQ (c) | - | 24 | 4 |
| 14 | 1a | Pd(OAc)2 | p-BQ (c) | L1 | 14 | 5 |
| 15 | 1a | Pd(OAc)2 | p-BQ (c) | L2 | 24 | 24 |
| 16 | 1b | Pd(OAc)2 | p-BQ (c) | - | 24 | 10 |
| 17 | 1b | Pd(OAc)2 | p-BQ (c) | L1 | 24 | 19 |
| 18 | 1b | Pd(OAc)2 | p-BQ (c) | L2 | 24 | 8 |
| 19 | 1a | Pd(dba)2 | PhCO3tBu (b) | - | 24 | 28 |
| 20 | 1a | Pd(dba)2 | Cu(OAc)2 (c) | - | 24 | 54 |
| 21 | 1a | Pd(dba)2 | p-BQ (c) | - | 24 | 15 |
| 22 | 1a | Pd(dba)2 | F+ (c) | - | 24 | 18 |
| 23 | 1a | PdCl2(CH3CN)2 | PhCO3tBu (b) | - | 24 | 56 |
| 24 | 1a | PdCl2(CH3CN)2 | Cu(OAc)2 (c) | - | 48 | 20 |
| 25 | 1b | PdCl2(CH3CN)2 | PhCO3tBu (b) | - | 3.5 | 55 |
| 26 | 1b | PdCl2(CH3CN)2 | F+ (c) | - | 24 | 27 |
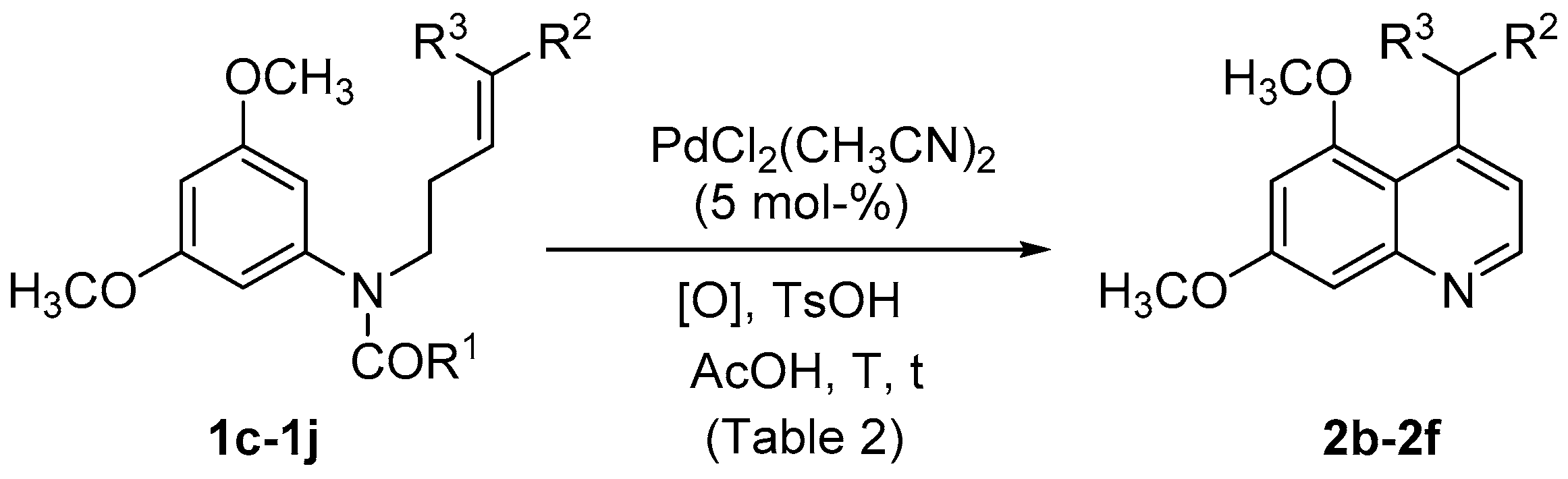
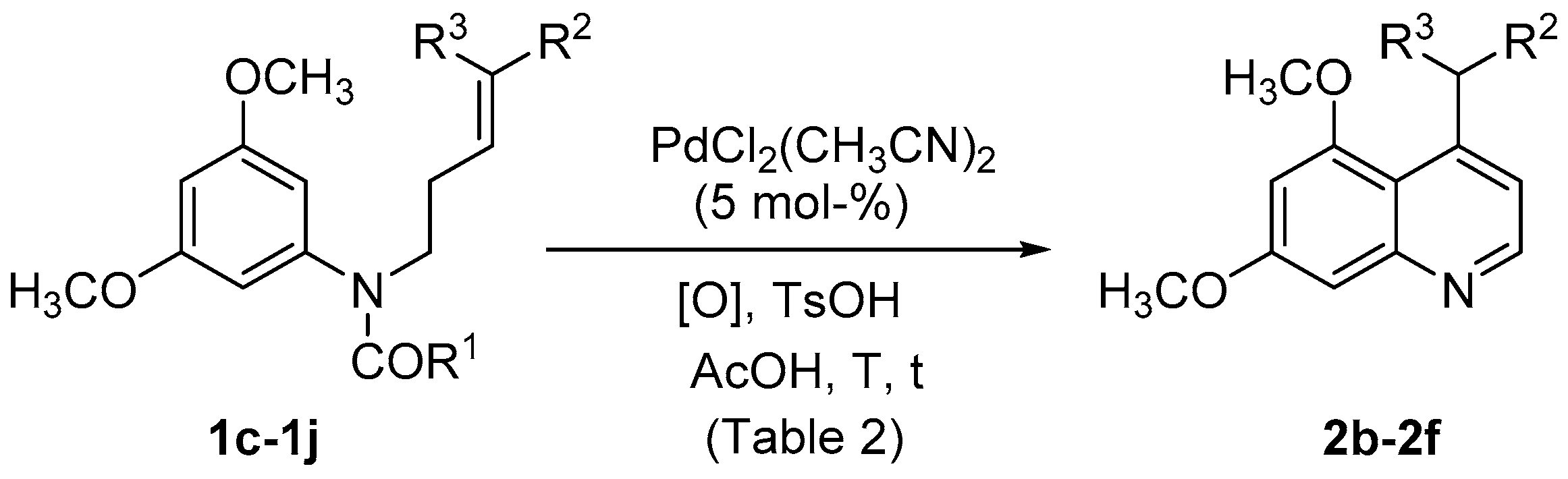
| Entry | Substrate | R1 | R2 | R3 | [O] | T (°C) | t (h) | 2 | Yield (%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1d | CH3 | SO2Ph | H | PhCO3tBu (a) | rt | 24 | 2b | (c) |
| 2 | 1d | CH3 | SO2Ph | H | PhCO3tBu (a) | 70 | 24 | 2b | (c),(d) |
| 3 | 1d | CH3 | SO2Ph | H | F+ (a) | rt | 24 | 2b | (c),(d) |
| 4 | 1d | CH3 | SO2Ph | H | F+ (a) | 70 | 24 | 2b | 44 |
| 5 | 1c | OCH3 | SO2Ph | H | F+ (a) | 70 | 24 | 2b | 61 |
| 6 | 1e | OCH3 | CO2CH3 | H | F+ (a) | 70 | 19 | 2c | 54 |
| 7 | 1f | CH3 | CO2CH3 | H | F+ (a),(b) | 70 | 41 | 2c | 54 |
| 8 | 1f | CH3 | CO2CH3 | H | PhCO3tBu (a) | 70 | 47 | 2c | 32 |
| 9 | 1g | OCH3 | CO2CH3 | CH3 | F+ (a) | 70 | 21 | - | (d) |
| 10 | 1h | OCH3 | CO2CH2CF3 | H | F+ (a) | 70 | 21 | 2d | 46 |
| 11 | 1i | OCH3 | CO2(CH2)11CH3 | H | F+ (a) | 70 | 21 | 2e | 50 |
| 12 | 1j | OCH3 | CO2CH2Ph | H | F+ (a) | 70 | 21 | 2f | 62 |
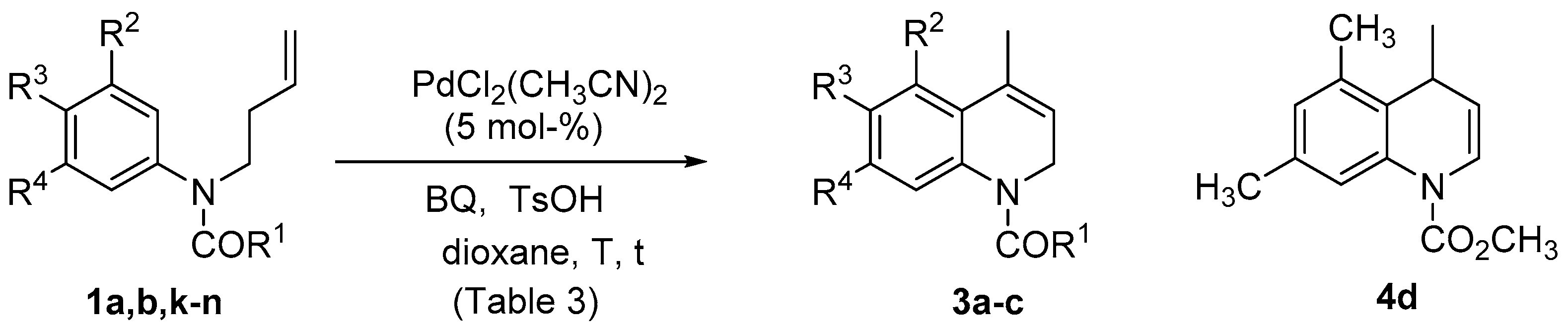
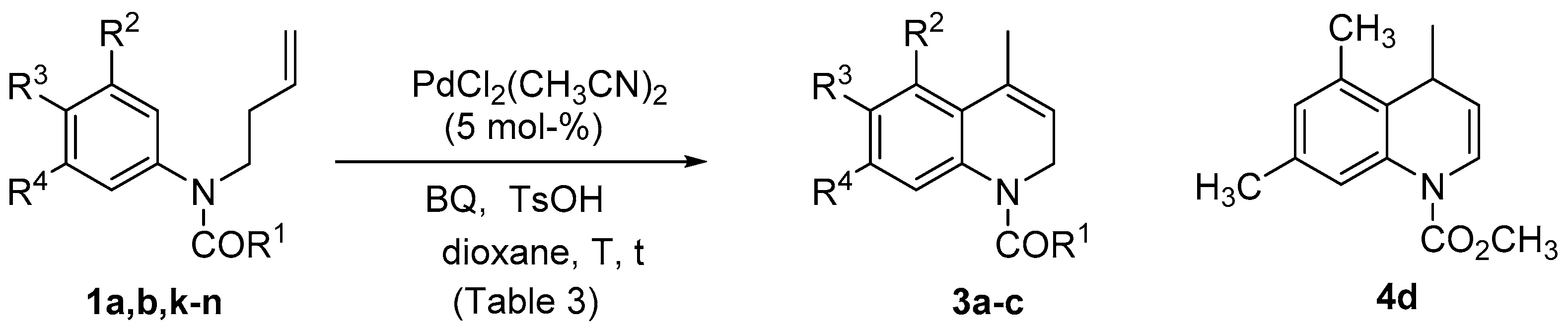
| Entry | Substrate | R1 | R2 | R3 | R4 | T (°C) | t (h/min) | Product | Yield (%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1a | OCH3 | OCH3 | H | OCH3 | rt | 7.5 h | 3a | 74 |
| 2 | 1a | OCH3 | OCH3 | H | OCH3 | 70 | 10 min | 3a | 89 |
| 3 | 1b | CH3 | OCH3 | H | OCH3 | rt | 25 h | 3b | 62 |
| 4 | 1b | CH3 | OCH3 | H | OCH3 | rt (a) | 23 h | - | (c) |
| 5 | 1b | CH3 | OCH3 | H | OCH3 | rt (b) | 23 h | - | (c) |
| 6 | 1k | OCH3 | OCH3 | OCH3 | OCH3 | rt | 24 h | - | (c) |
| 7 | 1k | OCH3 | OCH3 | OCH3 | OCH3 | 70 | 2 h | 3c | 33 |
| 8 | 1k | OCH3 | OCH3 | OCH3 | OCH3 | 70 (d) | 2 h | 3c | 40 |
| 9 | 1k | OCH3 | OCH3 | OCH3 | OCH3 | 70 (d) | 7 h | 3c | 11 |
| 10 | 1l | OCH3 | H | OCH2O | 70 (d) | 24 h | - | (c) | |
| 11 | 1m | OCH3 | H | OCH3 | OCH3 | 70 (d) | 24 h | - | (c) |
| 12 | 1n | OCH3 | CH3 | H | CH3 | 70 (d) | 24 h | 4d | 32 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carral-Menoyo, A.; Ortiz-de-Elguea, V.; Martinez-Nunes, M.; Sotomayor, N.; Lete, E. Palladium-Catalyzed Dehydrogenative Coupling: An Efficient Synthetic Strategy for the Construction of the Quinoline Core. Mar. Drugs 2017, 15, 276. https://doi.org/10.3390/md15090276
Carral-Menoyo A, Ortiz-de-Elguea V, Martinez-Nunes M, Sotomayor N, Lete E. Palladium-Catalyzed Dehydrogenative Coupling: An Efficient Synthetic Strategy for the Construction of the Quinoline Core. Marine Drugs. 2017; 15(9):276. https://doi.org/10.3390/md15090276
Chicago/Turabian StyleCarral-Menoyo, Asier, Verónica Ortiz-de-Elguea, Mikel Martinez-Nunes, Nuria Sotomayor, and Esther Lete. 2017. "Palladium-Catalyzed Dehydrogenative Coupling: An Efficient Synthetic Strategy for the Construction of the Quinoline Core" Marine Drugs 15, no. 9: 276. https://doi.org/10.3390/md15090276