
Klyflaccicembranols A–I, New Cembranoids from the Soft Coral Klyxum flaccidum

 ,
,  ,
, 
Abstract
:1. Introduction
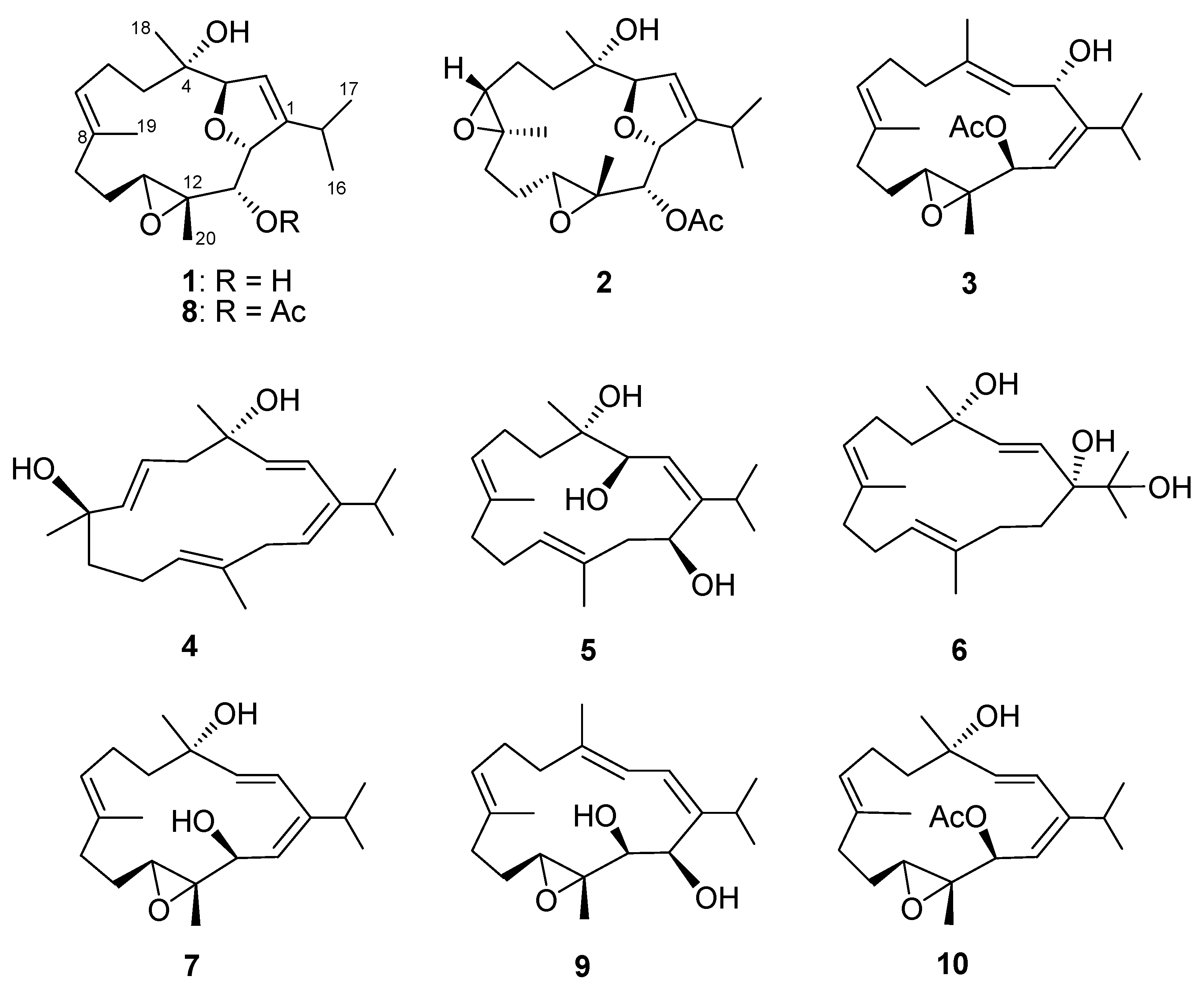
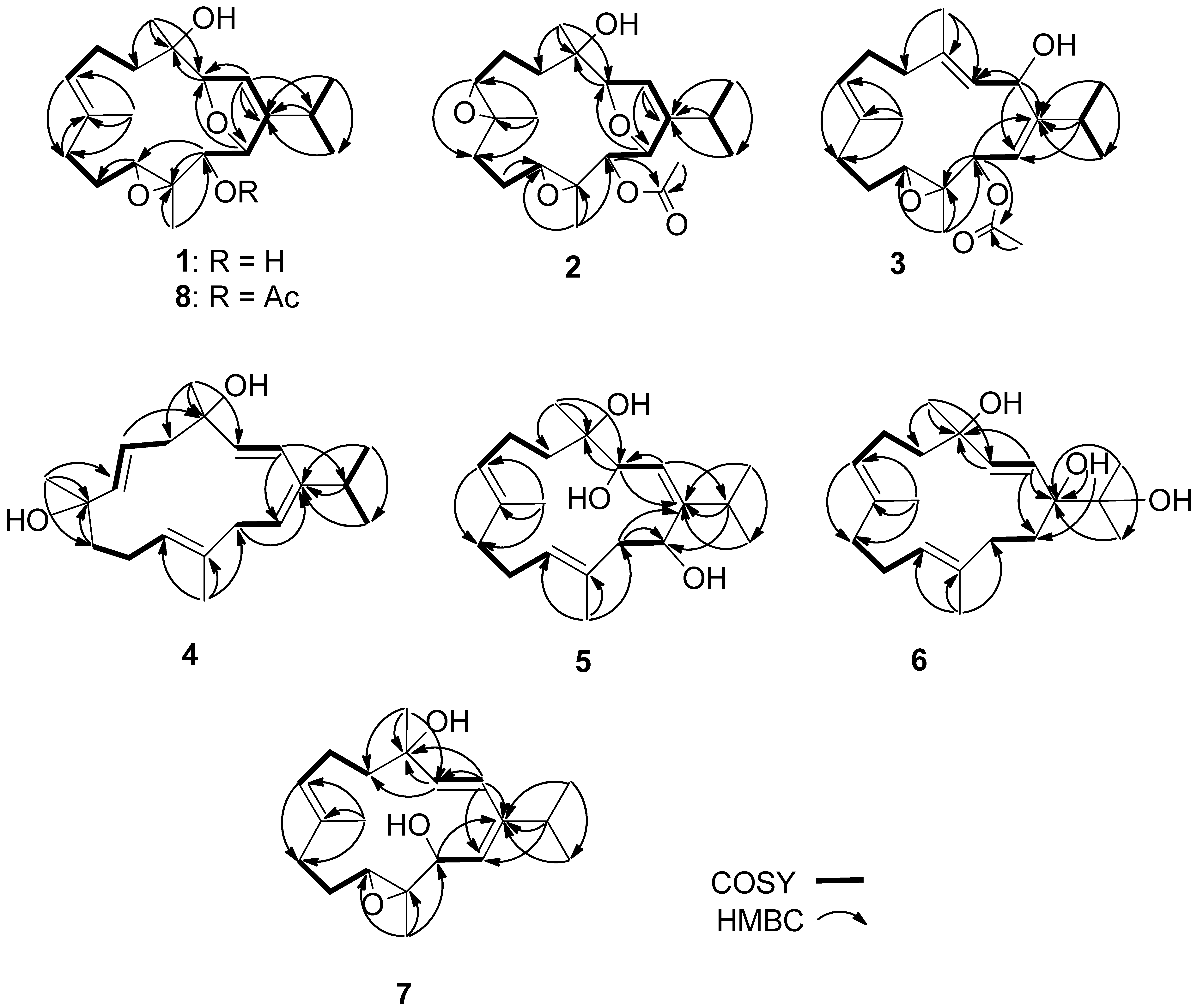
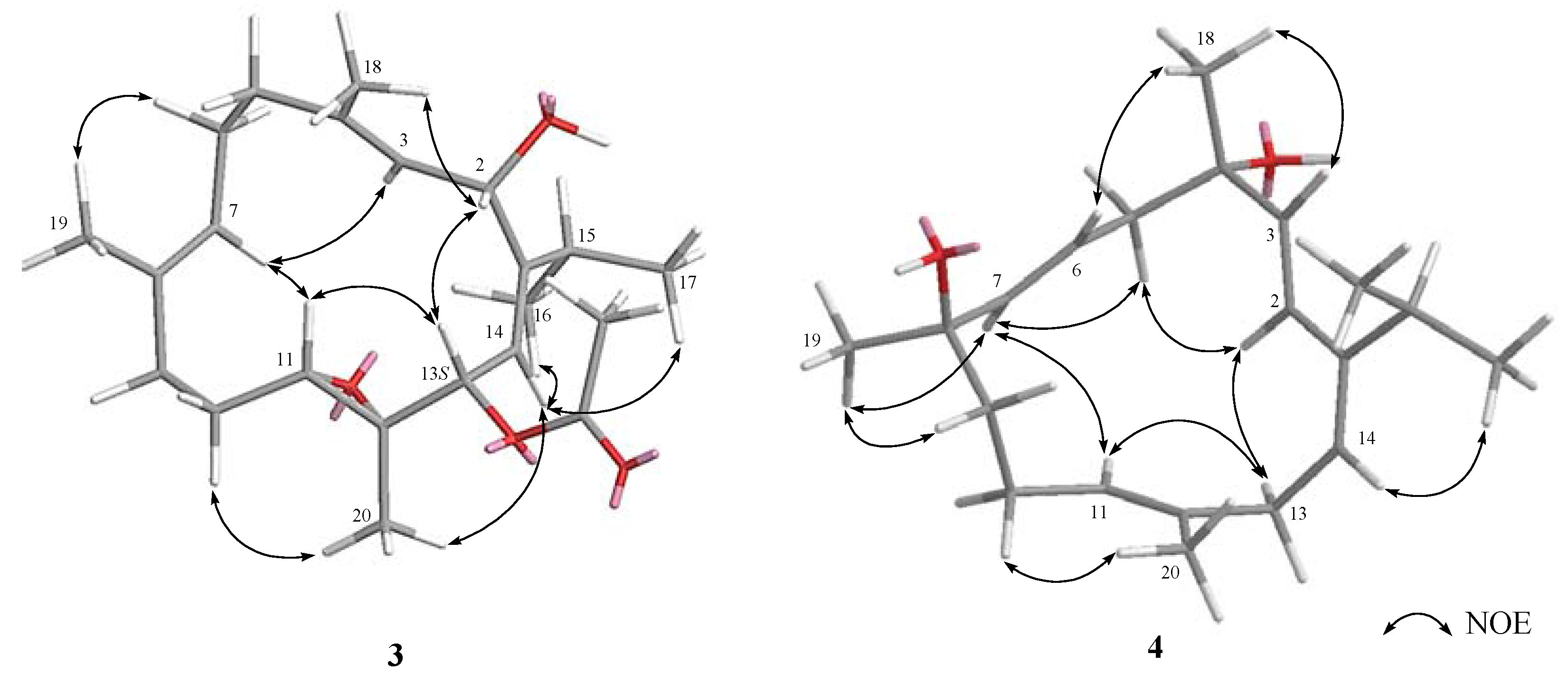
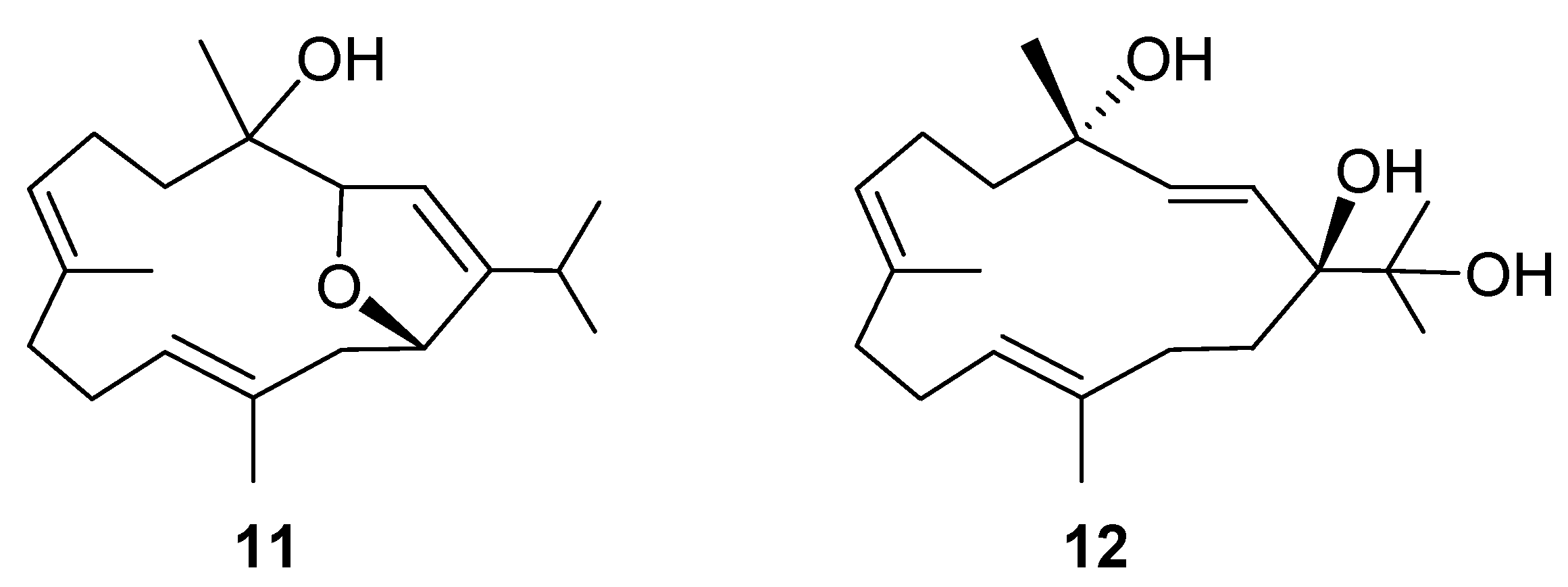
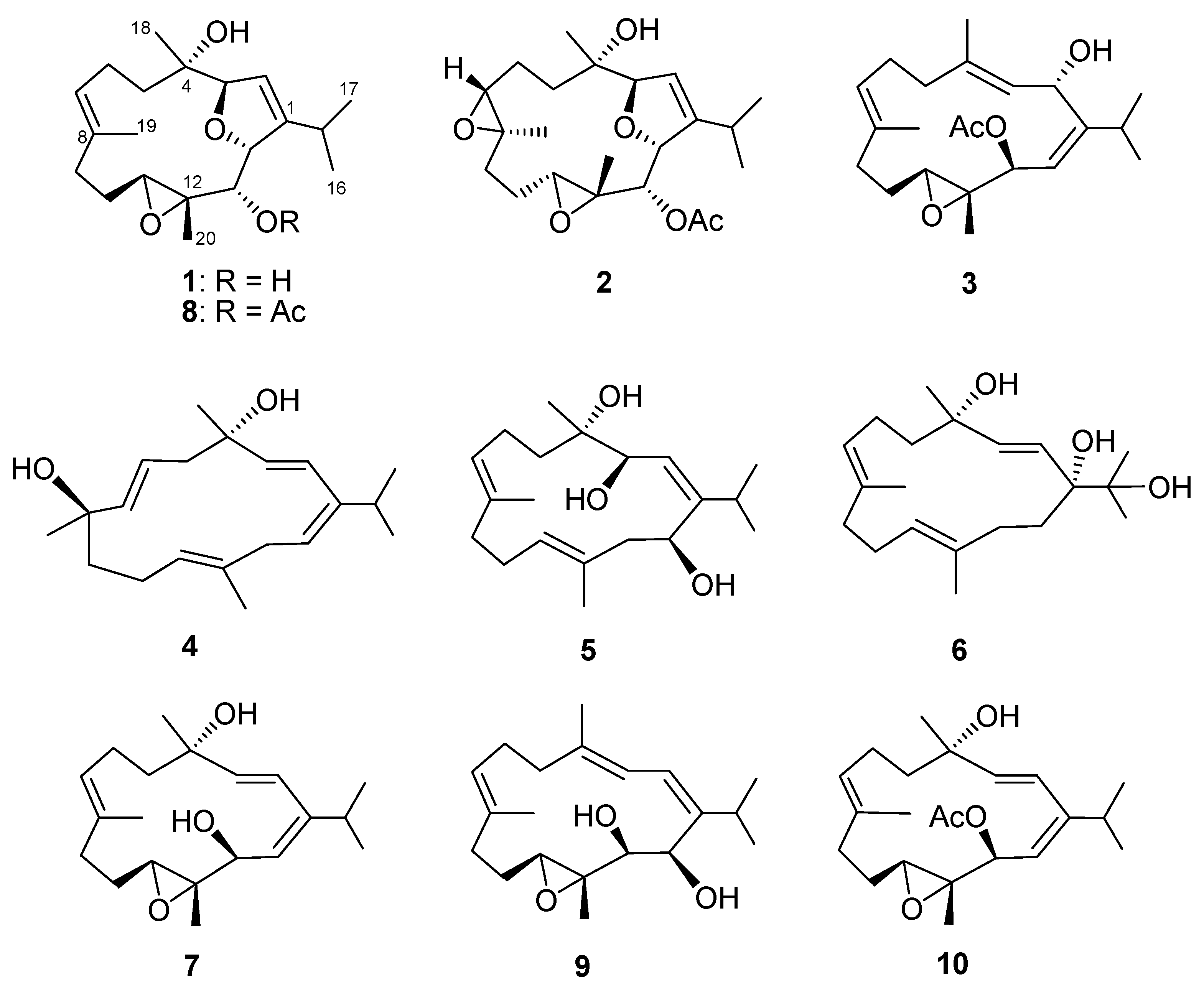
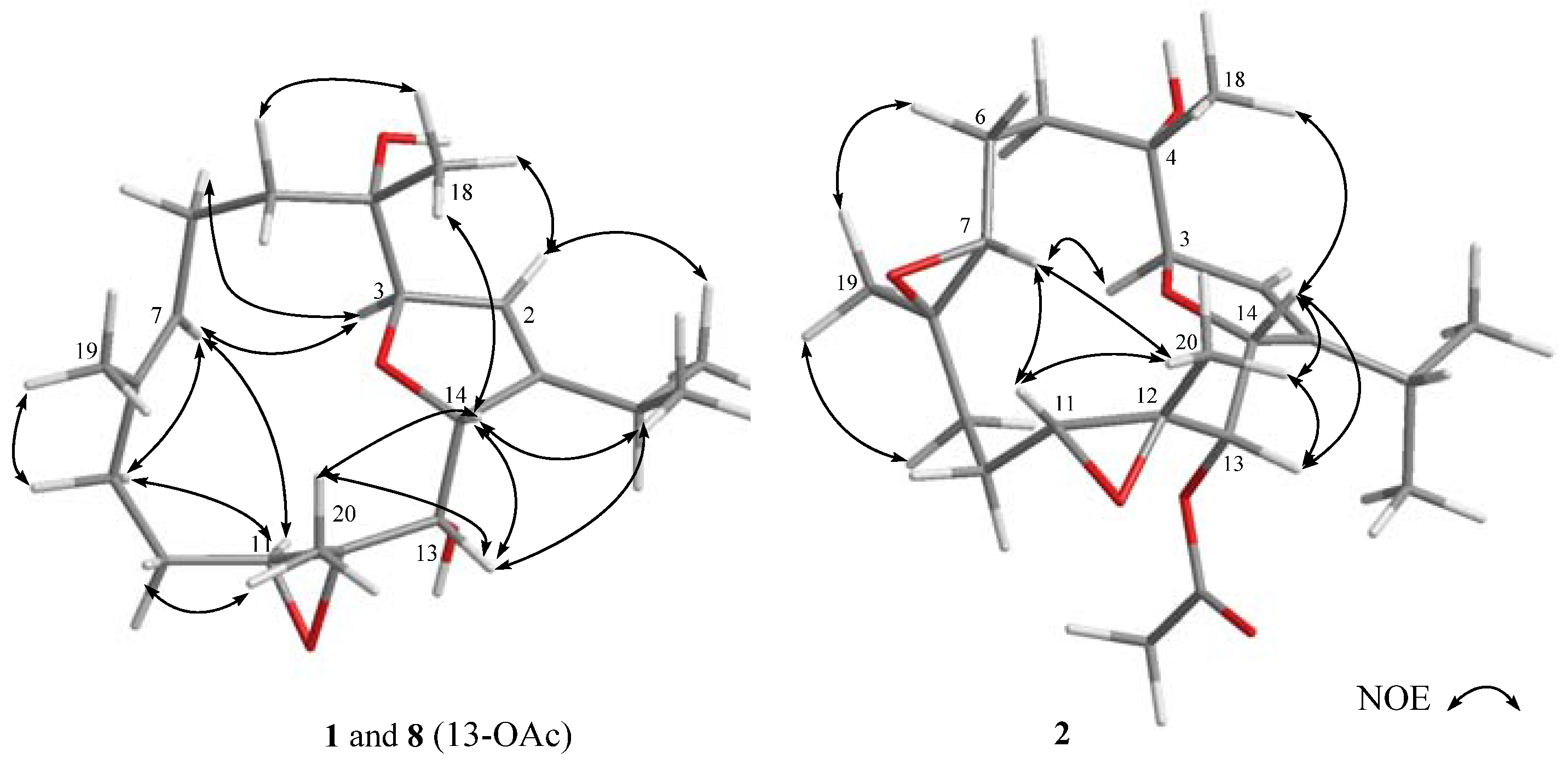
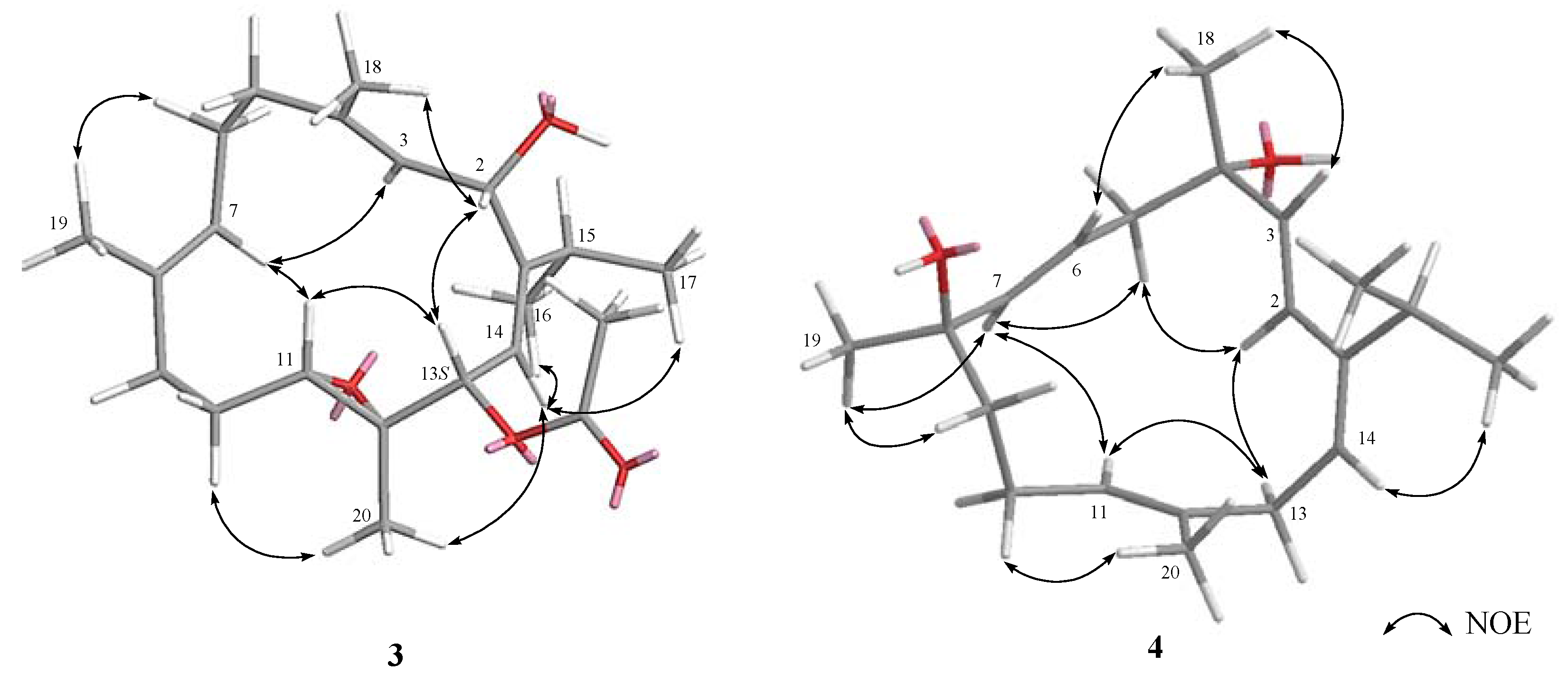
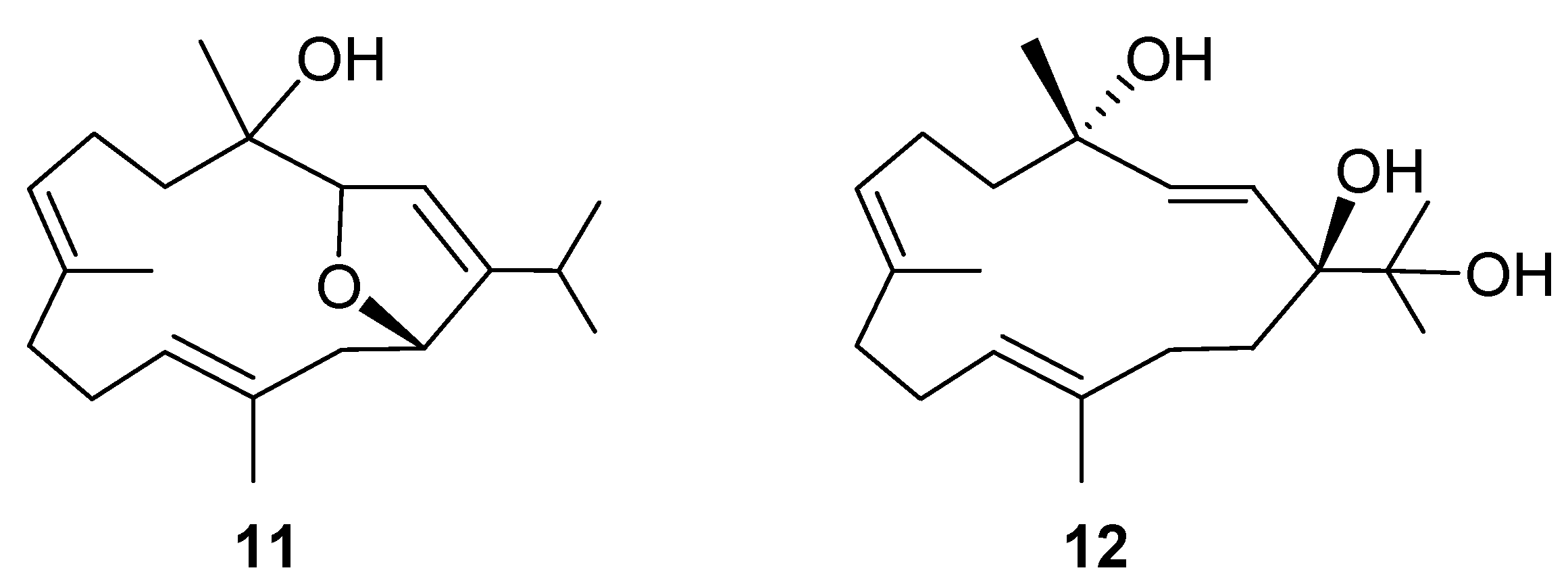
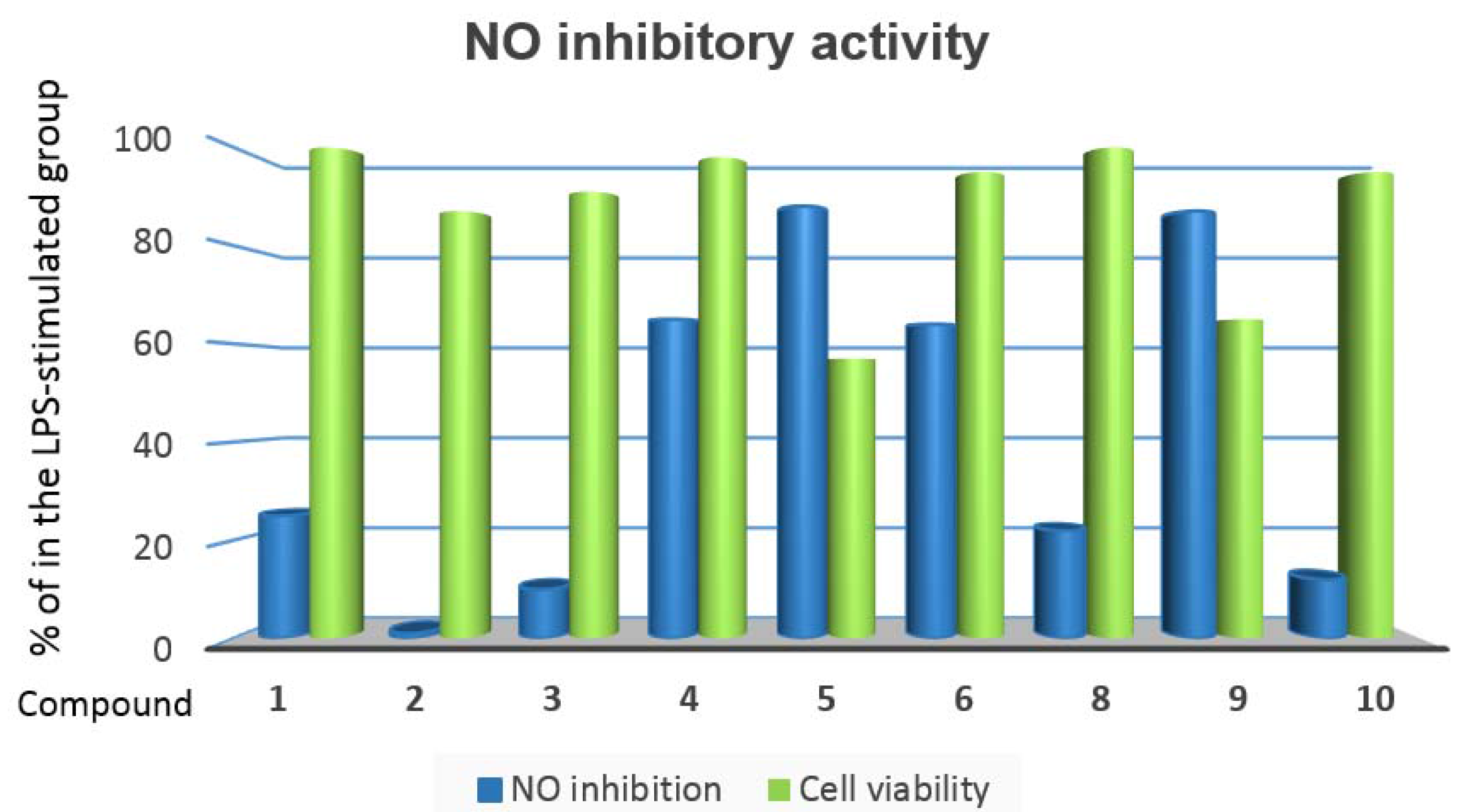
2. Results and Discussion
3. Materials and Methods
3.1. General Procedures
3.2. Animal Material
3.3. Extraction and Separation
3.3.1. Klyflaccicembranol A (1)
3.3.2. Klyflaccicembranol B (2)
3.3.3. Klyflaccicembranol C (3)
3.3.4. Klyflaccicembranol D (4)
3.3.5. Klyflaccicembranol E (5)
3.3.6. Klyflaccicembranol F (6)
3.3.7. Klyflaccicembranol G (7)
3.3.8. Klyflaccicembranol H (8)
3.3.9. Klyflaccicembranol I (9)
3.3.10. Hydrolysis of Klyflaccicembranol H (8)
3.3.11. Preparation of (S)- and (R)-MTPA Esters of 1
3.4. Cytotoxicity Assay
3.5. Nitric Oxide Inhibitory Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blunt, J.W.; Copp, B.R.; Hu, W.P.; Munro, M.H.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2016, 33, 382–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.W.; Chao, C.H.; Su, J.H.; Wen, Z.H.; Sung, P.J.; Sheu, J.H. Anti-inflammatory eunicellin-based diterpenoids from the cultured soft coral Klyxum simplex. Org. Biomol. Chem. 2010, 8, 2363–2366. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.C.; Strangman, W.K.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Thalassospiramides A and B, immunosuppressive peptides from the marine bacterium Thalassospira sp. Org. Lett. 2007, 9, 1525–1528. [Google Scholar] [CrossRef] [PubMed]
- Kijjoa, A.; Sawangwong, P. Drugs and cosmetics from the sea. Mar. Drugs 2004, 2, 73–82. [Google Scholar] [CrossRef]
- Chill, L.; Berrer, N.; Benayahu, Y.; Kashman, Y. Eunicellin diterpenes from two Kenyan soft corals. J. Nat. Prod. 2005, 68, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.L.; Su, J.H.; Wen, Z.H.; Hsu, C.H.; Chen, B.W.; Dai, C.F.; Kuo, Y.H.; Sheu, J.H. Simplexins A–I, eunicellin-based diterpenoids from the soft coral Klyxum simplex. J. Nat. Prod. 2009, 72, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.L.; Su, J.H.; Lu, Y.; Chen, B.W.; Huang, C.Y.; Wen, Z.H.; Kuo, Y.H.; Sheu, J.H. Simplexins J–O, eunicellin-based diterpenoids from a Dongsha Atoll soft coral Klyxum simplex. Bull. Chem. Soc. Jpn. 2011, 84, 626–632. [Google Scholar] [CrossRef]
- Chen, B.W.; Chao, C.H.; Su, J.H.; Tsai, C.W.; Wang, W.H.; Wen, Z.H.; Huang, C.Y.; Sung, P.J.; Wu, Y.C.; Sheu, J.H. Klysimplexins I–T, eunicellin-based diterpenoids from the cultured soft coral Klyxum simplex. Org. Biomol. Chem. 2011, 9, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.J.; Chen, B.W.; Wen, Z.H.; Huang, C.Y.; Dai, C.F.; Su, J.H.; Wu, Y.C.; Sheu, J.H. Klymollins A–H, bioactive eunicellin-based diterpenoids from the formosan soft coral Klyxum molle. J. Nat. Prod. 2011, 74, 2467–2471. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.Y.; Hsu, F.J.; Tai, C.J.; Wei, W.C.; Yang, N.S.; Sheu, J.H. Klymollins T–X, bioactive eunicellin-based diterpenoids from the soft coral Klyxum molle. Mar. Drugs 2014, 12, 3060–3071. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.W.; Wu, Y.C.; Chiang, M.Y.; Su, J.H.; Wang, W.H.; Fan, T.Y.; Sheu, J.H. Eunicellin-based diterpenoids from the cultured soft coral Klyxum simplex. Tetrahedron 2009, 65, 7016–7022. [Google Scholar] [CrossRef]
- Tseng, W.R.; Huang, C.Y.; Tsai, Y.Y.; Lin, Y.S.; Hwang, T.L.; Su, J.H.; Sung, P.J.; Dai, C.F.; Sheu, J.H. New cytotoxic and anti-inflammatory steroids from the soft coral Klyxum flaccidum. Bioorg. Med. Chem. Lett. 2016, 26, 3253–3257. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.R.; Huang, C.Y.; Chen, B.W.; Tsai, Y.Y.; Shih, S.P.; Hwang, T.L.; Dai, C.F.; Wang, S.Y.; Sheu, J.H. New bioactive steroids from the soft coral Klyxum flaccidum. RSC Adv. 2015, 5, 12546–12554. [Google Scholar] [CrossRef]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, G.R.; Caton, M.C.; Nova, M.P.; Parandoosh, Z. Assessment of the Alamar Blue assay for cellular growth and viability in vitro. J. Immunol. Methods 1997, 204, 205–208. [Google Scholar] [CrossRef]
- Ahmed, A.F.; Wen, Z.H.; Su, J.H.; Hsieh, Y.T.; Wu, Y.C.; Hu, W.P.; Sheu, J.H. Oxygenated cembranoids from a Formosan soft coral Sinularia gibberosa. J. Nat. Prod. 2008, 71, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Nakagawa, T.; Mitsuhashi, H. Marine terpenes and terpenoids. I. Structures of four cembrane-type diterpenes: Sarcophytol-A, sarcophytol-A acetate sarcophytol-B, and sarcophytonin-A, from the soft coral, Sarcophyton glaucum. Chem. Pharm. Bull. 1979, 27, 2382–2387. [Google Scholar] [CrossRef]
- Duh, C.Y.; Hou, R.S. Cytotoxic cembranoids from the soft corals Sinularia gibberosa and Sarcophyton trocheliophorum. J. Nat. Prod. 1996, 59, 595–598. [Google Scholar] [CrossRef]
- Barfield, M.; Spear, R.J.; Sternhell, S. Interproton spin-spin coupling across a dual path in 2,5-dihydrofurans and phthalans. J. Am. Chem. Soc. 1975, 97, 5160–5167. [Google Scholar] [CrossRef]
- Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. High-field FT NMR application of Mosher’s method. The absolute configurations of marine terpenoids. J. Am. Chem. Soc. 1991, 113, 4092–4096. [Google Scholar] [CrossRef]
- Randazzo, A.; Bifulco, G.; Giannini, C.; Bucci, M.; Debitus, C.; Cirino, G.; Gomez-Paloma, L. Halipeptins A and B: Two novel potent anti-inflammatory cyclic depsipeptides from the Vanuatu marine sponge Haliclona species. J. Am. Chem. Soc. 2001, 123, 10870–10876. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Kondo, K.; Osabe, K.; Mitsuhashi, H. Marine terpenes and terpenoids. V. Oxidation of sarcophytol A, a potent anti-tumor-promoter from the soft coral Sarcophyton glaucum. Chem. Pharm. Bull. 1988, 36, 2331–2341. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.T.; Wang, S.K.; Duh, C.Y. Cembranoids from the Dongsha Atoll soft coral Lobophytum crassum. Mar. Drugs 2011, 9, 2705–2716. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Hirase, T. Marine Terpenes and Terpenoids. XI: Structures of new dihydrofuranocembranoids isolated from a Sarcophyton sp. Soft coral of Okinawa. Chem. Pharm. Bull. 1990, 38, 2442–2445. [Google Scholar] [CrossRef]
- Ahmed, A.F.; Tai, S.H.; Wen, Z.H.; Su, J.H.; Wu, Y.C.; Hu, W.P.; Sheu, J.H. A C-3 methylated isocembranoid and 10-oxocembranoids from a formosan soft coral, Sinularia grandilobata. J. Nat. Prod. 2008, 71, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Kashman, Y.; Carmely, S.; Groweiss, A. Further cembranoid derivatives from the Red Sea soft corals Alcyonium flaccidum and Lobophytum crassum. J. Org. Chem. 1981, 46, 3592–3596. [Google Scholar] [CrossRef]
- Huang, H.C.; Ahmed, A.F.; Su, J.H.; Chao, C.H.; Wu, Y.C.; Chiang, M.Y.; Sheu, J.H. Crassolides A–F, cembranoids with a trans-fused lactone from the soft coral Sarcophyton crassocaule. J. Nat. Prod. 2006, 69, 1554–1559. [Google Scholar] [CrossRef] [PubMed]
- Bryan, N.S.; Grisham, M.B. Methods to detect nitric oxide and its metabolites in biological samples. Free Radic. Biol. Med. 2007, 43, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.H.; Kuo, P.M.; Chien, S.C.; Shyur, L.F.; Wang, S.Y. Effects of Chamaecyparis formosensis Matasumura extractives on lipopolysaccharide-induced release of nitric oxide. Phytomedicine 2007, 14, 675–680. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | 1 a | 2 b | 3 c | 4 a | 5 c | 6 b | 7 c | 8 c | 9 a |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 147.8 (C) | 147.7 (C) | 155.9 (C) | 146.3 (C) | 154.6 (C) | 80.9 (C) | 146.0 (C) | 147.4 (C) | 146.9 (C) |
| 2 | 121.0 (CH) d | 121.2 (CH) | 67.2 (CH) | 124.3 (CH) | 122.7 (CH) | 129.2 (CH) | 123.7 (CH) | 121.2 (CH) | 120.8 (CH) |
| 3 | 89.7 (CH) | 91.1 (CH) | 126.3 (CH) | 136.2 (CH) | 70.7 (CH) | 138.0 (CH) | 138.3 (CH) | 89.1 (CH) | 121.0 (CH) |
| 4 | 74.2 (C) | 74.6 (C) | 137.6 (C) | 72.9 (C) | 75.2 (C) | 71.9 (C) | 73.0 (C) | 74.6 (C) | 136.6 (C) |
| 5 | 41.6 (CH2) | 40.5 (CH2) | 39.0 (CH2) | 45.5 (CH2) | 38.6 (CH2) | 43.8 (CH2) | 39.0 (CH2) | 41.3 (CH2) | 39.1 (CH2) |
| 6 | 22.1 (CH2) | 25.2 (CH2) | 24.2 (CH2) | 121.3 (CH) | 22.3 (CH2) | 22.3 (CH2) | 24.2 (CH2) | 21.8 (CH2) | 25.6 (CH2) |
| 7 | 127.0 (CH) | 64.6 (CH) | 125.2 (CH) | 141.4 (CH) | 127.0 (CH) | 128.6 (CH) | 127.6 (CH) | 126.3 (CH) | 126.5 (CH) |
| 8 | 132.1 (C) | 59.9 (C) | 133.2 (C) | 72.5 (C) | 133.9 (C) | 132.7 (C) | 131.9 (C) | 132.6 (C) | 133.8 (C) |
| 9 | 36.8 (CH2) | 36.7 (CH2) | 36.9 (CH2) | 43.6 (CH2) | 39.0 (CH2) | 39.0 (CH2) | 36.7 (CH2) | 36.8 (CH2) | 36.6 (CH2) |
| 10 | 25.1 (CH2) | 23.6 (CH2) | 24.3 (CH2) | 23.5 (CH2) | 24.2 (CH2) | 23.8 (CH2) | 24.3 (CH2) | 24.5 (CH2) | 24.8 (CH2) |
| 11 | 59.7 (CH) | 59.0 (CH) | 59.7 (CH) | 126.7 (CH) | 125.6 (CH) | 126.9 (CH) | 61.3 (CH) | 59.2 (CH) | 59.9 (CH) |
| 12 | 61.9 (C) | 60.3 (C) | 61.3 (C) | 132.8 (C) | 131.9 (C) | 136.1 (C) | 64.7 (C) | 60.4 (C) | 62.9 (C) |
| 13 | 72.8 (CH) | 71.9 (CH) | 71.6 (CH) | 37.3 (CH2) | 44.1 (CH2) | 36.1 (CH2) | 71.6 (CH) | 72.7 (CH) | 75.2 (CH) |
| 14 | 85.8 (CH) | 85.0 (CH) | 115.6 (CH) | 122.0 (CH) | 70.6 (CH) | 29.9 (CH2) | 122.9 (CH) | 84.9 (CH) | 68.1 (CH) |
| 15 | 26.6 (CH) | 25.7 (CH) | 27.6 (CH) | 31.6 (CH) | 27.8 (CH) | 75.1 (C) | 32.1 (CH) | 25.7 (CH) | 28.2 (CH) |
| 16 | 22.2 (CH3) | 20.9 (CH3) | 23.7 (CH3) | 22.6 (CH3) | 22.8 (CH3) | 24.5 (CH3) | 22.1 (CH3) | 22.2 (CH3) | 24.0 (CH3) |
| 17 | 21.2 (CH3) | 20.6 (CH3) | 24.5 (CH3) | 22.6 (CH3) | 23.5 (CH3) | 24.7 (CH3) | 22.3 (CH3) | 20.9 (CH3) | 25.4 (CH3) |
| 18 | 23.1 (CH3) | 22.2 (CH3) | 15.7 (CH3) | 29.7 (CH3) | 25.1 (CH3) | 27.8 (CH3) | 30.0 (CH3) | 22.6 (CH3) | 16.7 (CH3) |
| 19 | 16.7 (CH3) | 16.8 (CH3) | 15.2 (CH3) | 29.6 (CH3) | 15.1 (CH3) | 14.7 (CH3) | 15.1 (CH3) | 16.7 (CH3) | 15.2 (CH3) |
| 20 | 15.5 (CH3) | 16.0 (CH3) | 15.2 (CH3) | 17.6 (CH3) | 17.1 (CH3) | 14.8 (CH3) | 15.5 (CH3) | 16.0 (CH3) | 16.1 (CH3) |
| OAc | 169.9 (C) | 170.7 (C) | 169.9 (C) | ||||||
| 21.0 (CH3) | 21.1 (CH3) | 20.6 (CH3) |
| # | 1 a | 2 b | 3 c | 4 a | 5 c |
|---|---|---|---|---|---|
| 2 | 5.59 br s | 5.65 br s | 5.70 d (10.0) | 6.19 d (16.4) | 5.41 d (7.6) |
| 3 | 4.61 d (4.8) d | 4.78 d (5.0) | 5.25 d (10.0) | 5.83 d (16.4) | 4.34 d (7.6) |
| 5 | 1.48 m; 1.85, m | 1.82 m; 1.92 m | 2.10 m; 2.24 m | 2.31 2H, d (6.8) | 1.55 m; 1.86 m |
| 6 | 2.02 m; 2.15, m | 1.59 m; 1.86 m | 2.10 m; 2.27 m | 5.56 dd (15.6, 6.8) | 2.11 m; 2.34 m |
| 7 | 5.38 dd (5.2, 5.2) | 3.07 dd (6.0, 2.5) | 4.83 br d (6.0) | 5.52 d (15.6) | 4.99 dd (6.0, 6.0) |
| 9 | 2.04 m; 2.09 m | 2.10 m; 1.39 m | 2.16 m; 2.20 m | 1.58 m; 1.67 m | 1.98 m; 2.15 m |
| 10 | 1.76 m; 1.83 m | 1.54 m; 1.95 m | 1.61 m; 1.86 m | 2.01 m; 2.37 m | 2.14 m; 2.18 m |
| 11 | 3.15 dd (6.4, 2.0) | 2.98 d (7.5) | 2.47 dd (7.2, 2.0) | 5.11 dd (7.2, 7.2) | 4.93 dd (6.8, 6.0) |
| 13 | 3.58 br s | 5.21 s | 5.52 d (10.4) | 2.71 2H, d (8.0) | 2.27 m; 2.37 m |
| 14 | 4.78 br d (4.8) | 5.05 d (5.0) | 5.03 d (10.4) | 5.48 dd (8.0, 5.6) | 4.78 dd (5.6, 5.6) |
| 15 | 2.25 sept (6.8) | 2.17 m | 2.78 sept (6.8) | 2.53 sept (6.8) | 2.48 m |
| 16 | 0.94 3H, d (6.8) | 1.05 3H, d (6.5) | 1.05 3H, d (6.8) | 1.09 3H, d (6.8) | 1.06 3H, d (6.8) |
| 17 | 1.12 3H, d (6.8) | 1.10 3H, d (6.5) | 1.09 3H, d (6.8) | 1.10 3H, d (6.8) | 1.12 3H, d (6.8) |
| 18 | 0.98 3H, s | 1.05 3H, s | 1.80 3H, s | 1.27 3H, s | 1.14 3H, s |
| 19 | 1.48 3H, s | 1.24 3H, s | 1.55 3H, s | 1.16 3H, s | 1.55 3H, s |
| 20 | 1.18 3H, s | 1.42 3H, s | 1.22 3H, s | 1.60 3H, s | 1.68 3H, s |
| OAc | 1.91 3H, s | 2.10 3H, s |
| # | 6 a | 7 b | 8 b | 9 c |
|---|---|---|---|---|
| 2 | 5.61 d (16.0) d | 6.24 d (16.0) | 5.60 br s | 6.25 d (11.2) |
| 3 | 6.10 d (16.0) | 5.75 d (16.0) | 4.57 d (5.2) | 5.85 d (11.2) |
| 5 | 1.51 m; 2.01 m | 2.10 m; 2.24 m | 1.63 m; 1.89 m | 2.00 2H, m |
| 6 | 2.22 m; 2.39 m | 2.10 m; 2.27 m | 2.16 m; 2.18 m | 1.97 m, 2.06 m |
| 7 | 5.34 dd (7.5, 7.5) | 5.03 dd (6.0, 6.0) | 5.36 dd (5.5, 5.5) | 5.03 dd (6.0, 6.0) |
| 9 | 1.95 m; 2.20 m | 2.16 m; 2.20 m | 2.04 m; 2.09 m | 1.95 m, 2.11 m |
| 10 | 2.07 m; 2.24 m | 1.61 m; 1.86 m | 1.76 m; 1.83 m | 1.43 2H, m |
| 11 | 5.19 br d (9.0) | 2.67 m | 2.89 dd (6.0, 3.6) | 3.08 dd (6.0, 6.0) |
| 13 | 2.13 m; 2.19 m | 4.59 dd (8.0, 8.0) | 5.16 d (2.0) | 3.72 d (6.0) |
| 14 | 1.63 m; 2.11 m | 5.10 d (8.0) | 5.00 dd (5.2, 2.0) | 4.60 d (6.0) |
| 15 | 2.52 m | 2.13 m | 2.76 m | |
| 16 | 1.21 3H, s | 1.04 3H, d (6.8) | 1.03 3H, d (6.8) | 1.06 d (6.8) |
| 17 | 1.13 3H, s | 1.07 3H, d (6.8) | 1.08 3H, d (6.8) | 1.28 d (6.8) |
| 18 | 1.40 3H, s | 1.35 3H, s | 1.00 3H, s | 1.59 3H, s |
| 19 | 1.62 3H, s | 1.60 3H, s | 1.59 3H, s | 1.32 3H, s |
| 20 | 1.67 3H, s | 1.42 3H, s | 1.39 3H, s | 1.34 3H, s |
| 15-OH | 2.60 s | |||
| 13-OH | 1.74 d (8.0) | |||
| OAc | 2.06 3H,s |
| Compound | HT-29 | A549 | K562 | P388 |
|---|---|---|---|---|
| 1 | – a | – a | – a | – a |
| 2 | – a | 16.5 | 34.6 | – a |
| 3 | – a | – a | – a | – a |
| 4 | – a | – a | 44.9 | – a |
| 5 | – a | – a | – a | – a |
| 6 | – a | 21.4 | – a | – a |
| 8 | – a | 49.4 | 47.4 | 34.6 |
| 9 | 41.9 | – a | – a | 25.9 |
| 10 | – a | – a | – a | – a |
| Fluorouracil | 8.5 | 110 | 31.5 | 5.5 |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, A.F.; Tsai, C.-R.; Huang, C.-Y.; Wang, S.-Y.; Sheu, J.-H. Klyflaccicembranols A–I, New Cembranoids from the Soft Coral Klyxum flaccidum. Mar. Drugs 2017, 15, 23. https://doi.org/10.3390/md15010023
Ahmed AF, Tsai C-R, Huang C-Y, Wang S-Y, Sheu J-H. Klyflaccicembranols A–I, New Cembranoids from the Soft Coral Klyxum flaccidum. Marine Drugs. 2017; 15(1):23. https://doi.org/10.3390/md15010023
Chicago/Turabian StyleAhmed, Atallah F., Chia-Ruei Tsai, Chiung-Yao Huang, Sheng-Yang Wang, and Jyh-Horng Sheu. 2017. "Klyflaccicembranols A–I, New Cembranoids from the Soft Coral Klyxum flaccidum" Marine Drugs 15, no. 1: 23. https://doi.org/10.3390/md15010023