The natural products isolated from
Aplidium species can be classified into two groups: nitrogenous and non-nitrogenous compounds. Numerous non-nitrogen containing metabolites have been isolated, mainly linear or cyclic prenyl hydroquinones and quinones, which are known as meroterpenes and present cytotoxic activities [
3,
11,
12]. Although, there are examples of metabolites that contains nitrogen in the prenylated quinone moiety, such as conicaquinone A, which has an unusual 1,1-dioxo-1,4-thiazine ring added to the quinone moiety. These meroterpenes are the focus of the current review, which describes the structures, biological activities and
13C-NMR data of 53 prenylated hydroquinones and quinones isolated from
Aplidium, thus, highlighting the structural diversity generated in this class of natural products and their potential in drug discovery. A large number of compounds (48) have been examined by
13C-NMR spectroscopy so considerable
13C chemical shift data have accumulated; however, a compilation of
13C data for meroterpenes from
Aplidium has not been available to date. The assignment of carbon from known compounds is simple and straightforward, providing
13C data of appropriate model compounds are available. Thus, we provide a tabulation of
13C-NMR data to provide easy access to previously published data on meroterpenes. This paper reports also a brief description of the most characteristics
1H-NMR chemical shift data.
Prenyl quinones and hydroquinones are metabolites of mixed biogenesis that originate from intra- and inter-molecular cyclizations and/or rearrangements, thus, producing macrocyclic or polycyclic skeletons that are often linked to amino acids or taurine residues [
11]. In this report, we have focused on quinones and hydroquinones from Ascidians of the genus
Aplidium and their related compounds: rossinones, longithorones, longithorols, floresolides, scabellones, conicaquinones, aplidinones, thiaplidiaquinones, and conithiaquinones. The described compounds were isolated from organisms belonging to 10 identified and three unidentified species of
Aplidium. The determination of their structures and biological activities are also summarized, and a compilation of
13C-NMR spectral data is provided. The intent of the present review is to provide the researchers who have isolated a prenyl quinones and hydroquinones from
Aplidium with a quick means of deciding whether the compound is known or new, and to allow them to establish a structure by comparison of
13C-NMR data. Additionally,
1H-NMR spectroscopic studies are often sufficient to establish structures unequivocally; although, there are isomeric molecules that
13C-NMR data were important to resolve structure problems.
Aplidium are clearly prolific producers of bioactive prenylated quinones and hydroquinones in the marine environment, and many other
Aplidium species need to be investigated.
1.1. Quinones
Quinones can be derived by the oxidation of appropriate phenolic compounds, with 1,2-dihydroxybenzenes and 1,4-dihydroxybenzenes, yielding
ortho-quinones and
para-quinones respectively. Therefore, quinones can be formed from phenolics compounds by either the acetate or shikimate pathways, affording a catechol or quinol system. A range of quinone derivatives and related structures that contain a terpenoid fragment or shikimate-derived portion are also wide spread. For example, ubiquinones (coenzyme Q) have important biochemical functions in electron transport systems for respiration [
13].
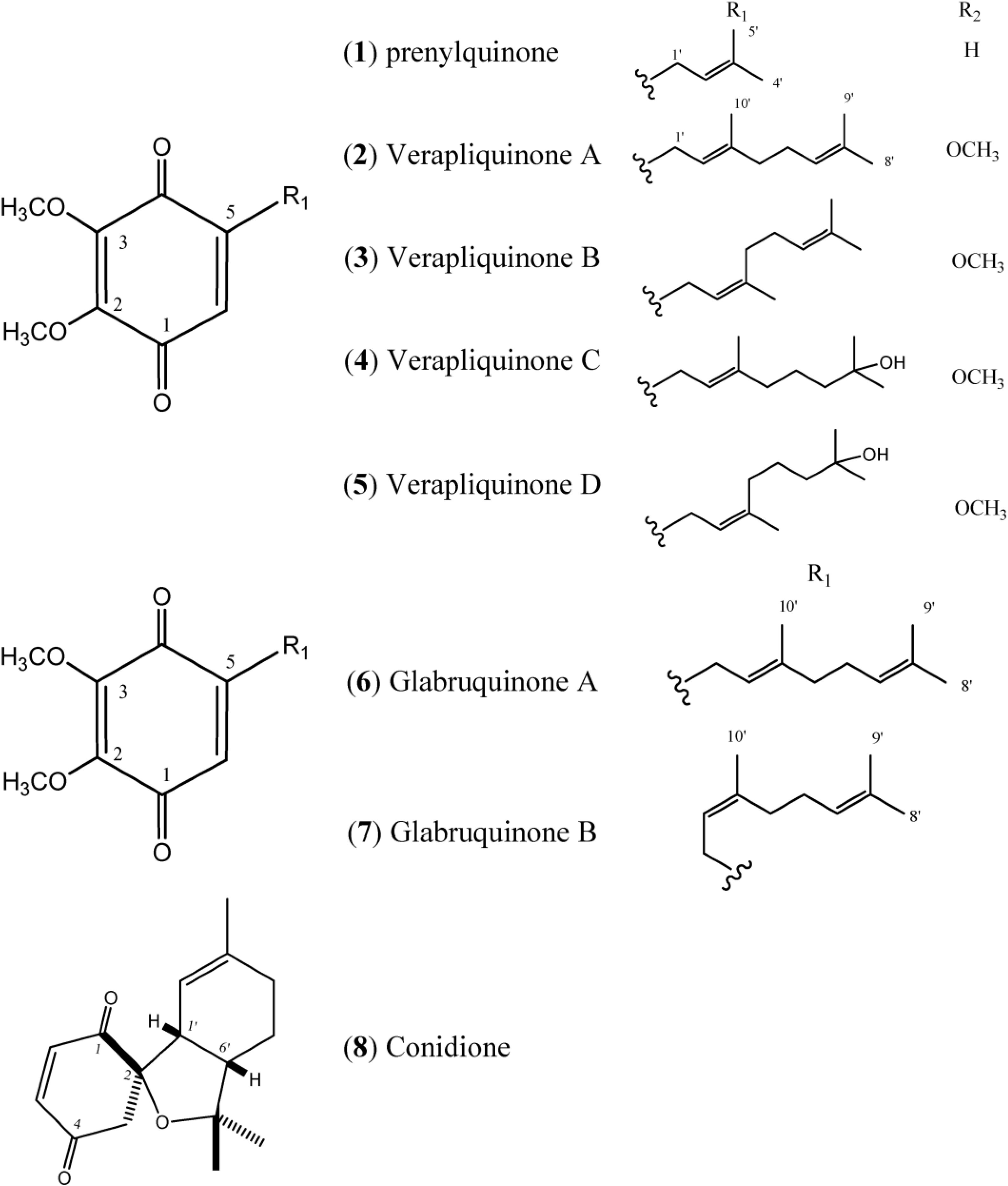
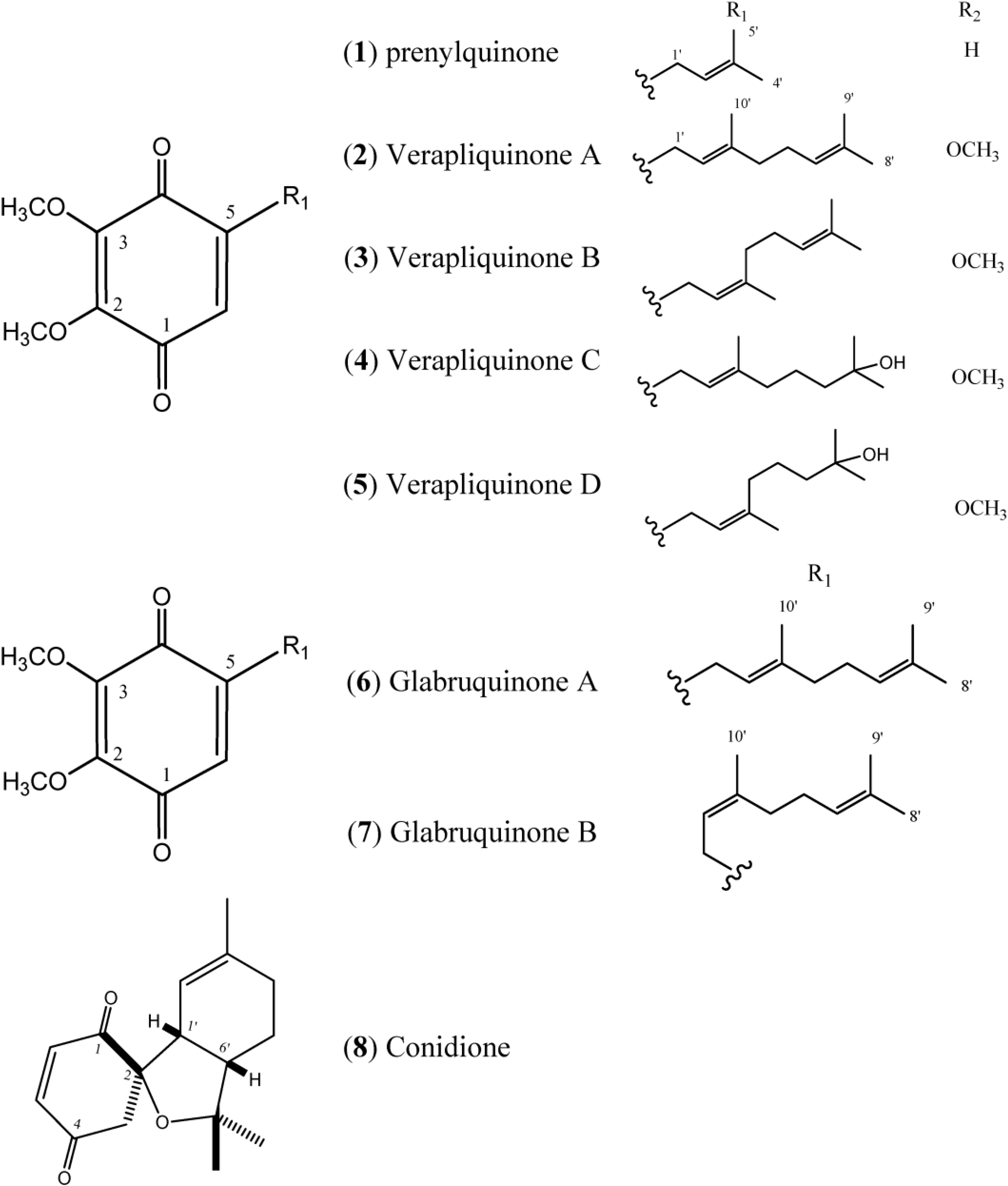
Figure 1 describes quinones that occur in
Aplidium.
Figure 1.
Structures of Quinones (1–8).
Figure 1.
Structures of Quinones (1–8).
The simple linear Prenylquinone (
1) was isolated from
Aplidium californicum [
14]. The verapliquinones A–D (
2–
5) were isolated from an unidentified
Aplidium sp. (Ascidiacea) collected off the Breton coast. Verapliquinones A/B and C/D were characterized in a mixture of
E,
Z-isomers by NMR spectroscopy. The NMR data revealed that Verapliquinone B and D had a neryl group at C-2 instead of geranyl [
15]. Davis and co-workers [
16] reported a simple and versatile route to 1,4-benzoquinones based on the Claisen rearrangement, and applied to the synthesis of verapliquinones A and B, which had not previously been synthesized [
16]. Chan and co-workers (2011) also reported verapliquinone A from
A. scabellum [
17].
Glabruquinone A (
6) (or desmethylubiquinone Q
2) and Glabruquinone B (
7) were isolated from
Aplidium glabrum and synthesized. The main difference between
6 and
7 is that compound
6 contains a geranyl side chain instead of neryl as in
7 [
18]. Glabruquinone A (
6) displayed cancer preventive activity in the anchorage-independent transformation assay against mouse JB6 P
+ Cl 41 cells transformed with an epidermal growth factor, with an inhibition of the number of colonies C
50 (INCC
50) value of 7.3 µM. The INCC
50 values for
6 were 12.7, 17.5, and 50.5 µM against HCT-116, MEL-28, and HT-460 human tumor cells, respectively. Compound
6, at 10 µM, increased the UVB-induced p53 transcriptional activity of JB6 P
+ Cl 41 cells [
18,
19]. Glabruquinone A was also evaluated
in vivo on mice inoculated with Ehrlich carcinoma tumors and found to inhibit tumorgrowth. Compound
6 inhibited the phenotype expression of HT-460, HCT-116, and SK-MEL-28 human tumor cells and induced apoptosis of these cell lines, as well as that of HL-60 and THP-1 tumor cells [
20].
Conidione (
8), a cyclic diprenyl quinone, was isolated from
A. conicum but was unstable, and this instability prevented bioassays from being performed [
21].
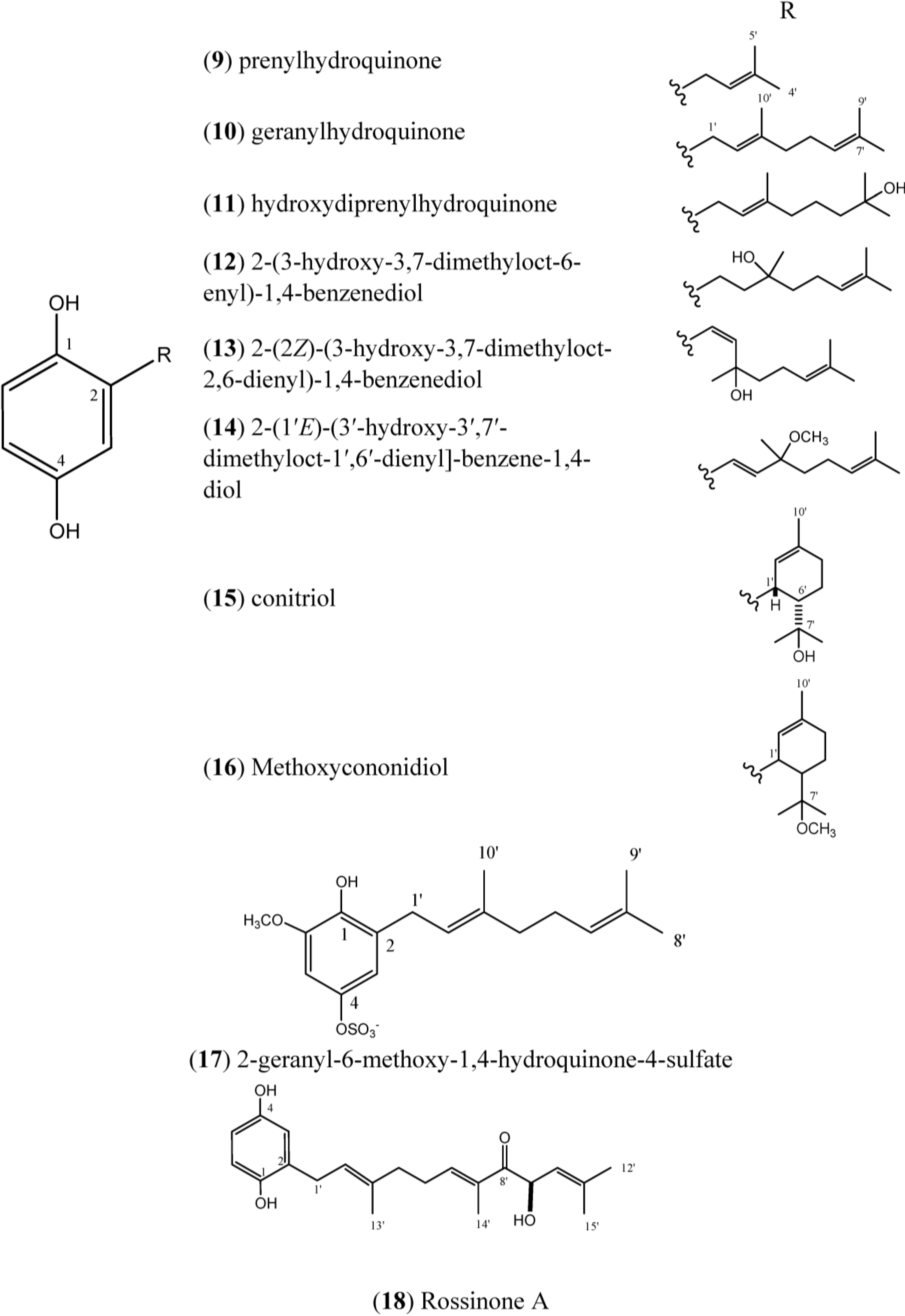
1.2. Hydroquinones
Prenylhydroquinone (
9),
Figure 2, isolated from
A. californicum, exhibited activity
in vivo against P388 lymphocytic leukemia. The potential cancer protective properties of prenylhydroquinone (
9) were also evaluated by employing a modified-Ames assay for mutagenicity against
Salmonella typhimurium; when prenylhydroquinone was added to experiments, the mutagenic effects of the carcinogens were drastically reduced [
14].
Compound
9 was able to form stable semiquinone radicals according to Cotelle and collaborators, and in presence of glutathione,
9 was involved in a redox cycle with the consumption of oxygen. This process triggered the formation of free radicals and decreased the glutathione content, which is considered to be one of the major defenses against oxidative damage. Even although not fully elucidated, the antitumor properties of
9 can be correlated with its redox properties and reactivity toward glutathione [
22]. Prenylhydroquinone also inhibits superoxide anion production in rat alveolar macrophages and in the xanthine/xanthine oxidase system. The antioxidant activity of
9 may be attributed to a direct reaction of the superoxide anion rather than to an enzymatic inhibition or a membrane signal transfer [
23].
Figure 2.
Structures of Hydroquinones (9–18).
Figure 2.
Structures of Hydroquinones (9–18).
Prenylhydroquinone (
9) and geranylhydroquinone (
10), isolated from
Aplidium sp., have exibited antiproliferative activity (IC
50: 41 and 9.5 μM, respectively) in a P388 murine leukemia cell-line. Geranylhydroquinone (
10) at doses up to 30 μM was inactive against the solid tumor cell lines A375 (human melanoma), A549 (human breast), HepG2 (human hepatic), and HT-29 (human colon), as well as normal human liver cells (WRL-68) [
24]. Both prenylhydroquinone (
9) and geranylhydroquinone (
10) exhibited anti-inflammatory activity in an
in vitro anti-inflammatory assay with activated human peripheral blood neutrophils by inhibing superoxide production. Compound
10 was also tested in the DPPH radical scavenging assay, and was considered inactive [
24].
Geranylhydroquinone (
10) has been reported to occur in various species of
Aplidium. It has been isolated from two unidentified
Aplidium sp [
25,
26],
A. antillense [
27],
A. nordmani [
28],
A. savignyi [
29], and
A. coninum [
21]. Compound
10 has also been isolated from plants [
30,
31,
32].
Geranylhydroquinone exhibited cytotoxicity against the leukemia cell lines of Rous sarcoma and mammary cincinoma
in vivo [
33]. Cytotoxic activity was also observed for
10 against P-388 leukemia (IC
50 0.034 µg/mL) and KB human epidermoid carcinoma cells (IC
50 4.3 µg/mL). Geranylhydroquinone (
10) has also demonstrated antibacterial activity, with a minimum inhibitory concentration (MIC) of 64 µg/mL against
Staphylococus aureus and
S. faecalis, and an MIC of 128 µg/mL against
Serratia marcescens. The minimum bactericide concentrations (MBCs) were determined to be 2, 1, and ˃4 µg/mL, respectively [
27]. Additionally, Compound
10 was more potent than two standard antioxidants in terms of its inhibitory effects on lipid peroxide formation in rat liver microsomes and on soy-bean 15-lipoxygenase [
34].
Geranylhydroquinone (
10) and hydroxydiprenylhydroquinone (
11) displayed significant cytotoxicity against four tumor cell lines, in particular, against P-388 mouse lymphoma suspension culture (IC
50 0.81 and 4.5 µM, respectively) indicating that the hydroxylation of the prenyl chain in
11 may result in a marginal decrease in its cytotoxicity [
26].
The compound 2-(3ʹ-hydroxy-3ʹ,7ʹ-dimethyloct-6ʹ-enyl)-1,4-benzenediol (
12) was isolated from de
A. conicum and
A. savignyi [
14,
22]. The compound 2-(2ʹ
Z)-(3ʹ-hydroxy-3ʹ,7ʹ-dimethyloct-2ʹ,6ʹ-dienyl)-1,4-benzenediol (
13) was isolated from
A. savignyi [
29].
The compounds 2-(1ʹ
E)-(3ʹ-hydroxy-3ʹ,7ʹ-dimethyloct-1ʹ,6ʹ-dienyl]-benzene-1,4-diol (
14) and conitriol (
15) were isolated from
A. conicum but exhibited chemical instability. This instability prevented pharmacological assays from being conducted [
21].
Methoxyconidiol (
16) was isolated from
A. aff. densum [
35] and its effects were tested on sea urchin embryos during cell division; compound
16 disturbed the mitotic spindle assembly leading to a cell cycle arrest during the metaphase/anaphase transition [
36]. The antibacterial activity of
16 on
Escherichia coli and
Micrococcus luteus was also evaluated by microtiter broth dilution method; it had no antibacterial effect [
37]. Methoxyconidiol inhibited egg division, with IC
50 values of 0.80 μM for
Paracentrotus lividus eggs and 4.30 μM for
Sphaerechinus granularis eggs. When tested against the human carcinoma cell lines: MCF7 (breast), PA1 (ovary), PC3 (prostate), CEM-WT (acute lymphoblastic leukemia), and L-929 murine immortalized cells, as well as normal human fibroblasts,
16 was non-toxic to all these cell lines with an IC
50 > 100 µM [
35,
37]. Methoxyconidiol caused antimitotic action during the first division of sea urchin embryos, and the mechanism of action of methoxyconidiol may be mediated by the disruption of microtubule dynamics [
29]. Hence, Simon-Levert
et al. 2010 [
37] concluded that methoxyconidiol was ineffective against human cancer cells but effective in sea urchin cells. This finding could be explained by a difference between these two type of cells in either membrane permeability and/or intracellular transport of
16.
The compound 2-geranyl-6-methoxy-1,4-hydroquinone-4-sulfate (
17) was isolated from
A. scabellum and inhibited superoxide production by PMA-stimulated human neutrophils
in vitro, with an IC
50 of 21 μM. To determine the effect of different treatments on cell survival, drug-treated neutrophils were stained with the fluorescent markers for necrosis (propidium iodide) and apoptosis (Annexin V-FITC), and analyzed by flow cytometry. Treatment with
17 had no effect on neutrophil viability, but the results indicated that
17 did inhibit neutrophil superoxide production [
17].
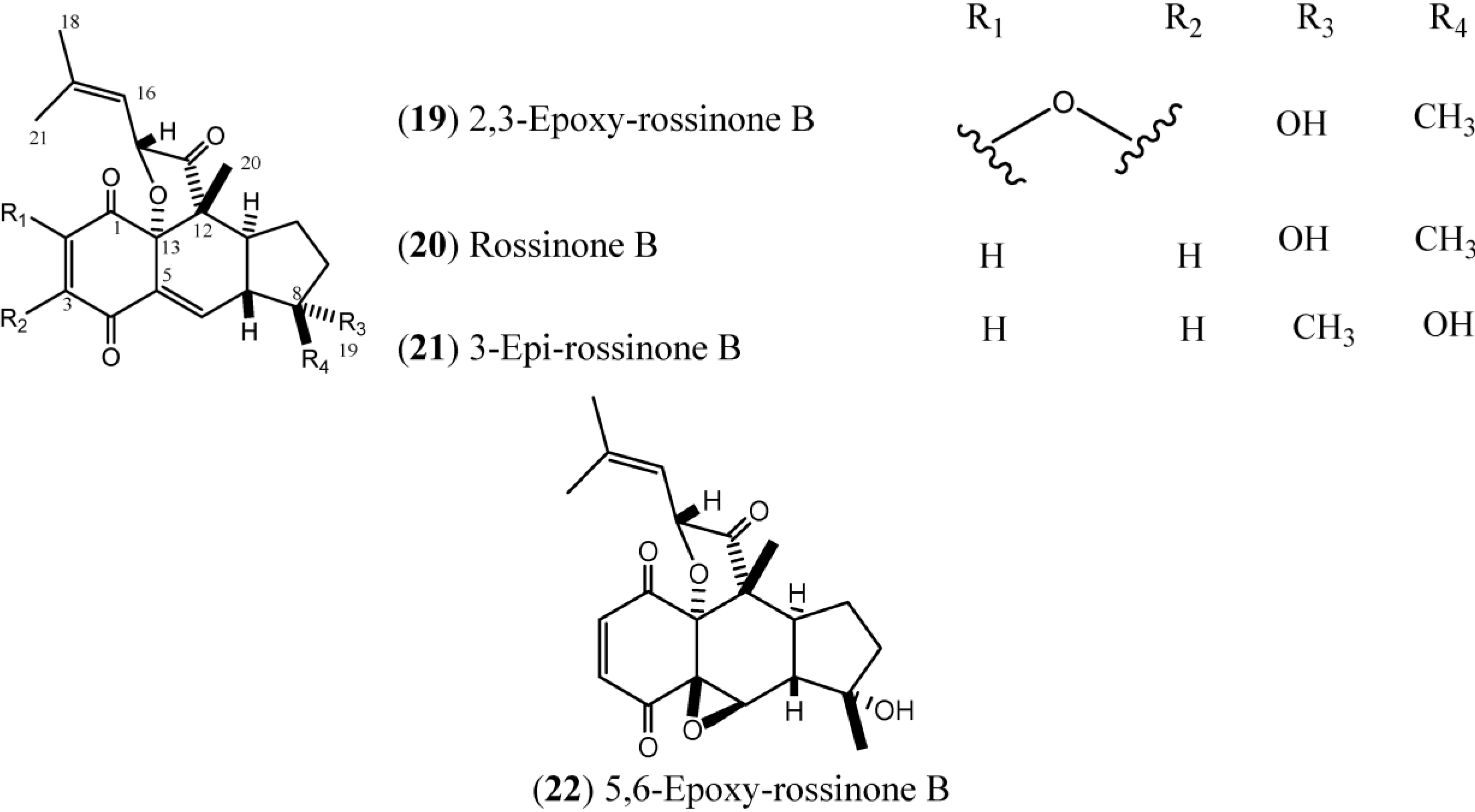
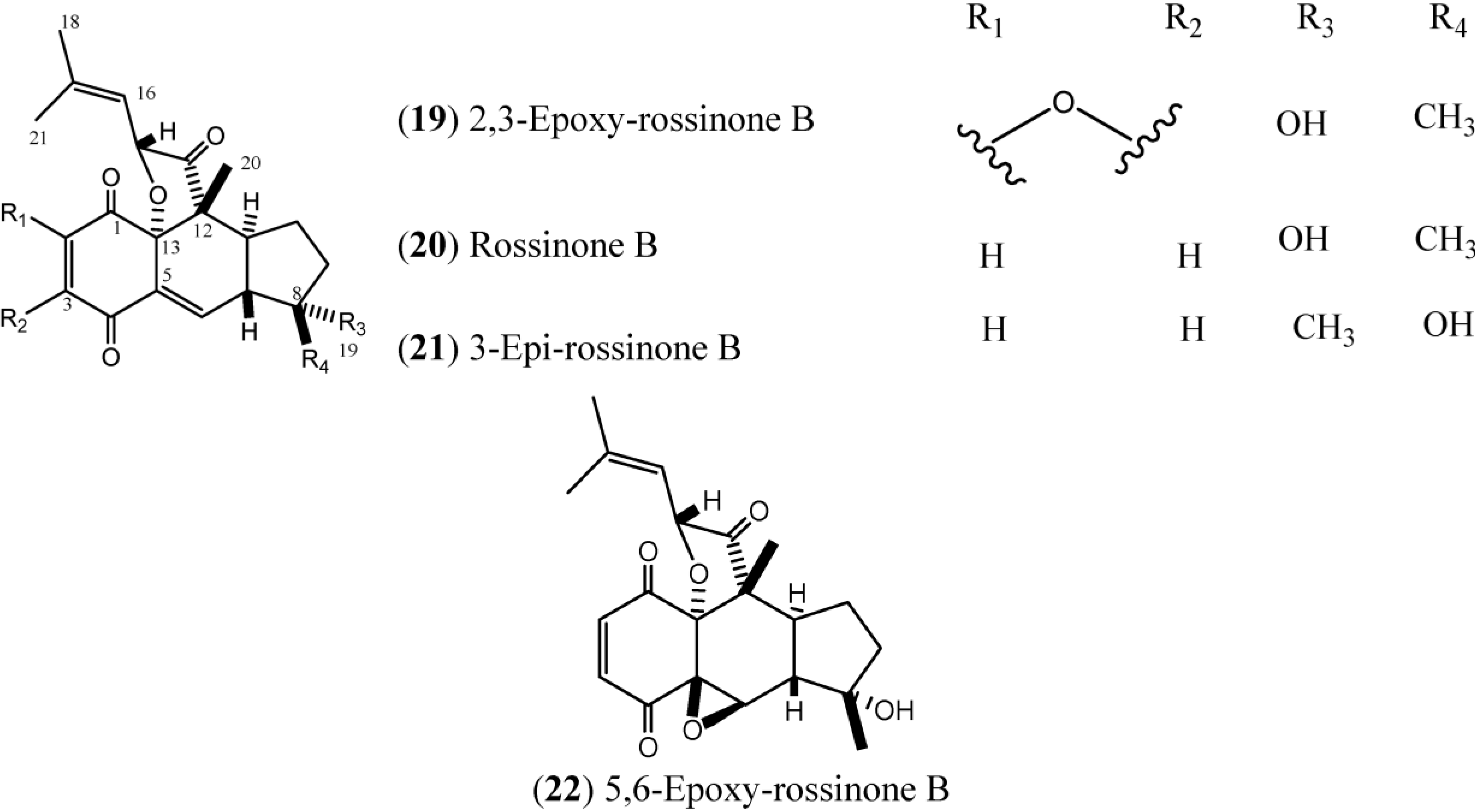
1.3. Rossinones
Rossinones (
Figure 3), particularly rossinone B (
20) and its derivatives, are linearly fused in a 6,6,5-ring core of rossinone B, which is an extremely rare skeleton, known for only three plant-derived natural products. Rossinone B was isolated for the first time from an unidentified Antarctic species of
Aplidium and then from
A. fuegiense [
24,
38]. Therefore, the isolation of
20 extended the evolutionary range of the requisite biosynthetic terpene cyclase(s) from Plant kingdom to Animalia [
24]. To date, five rossinones (
18–
22) have been discovered.
Figure 3.
Structures of Rossinones (19–22).
Figure 3.
Structures of Rossinones (19–22).
Rossinones A (
18) and B (
20) exhibited anti-inflammatory activity in an
in vitro anti-inflammatory assay with activated human peripheral blood neutrophils by inhibiting superoxide production. However, in the DPPH radical scavenging assay,
18 and
20 were found to be inactive (at doses up to 30 μM), indicating that these rossinones are considerably less effective as superoxide scavengers than as suppressors of superoxide production by neutrophils. Rossinones A and B have exhibited selective antiviral activity against the DNA virus HSV-1,
versus the RNA virus PV-1, with both compounds exhibiting antiviral activity at 2 μg/disk. Both compounds also exhibited antimicrobial activities against
Bacillus subtilis and the fungus
Trichophyton mentagrophytes [
24].
Rossinone B (
20) exhibited potent antiproliferative activity toward the P388 murine leukemia cell-line (IC
50: 0.084 μM), while rossinone A (
18) was less active (IC
50: 0.39 μM). Rossinone A (
18) was inactive against the solid tumor cell lines A375 (human melanoma), A549 (human breast), HepG2 (human hepatic), and HT-29 (human colon) at doses up to 30 μM, while rossinone B (
20) presented good activity toward the SH-SY5Y neuroblastoma cell line (IC
50: 1.6 μM) and modest activity against the A375, A549, and HT-29 cell lines with IC
50 values of 11, 30, and 30 μM, respectively. Additionally,
18 and
20 exhibited antiproliferative activity against normal human liver cells (WRL-68) at concentrations up to 30 μM [
24].
The rossinones 2,3-epoxy-rossinone B, rossinone B, 3-epi-rossinone B, and 5,6-epoxy-rossinone (
19–
22) isolated from
A. fuegiense, mainly rossinone B was proven to be involved in the whole-colony chemical defense of
A. fuegiense, repelling sea stars and amphipods [
39].
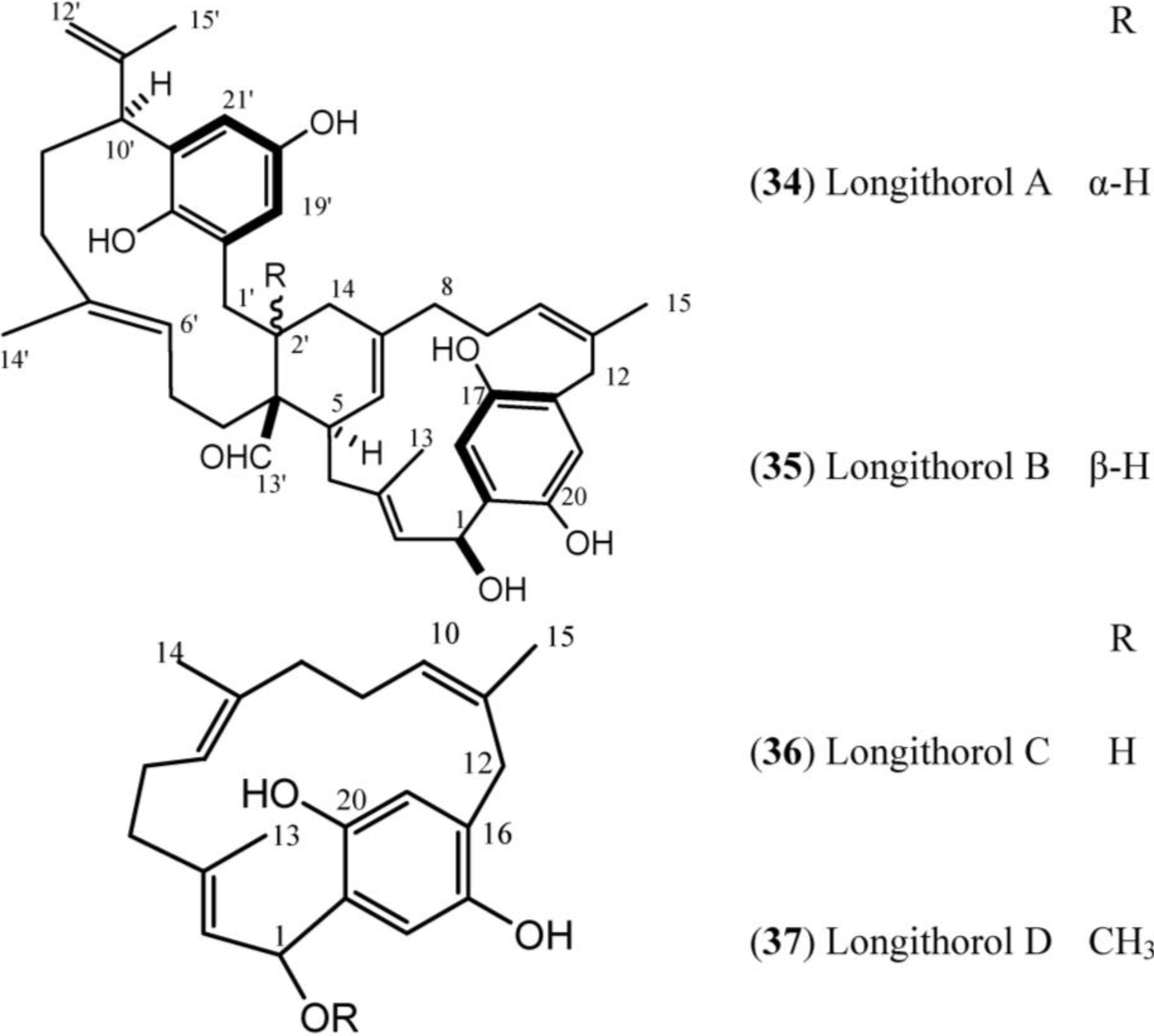
1.4. Longithorones, Longithorols and Floresolides
Longithorones (
Figure 4) and longithorols (
Figure 5) are unique farnesylated quinones/hydroquinones isolated from
A. longithorax (Monniot). Their complex structures are characterized by the presence of a metacycophane and/or paracyclophane system built in a farnesyl quinone or hydroquinone formally by the rarely encountered cyclization of farnesyl quinones/hydroquinones [
12]. One special characteristic of those compounds is the atropisomerism that is caused by the restricted rotations of their macrocyclic rings. Eleven compounds have been isolated (longithorones A–K), including monomeric prenylated quinones (
24–
26) and dimeric prenylated quinones (
23 and
27–
31), and the cyclofarnesylated quinones (longithorones J and K;
32–
33) [
40,
41]. The biosynthesis of dimeric longithorones, which have been supposed to originate by both intra- and intermolecular Diels-Alder reactions, has been speculated about. Fusion of the two farnesyl-quinone units can be envisioned as arising via a Diels-Alder cycloaddition of suitably unsaturated precursors, whereas rings B and C could arise by a transannular Diels-Alder reaction. The co-isolation of the monomers provides some support for this proposal [
12].
Longithorone A (
23) displayed cytotoxicity against P388 murine leukemia cells with an IC
50 of ~10 mg·mL
−1 [
42]. In addition, the longithorone J (
32) was tested for cytotoxicity against the cell lines SHSY5Y (human neuroblastoma), HEK293T (SV40 T antigen transformed human embryonal kidney cells) and A549 (human non-small cell lung carcinoma). Compound
32 did not exhibit any cytotoxicity in the A549 cell assay when tested at 2 and 20 mg/mL. However,
32 displayed minimal activity at 20 mg/mL against the SHSY5Y and HEK293T cells, with cell deaths of 28% and 16%, respectively [
43].
Zacarian and collaborators synthesized the highly rigid macrocyclic carbon skeleton of longithorone C (
25) by exploiting quadrupolar interactions as synthetic strategy [
44]. Despite the fact that cyclophanes have been extensively synthesized and evaluated because of their unique physical and chemical properties, examples of the isolation and total synthesis of cyclophane-containing natural products are rare and challenging. This has attracted the interest of synthetic chemists in search of new and efficient strategies for syntheses with a reduced number of steps [
45,
46].
Figure 4.
Structures of Longithorones (23–33).
Figure 4.
Structures of Longithorones (23–33).
Figure 5.
Structures of Longithorol (34–37).
Figure 5.
Structures of Longithorol (34–37).
Longithorols A (
34) and B (
35) are prenylated paracyclophane and metacyclophane hydroquinones, and longithorols C (
36) and D (
37) are para-substituted cyclofarnesylated hydroquinones. The hydroquinones (longithorols A–D,
34–
37) were also isolated from
A. longithorax, moreover
34 and
35 were isolated as their pentaacetate forms because of their instability [
47,
48].
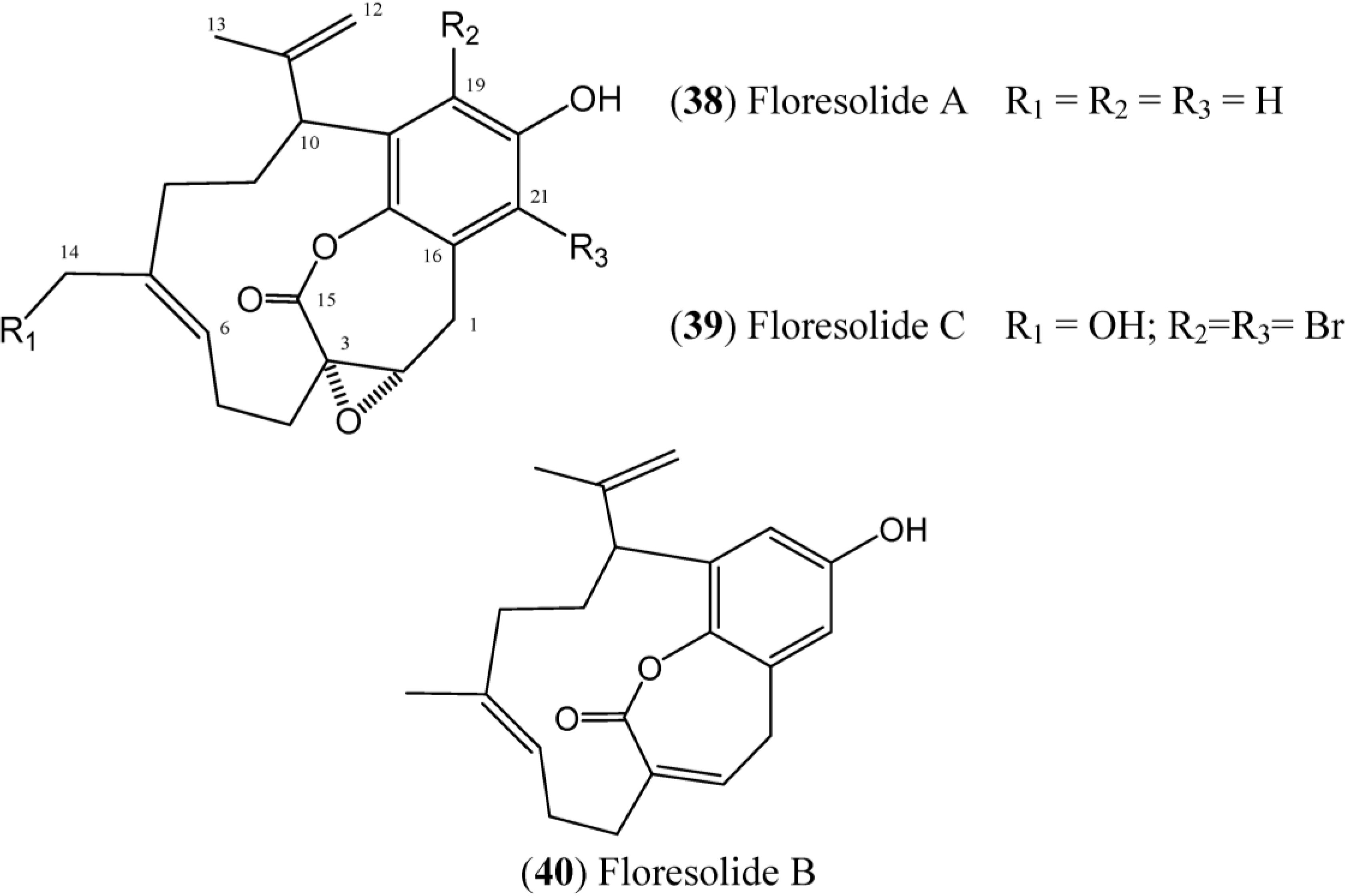
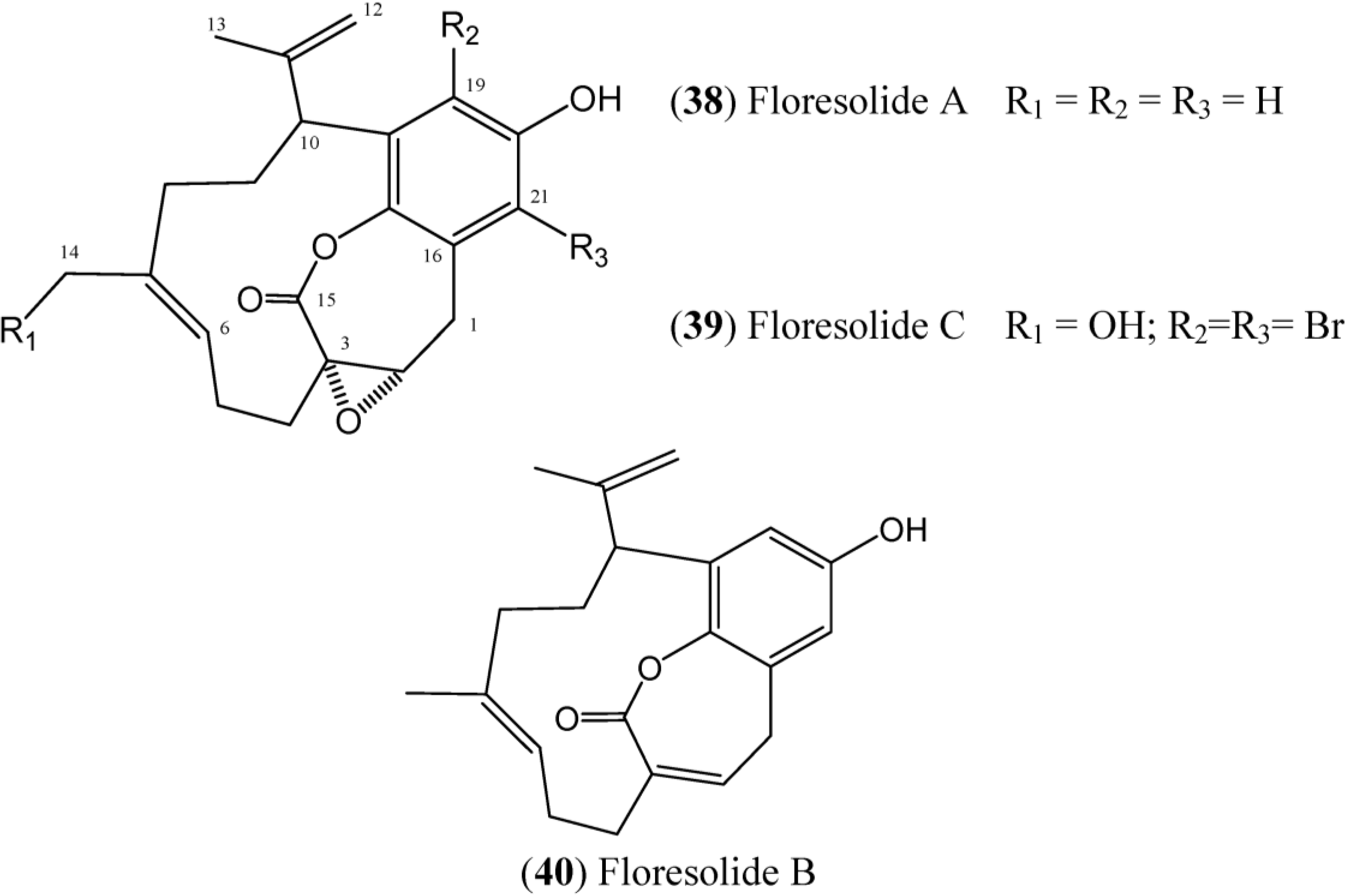
Floresolides are monomeric cyclofarnesylated hydroquinones with an endocyclic ε-lactone. They are members of longithorone/longithorol class of meroterpenes. Floresolides A–C (
38–
40) (
Figure 6) have been isolated from
Aplidium sp. collected in Indonesian. All of these floresolides exhibited moderate cytotoxicity against KB tumor cells [
49]. The synthesis of floresolide B (
40) hydroquinone lactone core was performed by employing ring-closing metathesis approach by Briggs and Dudley [
50]. The total synthesis of racemic floresolide B was reported by Nicolaou and Xu, who used an olefin metathesis-based strategy for the formation of the macrocyclic lactone portion [
51,
52].
Figure 6.
Structures of floresolides floresolides (38–40).
Figure 6.
Structures of floresolides floresolides (38–40).
1.7. Structural Elucidation
A compilation of the
13C chemical shifts of the quinones (
2–
4 and
6–
8), hydroquinones (
9–
12 and
14–
17), rossinones (
18–
22), longithorones (
23–
29 and
32–
33), longithorols (
34–
37), floresolides (
38–
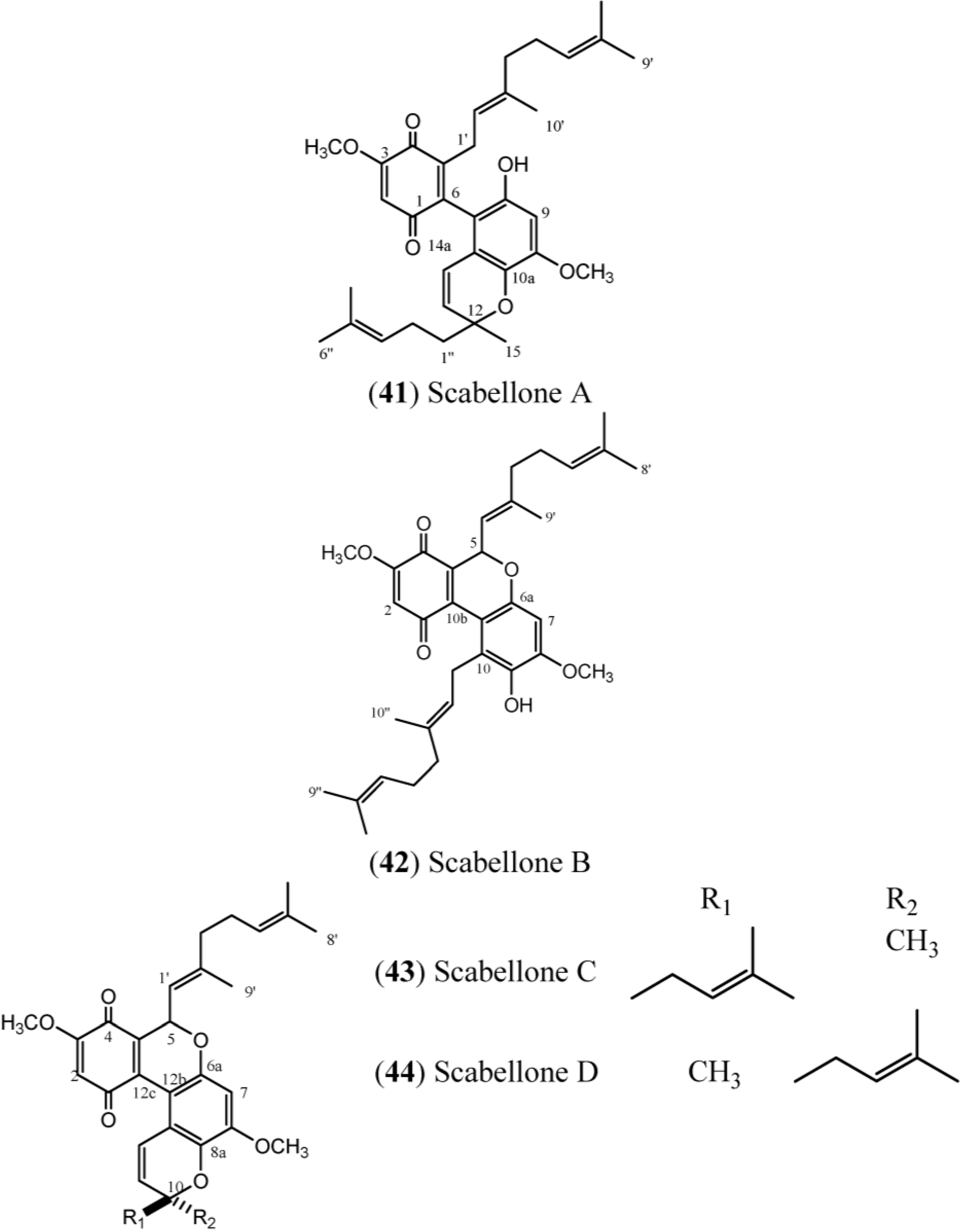
40), scabellones (
41–
44), conicaquinones (
45–
46), aplidinones (
47–
49), thiaplidiaquinones (
50–
51), and conithiaquinones (
52–
53), is given in
Table 1,
Table 2,
Table 3,
Table 4,
Table 5,
Table 6 and
Table 7. The solvent (A = CDCl
3, B = Me
2CO-
d6, C = C
6D
6, D = CD
3OD, E = CD
2Cl
2 and F = DMSO-
d6) and references are shown in the first line of the tables. Inspection of the
13C-NMR data of compounds
2–
4 and
6–
8 (prenylated quinones) compared with
9–
12 and
14–
17 (prenylated hydroquinones) (
Table 1 and
Table 2) reveals the introduction of hydroxyl groups in the C-1 and C-4 in
9–
12 and
14–
17 (instead of carbonyls group as in
2–
4 and
6–
8) results in upfield signals at the α carbon. Thus, the quinone family skeleton can be recognized by the typical carbonyl signals.
Table 1.
13C chemical shifts (δ in ppm) of quinones derivatives. Ref. = References, n.d. = not detected.
Table 1.
13C chemical shifts (δ in ppm) of quinones derivatives. Ref. = References, n.d. = not detected.
| Carbon | 2 | 3 | 4 | 6 | 7 | 8 |
|---|
| Solvent | A | A | A | A | A | A |
| Ref. | [15] | [15] | [15] | [18] | [18] | [21] |
| 1 | 182.2 | 182.2 | 187.7 | 184.3 | 184.3 | 197.9 |
| 2 | 146.4 | 146.4 | n.d. | 144.9 | 144.9 | 86.9 |
| 3 | 132.8 | 132.8 | 132.8 | 145.1 | 145.1 | 50.4 |
| 4 | 187.7 | 187.7 | 187.7 | 184.5 | 184.5 | 196.6 |
| 5 | 107.1 | 107.1 | 107.1 | 146.9 | 146.9 | 141.1 |
| 6 | 158.9 | 158.9 | 158.9 | 130.4 | 130.4 | 140.7 |
| 1ʹ | 27.1 | 26.9 | 27.2 | 27.1 | 27.2 | 41.8 |
| 2ʹ | 117.7 | 118.4 | 117.9 | 117.7 | 117.7 | 116.2 |
| 3ʹ | 140.0 | 140.0 | 139.9 | 140.1 | 140.1 | 139.0 |
| 4ʹ | 39.6 | 31.8 | 39.9 | 39.7 | 32.0 | 29.2 |
| 5ʹ | 26.4 | 26.3 | 22.5 | 26.5 | 26.5 | 21.9 |
| 6ʹ | 123.8 | 123.8 | 43.4 | 123.9 | 123.9 | 46.0 |
| 7ʹ | 131.8 | 131.8 | 70.9 | 131.9 | 131.9 | 84.7 |
| 8ʹ | 25.6 | 25.6 | 29.3 | 25.7 | 25.7 | 24.2 |
| 9ʹ | 17.7 | 16.1 | 29.3 | 17.7 | 17.7 | 23.7 |
| 10ʹ | 16.1 | 23.5 | 16.0 | 16.2 | 22.7 | 29.0 |
| OCH3 | 56.2 | 56.2 | 56.3 | 61.3 | 61.3 | - |
| OCH3 | - | - | - | 61.2 | 61.2 | - |
The prenylated quinone family skeleton can also be recognized by the typical
1H-NMR signals; for example, in the case of compound
2, the doublet of the triplet at δ
H 6.43 (H-3,
J = 2.8 and 2.2 Hz) and a doublet at δ
H 5.86 (H-5,
J = 2.8), and the presence of a methoxyl group at δ
H 3.80 s are typical of the pattern for a 2,6-substituted quinone nucleus [
15]. On the other hand, the prenyl hydroquinone core (2-substituted hydroquinone) can be evinced by the typical
1H-NMR signals for three aromatic hydrogens corresponding to two with an
ortho coupling at δ
H 6.62 (d,
J = 8.5 Hz) and δ
H 6.52 (dd,
J = 8.4 and 2.85 Hz) and one with a
meta coupling at δ
H 6.55 (d,
J = 2.5 Hz), as observed in
10 [
29].
Table 2.
13C chemical shifts (δ in ppm) of hydroquinones derivatives.
Table 2.
13C chemical shifts (δ in ppm) of hydroquinones derivatives.
| Carbon | 9 | 10 | 11 | 12 | 14a | 15 | 16 | 17 |
|---|
| Solvent | B | A | A | A | C | C | A | D |
| Ref. | [14] | [29] | [25] | [29] | [21] | [21] | [35] | [17] |
| 1 | 148.3 | 148.6 | 147.8 | 147.9 | 147.3 | 148.6 | 149.4 | 141.2 |
| 2 | 129.3 | 147.7 | 138.2 | 123.8 | 135.9 | 130.8 | 130.5 | 129.2 |
| 3 | 116.9 | 116.6 | 121.8 | 116.6 | 113.2 | 118.7 | 118.8 | 114.1 |
| 4 | 150.8 | 148.2 | 149.7 | 148.9 | 150.3 | 148.8 | 148.4 | 146.4 |
| 5 | 113.5 | 114.3 | 116.5 | 113.8 | 115.2 | 114.2 | 114.3 | 103.6 |
| 6 | 116.1 | 116.2 | 116.4 | 116.9 | 116.8 | 118.4 | 118.2 | 148.6 |
| 1ʹ | 28.9 | 30.0 | 29.0 | 41.4 | 125.2 | 34.3 | 34.4 | 29.1 |
| 2ʹ | 123.6 | 120.8 | 113.6 | 24.5 | 124.5 | 125.4 | 125.6 | 123.9 |
| 3ʹ | 132.1 | 128.1 | 128.1 | 73.8 | 77.6 | 133.7 | 133.8 | 136.7 |
| 4ʹ | 17.5 | 40.0 | 42.9 | 41.7 | 40.6 | 31.5 | 31.8 | 41.0 |
| 5ʹ | - | 26.7 | 22.4 | 22.9 | 23.0 | 20.7 | 20.6 | 27.9 |
| 6ʹ | - | 123.6 | 39.7 | 124.0 | n. d. | 49.1 | 47.4 | 125.5 |
| 7ʹ | - | 122.0 | 71.4 | 132.4 | 131.1 | 75.3 | 79.5 | 132.2 |
| 8ʹ | - | 26.6 | 29.2 | 25.7 | 25.8 | 32.5 | 25.7 | 25.9 |
| 9ʹ | - | 18.1 | 29.2 | 17.7 | 22.1 | 23.3 | 19.1 | 17.8 |
| 10ʹ | - | 15.5 | 16.2 | 26.6 | 17.7 | 22.9 | 23.3 | 16.3 |
| OCH3 | - | - | - | - | 49.9 | - | 49.1 | 56.6 |
Table 3.
13C chemical shifts (δ in ppm) of rossinones derivatives.
Table 3.
13C chemical shifts (δ in ppm) of rossinones derivatives.
| Carbon | 18 | Carbon | 19 | 20 | 21 | 22 |
|---|
| Solvent | A | Solvent | A | A | A | A |
| Ref. | [24] | Ref. | [38] | [38] | [38] | [38] |
| 1 | 147.4 | 1 | n.d. | 190.3 | 189.9 | 191.0 |
| 2 | 128.1 | 2 | 55.5 | 139.0 | 139.0 | 140.0 |
| 3 | 116.3 | 3 | 56.4 | 141.2 | 141.4 | 141.7 |
| 4 | 149.7 | 4 | 191.8 | 185.0 | 184.4 | 193.9 |
| 5 | 113.9 | 5 | 134.3 | 134.6 | 133.3 | 55.9 |
| 6 | 116.2 | 6 | 145.0 | 144.2 | 144.0 | 61.4 |
| 1ʹ | 28.7 | 7 | 49.7 | 50.5 | 50.5 | 48.5 |
| 2ʹ | 123.5 | 8 | 78.0 | 78.0 | 78.3 | 79.3 |
| 3ʹ | 135.5 | 9 | 40.2 | 40.2 | 40.5 | 39.8 |
| 4ʹ | 38.1 | 10 | 21.0 | 21.1 | 20.9 | 21.1 |
| 5ʹ | 26.8 | 11 | 39.3 | 39.5 | 39.5 | 34.4 |
| 6ʹ | 145.2 | 12 | 49.2 | 49.3 | 48.9 | 49.8 |
| 7ʹ | 134.0 | 13 | 83.8 | 82.8 | 82.8 | 83.7 |
| 8ʹ | 201.8 | 14 | 212.2 | 213.0 | 212.3 | 212.2 |
| 9ʹ | 69.8 | 15 | 77.4 | 77.2 | 77.4 | 77.0 |
| 10ʹ | 122.9 | 16 | 118.7 | 118.6 | 117.8 | 118.5 |
| 11ʹ | 138.6 | 17 | 142.3 | 142.5 | 143.0 | 142.7 |
| 12ʹ | 25.8 | 18 | 25.9 | 25.9 | 25.6 | 25.9 |
| 13ʹ | 15.8 | 19 | 27.1 | 27.1 | 26.7 | 27.4 |
| 14ʹ | 11.8 | 20 | 8.7 | 8.8 | 8.6 | 11.0 |
| 15ʹ | 18.3 | 21 | 18.9 | 18.7 | 18.6 | 18.7 |
Table 4.
13C chemical shifts of (δ in ppm) longithorones derivatives.
Table 4.
13C chemical shifts of (δ in ppm) longithorones derivatives.
| Carbon | 23 | 24 | 25 | 26 | 27 | 28 | 29 | 32 | 33 |
|---|
| Solvent | C | C | C | C | C | A | C | A | A |
| Ref. | [40] | [40] | [40] | [40] | [40] | [40] | [40] | [41] | [41] |
| 1 | 27.6 | 29.1 | 28.7 | 29.5 | 27.8 | 29.2 | 29.0 | 28.2 | 28.3 |
| 2 | 125.3 | 122.1 | 120.9 | 121.7 | 125.9 | 123.9 | 122.9 | 122.3 | 120.5 |
| 3 | 136.3 | 138.5 | 139.1 | 139.6 | 135.8 | 136.9 | 136.0 | 135.3 | 137.6 |
| 4 | 40.5 | 39.0 | 27.9 | 39.7 | 40.1 | 39.0 | 42.5 | 39.5 | 39.4 |
| 5 | 36.3 | 24.1 | 24.1 | 25.4 | 37.4 | 36.8 | 37.3 | 23.9 | 23.9 |
| 6 | 119.7 | 122.6 | 123.2 | 131.5 | 120.4 | 121.8 | 122.0 | 123.9 | 123.0 |
| 7 | 134.7 | 135.2 | 135.2 | 131.4 | 134.4 | 138.1 | 137.6 | 134.9 | 135.3 |
| 8 | 37.4 | 39.5 | 39.7 | 44.1 | 37.8 | 36.1 | 35.0 | 39.8 | 39.6 |
| 9 | 26.6 | 29.2 | 30.3 | 43.5 | 26.5 | 26.0 | 27.4 | 29.0 | 28.6 |
| 10 | 126.6 | 127.4 | 126.7 | 126.6 | 126.6 | 127.2 | 126.4 | 128.2 | 128.8 |
| 11 | 132.5 | 130.9 | 132.1 | 132.6 | 132.3 | 130.9 | 131.7 | 133.8 | 130.5 |
| 12 | 35.6 | 32.1 | 32.9 | 17.7 | 35.4 | 31.7 | 33.1 | 29.0 | 36.9 |
| 13 | 15.3 | 14.6 | 22.2 | 15.4 | 15.1 | 15.4 | 15.2 | 14.9 | 14.8 |
| 14 | 32.9 | 15.6 | 16.0 | 14.2 | 37.3 | 37.4 | 37.8 | 16.1 | 16.2 |
| 15 | 27.6 | 26.5 | 27.2 | 25.9 | 27.5 | 26.2 | 27.1 | 22.8 | 22.5 |
| 16 | 149.4 | 148.3 | 149.0 | 151.7 | 149.5 | 146.9 | 148.3 | 38.0 | 46.5 |
| 17 | 187.2 | 187.8 | 187.2 | 187.6 | 186.4 | 187.4 | 187.9 | 71.5 | 200.7 |
| 18 | 131.5 | 130.2 | 132.9 | 149.3 | 131.5 | 129.5 | 129.8 | 147.1 | 136.4 |
| 19 | 148.1 | 147.8 | 149.4 | 129.6 | 147.8 | 148.0 | 148.1 | 138.4 | 151.7 |
| 20 | 187.0 | 188.1 | 187.4 | 188.8 | 187.0 | 188.1 | 187.0 | 196.8 | 197.1 |
| 21 | 133.6 | 132.4 | 133.1 | 133.1 | 133.5 | 132.5 | 132.5 | 42.0 | 42.0 |
| 1ʹ | 37.8 | - | - | - | 29.5 | 28.3 | 28.7 | - | - |
| 2ʹ | 30.7 | - | - | - | 39.2 | 44.7 | 39.1 | - | - |
| 3ʹ | 49.4 | - | - | - | 52.8 | 52.5 | 53.7 | - | - |
| 4ʹ | 27.6 | - | - | - | 30.9 | 30.1 | 33.1 | - | - |
| 5ʹ | 38.1 | - | - | - | 21.1 | 19.7 | 22.4 | - | - |
| 6ʹ | 126.1 | - | - | - | 130.4 | 129.2 | 130.6 | - | - |
| 7ʹ | 143.8 | - | - | - | 135.4 | 135.1 | 134.6 | - | - |
| 8ʹ | 34.3 | - | - | - | 40.5 | 39.9 | 39.8 | - | - |
| 9ʹ | 29.9 | - | - | - | 32.2 | 32.1 | 31.4 | - | - |
| 10ʹ | 130.4 | - | - | - | 43.4 | 43.2 | 43.3 | - | - |
| 11ʹ | 132.2 | - | - | - | 147.3 | 147.8 | 147.0 | - | - |
| 12ʹ | 34.5 | - | - | - | 111.1 | 110.9 | 111.3 | - | - |
| 13ʹ | 205.5 | - | - | - | 21.4 | 206.3 | 203.8 | - | - |
| 14ʹ | 30.9 | - | - | - | 15.4 | 15.7 | 14.8 | - | - |
| 15ʹ | 27.5 | - | - | - | 204.6 | 21.4 | 21.2 | - | - |
| 16ʹ | 155.7 | - | - | - | 151.2 | 151.5 | 151.8 | - | - |
| 17ʹ | 200.7 | - | - | - | 187.0 | 186.1 | 187.4 | - | - |
| 18ʹ | 56.3 | - | - | - | 151.9 | 152.2 | 152.5 | - | - |
| 19ʹ | 52.9 | - | - | - | 134.0 | 133.8 | 131.8 | - | - |
| 20ʹ | 201.2 | - | - | - | 187.1 | 188.5 | 187.6 | - | - |
| 21ʹ | 139.2 | - | - | - | 130.4 | 129.9 | 130.9 | - | - |
| 1-OCH3 | | - | - | - | - | - | - | - | - |
Table 5.
13C chemical shifts of (δ in ppm) longithorols and floresolides derivatives.
Table 5.
13C chemical shifts of (δ in ppm) longithorols and floresolides derivatives.
| Carbon | 34 | 35 | 36 | 37 | 38 | 39 | 40 |
|---|
| Solvent | E | E | F | F | A | A | A |
| Ref. | [47] | [47] | [48] | [48] | [49] | [49] | [49] |
| 1 | 67.1 | 67.2 | 71.6 | 81.2 | 33.9 | 34.0 | 30.1 |
| 2 | 129.0 | 129.6 | 130.0 | 127.4 | 63.7 | 62.5 | 137.8 |
| 3 | 135.5 | 135.1 | 131.4 | 133.9 | 59.1 | 58.3 | 136.1 |
| 4 | 39.9 | 40.4 | 38.5 | 38.5 | 33.4 | 32.6 | 31.4 |
| 5 | 37.4 | 38.4 | 23.9 | 23.8 | 24.0 | 23.2 | 25.0 |
| 6 | 119.2 | 119.6 | 120.8 | 121.0 | 123.4 | 126.7 | 124.6 |
| 7 | 137.6 | 137.2 | 134.1 | 134.2 | 137.4 | 141.2 | 135.8 |
| 8 | 36.7 | 37.3 | 38.9 | 38.8 | 39.3 | 35.4 | 37.4 |
| 9 | 24.6 | 24.7 | 25.4 | 26.0 | 29.6 | 25.0 | 29.3 |
| 10 | 126.6 | 126.5 | 125.7 | 125.8 | 43.2 | 44.4 | 41.5 |
| 11 | 131.5 | 131.4 | 133.1 | 133.0 | 149.3 | 143.9 | 149.2 |
| 12 | 36.8 | 36.8 | 33.2 | 32.9 | 109.4 | 111.8 | 108.9 |
| 13 | 16.3 | 16.2 | 15.3 | 15.1 | 21.9 | 22.5 | 22.4 |
| 14 | 39.1 | 37.8 | 16.4 | 16.1 | 14.9 | 58.3 | 13.6 |
| 15 | 27.5 | 27.5 | 27.4 | 27.3 | 169.1 | 167.8 | 166.6 |
| 16 | 133.7 | 133.7 | 125.7 | 126.3 | 137.3 | 128.8 | 135.7 |
| 17 | 148.0 | 148.0 | 147.4 | 147.1 | 142.7 | 142.9 | 144.2 |
| 18 | 120.9 | 120.8 | 112.4 | 114.3 | 128.9 | 133.7 | 132.7 |
| 19 | 132.8 | 132.7 | 127.5 | 124.8 | 114.0 | 112.2 | 112.6 |
| 20 | 143.6 | 143.6 | 147.4 | 147.4 | 153.0 | 150.1 | 152.6 |
| 21 | 126.3 | 126.2 | 117.1 | 117.2 | 114.2 | 109.5 | 112.1 |
| 1ʹ | 31.6 | 32.6 | - | - | - | - | - |
| 2ʹ | 44.4 | 39.4 | - | - | - | - | - |
| 3ʹ | 53.1 | 52.9 | - | - | - | - | - |
| 4ʹ | 25.8 | 30.6 | - | - | - | - | - |
| 5ʹ | 19.9 | 20.9 | - | - | - | - | - |
| 6ʹ | 126.0 | 126.4 | - | - | - | - | - |
| 7ʹ | 133.9 | 133.9 | - | - | - | - | - |
| 8ʹ | 40.7 | 40.6 | - | - | - | - | - |
| 9ʹ | 31.1 | 31.3 | - | - | - | - | - |
| 10ʹ | 45.6 | 45.8 | - | - | - | - | - |
| 11ʹ | 149.5 | 149.3 | - | - | - | - | - |
| 12ʹ | 109.8 | 110.0 | - | - | - | - | - |
| 13ʹ | 208.7 | 205.9 | - | - | - | - | - |
| 14ʹ | 15.4 | 15.2 | - | - | - | - | - |
| 15ʹ | 22.4 | 22.5 | - | - | - | - | - |
| 16ʹ | 138.4 | 138.1 | - | - | - | - | - |
| 17ʹ | 143.0 | 142.8 | - | - | - | - | - |
| 18ʹ | 136.6 | 136.1 | - | - | - | - | - |
| 19ʹ | 120.8 | 120.8 | - | - | - | - | - |
| 20ʹ | 148.7 | 148.9 | - | - | - | - | - |
| 21ʹ | 119.4 | 119.4 | - | - | - | - | - |
| 1-OCH3 | - | - | - | 55.4 | - | - | - |
Table 6.
13C chemical shifts (δ in ppm) of scabellone derivatives.
Table 6.
13C chemical shifts (δ in ppm) of scabellone derivatives.
| Carbon | 41 | Carbon | 42 | Carbon | 43 | 44 |
|---|
| Solvent | A | Solvent | A | Solvent | A | A |
| Ref. | [17] | Ref. | [17] | Ref. | [17] | [17] |
| 1 | 186.4 | 1 | 182.6 | 1 | 185.4 | 184.9 |
| 2 | 107.6 | 2 | 107.3 | 2 | 107.1 | 107.2 |
| 3 | 158.3 | 3 | 157.8 | 3 | 158.3’ | 158.0 |
| 4 | 181.5 | 4 | 178.7 | 4 | 179.0 | 178.7 |
| 5 | 145.7 | 4a | 130.8 | 4a | 131.4 | 131.1 |
| 6 | 138.4 | 5 | 67.6 | 5 | 67.7 | 67.7 |
| 7 | 109.1 | 6a | 151.4 | 6a | 151.6 | 151.3 |
| 8 | 145.7 | 7 | 98.4 | 7 | 101.2 | 101.0 |
| 9 | 101.4 | 8 | 150.0 | 8 | 153.0 | 152.5 |
| 10 | 149.4 | 9 | 139.2 | 8a | 138.3 | 137.9 |
| 10a | 136.2 | 10 | 126.8 | 10 | 77.8 | 77.4 |
| 12 | 77.5 | 10a | 111.0 | 11 | 126.6 | 127.4 |
| 13 | 131.3 | 10b | 137.6 | 12 | 123.8 | 123.6 |
| 14 | 120.1 | 1ʹ | 116.9 | 12a | 120.2 | 120.5 |
| 14a | 120.4 | 2ʹ | 144.3 | 12b | 107.6 | 107.1 |
| 15 | 25.4 | 3ʹ | 39.7 | 12c | 133.9 | 133.6 |
| 1ʹ | 26.6 | 4ʹ | 26.3 | 13 | 24.5 | 25.9 |
| 2ʹ | 118.0 | 5ʹ | 123.6 | 1ʹ | 117.0 | 117.0 |
| 3ʹ | 138.1 | 6ʹ | 131.7 | 2ʹ | 144.3 | 144.0 |
| 4ʹ | 39.2 | 7ʹ | 17.6 | 3ʹ | 39.7 | 39.4 |
| 5ʹ | 26.3 | 8ʹ | 25.6 | 4ʹ | 26.2 | 26.1 |
| 6ʹ | 124.1 | 9ʹ | 17.2 | 5ʹ | 123.6 | 123.7 |
| 7ʹ | 131.2 | 1ʹʹ | 26.5 | 6ʹ | 131.7 | 131.5 |
| 8ʹ | 17.3 | 2ʹʹ | 124.2 | 7ʹ | 17.7 | 17.6 |
| 9ʹ | 25.4 | 3ʹʹ | 137.1 | 8ʹ | 25.6 | 25.8 |
| 10ʹ | 15.6 | 4ʹʹ | 39.8 | 9ʹ | 17.2 | 17.2 |
| 1ʹʹ | 39.6 | 5ʹʹ | 26.2 | 1ʹʹ | 41.1 | 38.5 |
| 2ʹʹ | 22.2 | 6ʹʹ | 123.8 | 2ʹʹ | 23.2 | 22.4 |
| 3ʹʹ | 124.0 | 7ʹʹ | 131.6 | 3ʹʹ | 124.2 | 124.3 |
| 4ʹʹ | 131.5 | 8ʹʹ | 17.5 | 4ʹʹ | 131.7 | 131.5 |
| 5ʹʹ | 17.3 | 9ʹʹ | 25.5 | 5ʹʹ | 17.7 | 17.6 |
| 6ʹʹ | 25.4 | 10ʹʹ | 16.5 | 6ʹʹ | 25.7 | 25.8 |
| 3-OCH3 | 56.2 | 3-OCH3 | 56.1 | 3-OCH3 | 56.2 | 56.5 |
| 10-OCH3 | 56.2 | 8-OCH3 | 56.1 | 8-OCH3 | 56.2 | 56.5 |
The presence in the
13C-NMR data of 4 olefinic resonances for two trisubstituted double bonds clearly suggests a linear diprenyl chain in
2,
3,
6,
7,
10, and
17. The configurations at C-2ʹ and C-3ʹ for the linear diprenyl chain (as in
2,
3,
4,
6,
7,
10,
11, and
17) can be established on the bases of the relatively low-field and high-field signals for the C-10ʹ of the (
Z) compound and the sterically more congested (
E) compound, respectively [
15,
18]. Thus, this δ value allows for the determination of whether the diprenylated substituent is a neryl or geranyl group.
Table 7.
13C chemical shifts (δ in ppm) of conicaquinones, aplidinones, thiaplidiaquinones, and conithiaquinones.
Table 7.
13C chemical shifts (δ in ppm) of conicaquinones, aplidinones, thiaplidiaquinones, and conithiaquinones.
| Carbon | 45 | 46 | Carbon | 47 | 48 | 49 | Carbon | 50 | 51 | Carbon | 52 | 53 |
|---|
| Solvent | A | A | Solvent | A | A | D | Solvent | A | A | Solvent | D | D |
| Ref. | [53] | [53] | Ref. | [54] | [54] | [54] | Ref. | [55] | [55] | Ref. | [58] | [58] |
| 1 | 25.3 | 25.7 | 2 | 49.0 | 48.6 | 49.0 | 1 | 111.4 | 113.8 | 2 | 48.3 | 48.2 |
| 2 | 135.6 | 133.3 | 3 | 40.5 | 40.1 | 41.0 | 2 | 149.7 | 148.9 | 3 | 39.4 | 39.3 |
| 3 | 122.9 | 122.2 | 4a | 144.3 | 149.5 | 151.0 | 3 | 120.0 | 133.4 | 4a | 146.1 | 146.4 |
| 4 | 28.7 | 29.3 | 5 | 179.3 | 173.1 | 174.5 | 4 | 132.3 | 118.4 | 5 | 181.5 | 181.5 |
| 5 | 34.8 | 36.5 | 6 | 126.7 | 108.9 | 110.0 | 4a | 145.9 | 148.1 | 5a | 148.0 | 150.0 |
| 6 | 147.3 | 151.5 | 7 | 157.3 | 147.8 | 149.0 | 6 | 67.2 | 67.4 | 6 | 37.5 | 37.5 |
| 7 | 135.5 | 133.2 | 8 | 174.2 | 175.4 | 176.6 | 6a | 138.6 | 137.4 | 6a | 47.5 | 48.1 |
| 8 | 125.8 | 128.2 | 8a | 109.2 | 109.2 | 109.0 | 7 | 175.1 | 175.2 | 7 | 21.1 | 21.7 |
| 9 | 130.6 | 127.1 | 1ʹ | 22.2 | 23.4 | 24.0 | 7a | 110.1 | 109.9 | 8 | 33.1 | 33.7 |
| 10 | 174.9 | 178.4 | 2ʹ | 119.2 | 119.5 | 124.8 | 9 | 48.6 | 48.6 | 9 | 82.7 | 82.4 |
| 11 | 146.7 | 113.4 | 3ʹ | 137.6 | 138.4 | 137.0 | 10 | 39.9 | 39.8 | 9a | 51.8 | 52.1 |
| 12 | 113.4 | 146.7 | 4ʹ | 39.7 | 39.5 | 40.5 | 11a | 144.0 | 144.3 | 10 | 65.1 | 74.4 |
| 13 | 178.9 | 174.8 | 5ʹ | 26.6 | 26.3 | 27.8 | 12 | 179.1 | 179.2 | 10a | 147.5 | 149.7 |
| 14 | 129.3 | 132.6 | 6ʹ | 124.1 | 123.8 | 125.0 | 12a | 128.0 | 128.0 | 11 | 180.5 | 179.7 |
| 15 | 126.0 | 126.5 | 7ʹ | 131.5 | 131.8 | 133.0 | 12b | 116.6 | 114.4 | 11a | 110.0 | 110.3 |
| 16 | 17.5 | 17.6 | 8ʹ | 25.6 | 25.7 | 26.1 | 1ʹ | 28.1 | 29.1 | 12-CH3 | 21.2 | 21.6 |
| 17 | 39.8 | 48.1 | 9ʹ | 16.1 | 16.2 | 16.5 | 2ʹ | 121.3 | 120.5 | 13-OCH3 | 48.5 | 48.5 |
| 18 | 48.2 | 39.8 | 10ʹ | 17.7 | 17.7 | 17.8 | 3ʹ | 136.8 | 138.7 | 16-OCH3 | - | 58.0 |
| - | - | - | 1ʹʹ | - | - | 41.5 | 4ʹ | 39.7 | 39.8 | 14-CH3 | 18.6 | 18.4 |
| - | - | - | 2ʹʹ | - | - | 51.5 | 5ʹ | 26.6 | 26.6 | 15-CH3 | 25.4 | 25.7 |
| - | - | - | OCH3 | 62.3 | - | - | 6ʹ | 124.1 | 123.9 | - | - | - |
| - | - | - | - | - | - | - | 7ʹ | 131.4 | 131.8 | - | - | - |
| - | - | - | - | - | - | - | 8ʹ | 25.7 | 25.7 | - | - | - |
| - | - | - | - | - | - | - | 9ʹ | 16.1 | 16.2 | - | - | - |
| - | - | - | - | - | - | - | 10ʹ | 16.1 | 17.7 | - | - | - |
| - | - | - | - | - | - | - | 1ʹʹ | 116.8 | 116.8 | - | - | - |
| - | - | - | - | - | - | - | 2ʹʹ | 145.3 | 145.0 | - | - | - |
| - | - | - | - | - | - | - | 3ʹʹ | 39.7 | 39.7 | - | - | - |
| - | - | - | - | - | - | - | 4ʹʹ | 26.2 | 26.2 | - | - | - |
| - | - | - | - | - | - | - | 5ʹʹ | 123.5 | 123.5 | - | - | - |
| - | - | - | - | - | - | - | 6ʹʹ | 131.7 | 131.8 | - | - | - |
| - | - | - | - | - | - | - | 7ʹʹ | 25.6 | 25.6 | - | - | - |
| - | - | - | - | - | - | - | 8ʹʹ | 17.3 | 17.4 | - | - | - |
| - | - | - | - | - | - | - | 9ʹʹ | 17.5 | 17.7 | - | - | - |
The lack of
13C-NMR resonance for the group of unsaturated carbon atoms, which were substituted by a δ-deshielded value (δ
C 70.9), as in
4 [
15], suggest the presence of a hydroxyl group. This finding was also observed for compounds
11 and
12.
Conidinone (
8), which has a cyclic diprenyl substituent, presented a 1H-NMR spectrum with signals at δ
H 6.71 (1H, d,
J = 10.3 Hz), δ
H 6.87 (1H, d,
J = 10.3 Hz), δ
H 2.88 (1H, d,
J = 16.7 Hz), and δ
H 2.81 (1H, d,
J = 16.7 Hz), defining the presence of a 2,2-disubstituted cyclohexenedione ring in the molecule. The
13C-NMR of
8 includes two α,β-unsaturated ketone carbonyl signals, as well as a trisubstituted and a disubstituted double bond [
21].
Compound
15 and
16 presented a substituted hydroquinone nucleus, together with a 3,4-disubstituted-1-methylcyclohexene ring, and 1-hydroxyisopropyl unit, which was attached to C-6ʹ of the cyclohexene ring upon observation of the ROESY spectrum and NOE experiment, respectively. The difference between
15 and
16 was the presence of a methoxy group at
16 [
31,
35].
Compound
17 produced
1H-NMR spectrum resonances attributable to two aromatic protons at δ
H 6.75 (1H, d,
J = 2.1 Hz, H-5) and δ
H 6.54 (1H, d,
J = 2.1 Hz, H-3), two olefinic protons at δ
H 5.29 (1H, t,
J = 6.6 Hz, H-2′) and δ
H 5.09 (1H, t,
J = 6.6 Hz, H-6′), three moderately deshielded methylene signals (δ
H 3.27, 2.08, and 2.00), one methoxyl signal at δ
H 3.83, and three allylic methyl singlets at δ
H 1.69, δ
H 1.65, and δ
H 1.58. Direct comparison of the
1H and
13C chemical shifts of the aromatic ring signals of
17 with a synthetic derivative of
17 (compound which had a hydroxyl group at C-4 instead of the sulfate) allow Chan and co-worker to identified an upfield shift of C-4 and downfield shifts of C-3 and C-5 in the
13C-NMR spectrum and downfield shifts of H-3 and H-5 in the
1H-NMR spectrum which were consistent with the placement of the sulfate group at the C-4 [
17].
The
13C-NMR data for Rossinone A (
18),
Table 3, allowed for the identifications of a triprenylated (farnesyl) hydroquinone-bearing substitution in the terminal prenyl unit. A α-hydroxy ketone group (δ
C 201.8) and a carbinol resonance at δ
C 69.8 in the side chain of
18 were established [
23].
The Rossinone family, mainly compounds
19–
22, have a cyclic triprenylated group. When comparing
19 and
20, the principal differences were in the chemical shift of the AB quartet due to the quinone protons resonating at higher fields (H-2: δ
H 3.75 (d,
J = 3.5 Hz) in
19 and δ
H 6.79 (d,
J = 10.5 Hz) in
20; H-3: δ
H 3.80 (d,
J = 3.5 Hz) in
19 and δ
H 6.90 (d,
J = 10.5 Hz) in
20). Additionally, the
13C-NMR spectrum of
19 contained signals at δ
C 55.5 (C-2) and δ
C 56.4 (C-3) in the place of the quinone resonances of
20 observed at δ
C 139.0 (C-2) and δ
C 141.2 (C-3) [
38].
The compound 3-
Epi-rossinone B (
21) was an isomer of rossinone B (
20). The analysis of the
1H-NMR revealed significant differences in the proton values of both H-7 and H-11 (H-7: δ
H 2.06 in
20 and δ
H 2.48 in
21; H-11: δ
H 2.67 in
20 compared to δ
H 2.10 in
21) that could be explained by the diverse steric influence of the hydroxyl group in these two compounds; the structure was confirmed by NOE experiments [
38].
For 5,6-Epoxy-rossinone B (
22), revealed in
1H-NMR spectrum the lack of the double bond in ring B when compared with
20, which is substituted by an epoxide ring in
22. The H-6 was attributed to a singlet at δ
H 3.80. In the
13C-NMR spectrum the values at δ
C 61.4 and δ
C 55.7, were assigned to C-6 and C-5, respectively. Additionally, due to the absence of the conjugated double bond, the C-4 quinone carbonyl in
22 was shifted down-field (δ
C 193.9) with respect to
20 (δ
C 185.0) [
38].
The complex and unprecedented structure of longithorone A (
23) was determined by crystal X-ray diffraction, while the enantioselective biomimetic synthesis of longithorone A was accomplished by Layton and coworkers [
59]. The
1H and
13C-NMR of (
23) (
Table 4) revealed that
23 contained eight double bonds and five carbonyl groups, including one aldehyde (δ
H 9.6 and δ
C 206.6), in accord with the presence of seven carbocycles to satisfy the unsaturation number revealed by the mass spectral data (C
42H
46O
5) [
42].
Longithorones B (
24) and C (
25) are optically active isomers with the double bond configurations 2
E and 2
Z in
24 and
25, respectively. The singlets at δ
H 6.23 (H-18) and δ
H 6.45 (H-21) for
24 and δ
H 6.38 (H-18 and H-21) for
25 are representative of
para-disubstituted benzoquinone core. The synthesis of Longithorone B was accomplished by Kato
et al. [
60].
The structures of longithorones E (
27) and F (
28) are very similar. However, the NOESY experiment for Longithorone F revealed that H-13 is spatially close to H-6, H-18, H-1α, and H-4a. On the other hand, the NOE experiment of
27 showed interactions between H-13 with H-6 and H-1β, but not with H-18. Thus, the dimeric longithorones E (
27) and F (
28) are atropoisomers with respect to the
para-disubstituted benzoquinone ring [
40]. In turn, longithorone F (
28) differs from longithorone G (
29) in its C-2ʹ, C-3ʹ, and C-10ʹ stereochemistry. The NOESY data for
29 revealed correlations between the aldehyde hydrogen H-13 with H-5 and H-2ʹ suggesting that these protons were all on the same face of the molecule. These correlations were not, however, observed in the NOE experiment for
28.
The farnesyl chains in longithorone J (
32) and K (
33) were characterized by three shielded olefinic methyl resonances, two of these (δ
C 14.9 and δ
C 16.9) assigned
E-geometry configuration and the other (δ
H 22.8) was assigned a
Z-geometry. The
1H and
13C-NMR spectra of longithorone J include an oxymethine proton at δ
H 4.84 (δ
C 71.5) in contrast to the presence of two ketone resonances at δ
C 200.7 and δ
C 196.8 in longithorone K (
33). The absolute stereochemistry of longithorone J (
32) has been determined by the advanced Mosher method, while the absolute stereochemistry for longithorone K (
33) was suggested by comparison with
32 and based on biosynthesis [
43].
A comparison of the
1H and
13C-NMR data for longithorols A–D (
34–
37) (
Table 5) with previous longithorones reveals that, in longithorols, signals for two substituted 1,4-hydroquinones (C-16 to C-21 and C-16ʹ to C-21ʹ) are present instead of signals for two substituted 1,4-quinones. In addition, an acetoxymethine group can be recognized by the signals at δ
H 6.61 (d,
J = 10.0 Hz) and δ
C 67.1 (for example, in
34), which replace the methylene group corresponding to C-1 in longithorones [
47]. The absolute stereochemistry of longithorol C (
36) has been determined by the advanced Mosher method [
48].
Floresolides skeleton can be recognized by the presence of an endocyclic ε-lactone with an α,β-epoxy-group (δ
C 63.7 and δ
C 59.1) in
38 and (δ
C 62.5 and δ
C 58.3) in
39, and a double bond at the C-2 and C-3 position (δ
C 137.8 and δ
C 136.1) in
40 (
Table 5). The terminal methylene group at the C-11 and C-12 positions is also typical in this structural family. The Floresolide
39 has a primary alcohol and a fully substituted benzene ring including two bromine atoms in its structure. Compound
40 has absolute stereostructure, confirmed by crystal-X-ray diffraction [
49].
Scabellones are conspicuous for the rare tricycle benzo[
c] chromene-7,10 dione core in their structure. The signals at δ
C 182.6 (C-1) and δ
C 178.7 (C-4) (
Table 6) define the presence of the quinonoid ring system, while the HMBC correlations from the aromatic proton resonance at δ
H 6.40 (H-7) to the
13C resonances at δ
C 150.0 (C-8), δ
C 139.2 (C-9), δ
C 151.4 (C-6a) and δ
C 111.0 (C-10a), together with the methoxyl group at δ
H 3.89 (C-8) and the signal at δ
C 126.8 (C-10), define the tetrasubstituted phenol in Scabellone B (
42), the active representative compound of this family [
17].
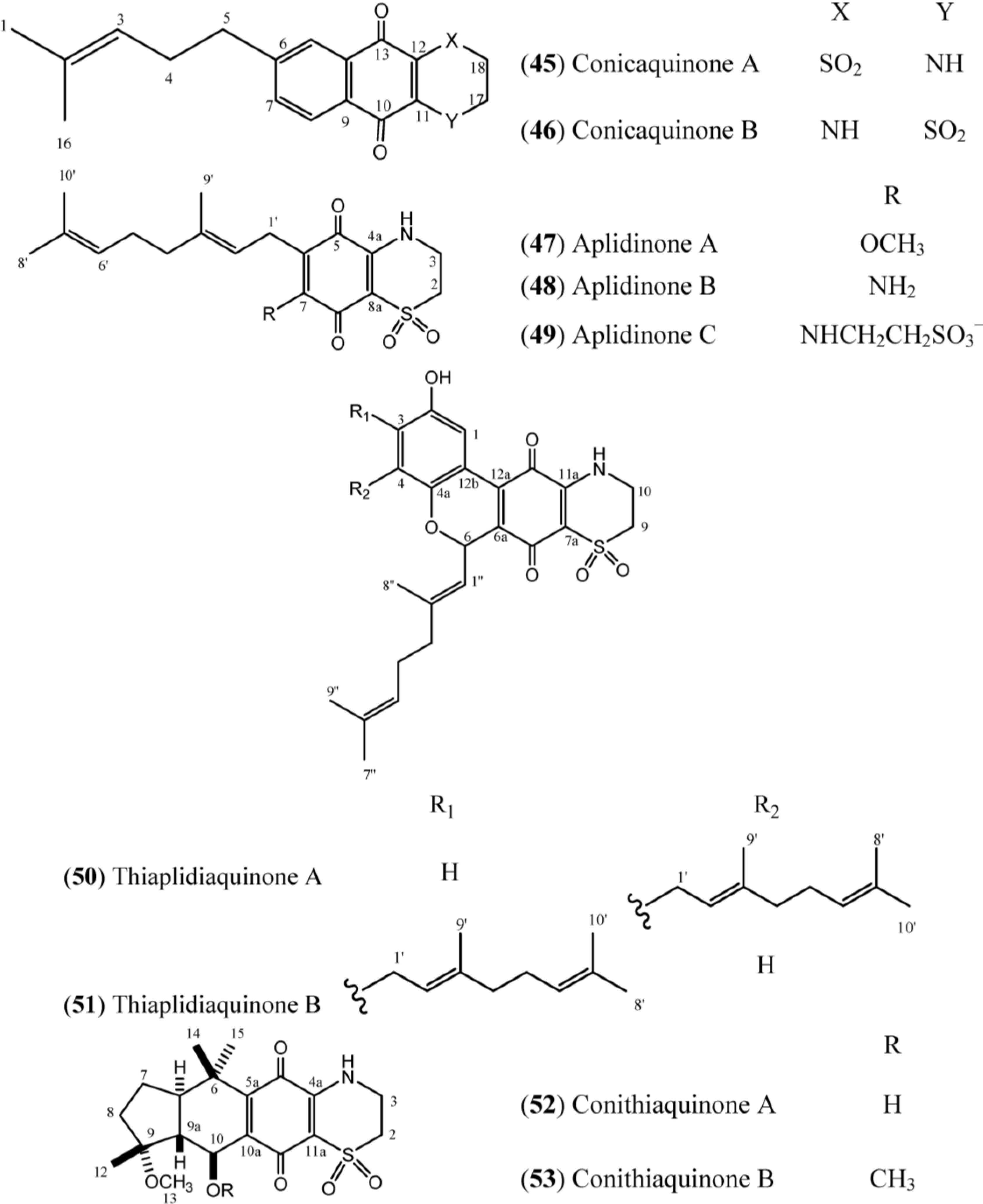
The structures of conicaquinones (
45–
46), aplidinones (
47–
49), thiaplidiaquinones (
50–
51) and conithiaquinones (
52–
53) have the presence of an unusual 1,1-dioxo-1,4-thiazine ring characterized by N-H (δ
H 6.78), two methylene protons at δ
H 4.10 (δ
C 39.8) and δ
H 3.36 (δ
C 48.2) in common, as exemplified in Conicaquinone A (
45) (
Table 7) [
53]. In turn, the aplidinones contain a geranyl chain that is attached to the system -NHCH
2CH
2SO
2− in addition to the quinone ring. Finally, the thiaplidiaquinones and conithiquinones contain a tetracyclic skeleton. Thiaplidiaquinones have the same tricyclic 6H-benzo[
c] chromene-7,10-dione described for Scabellone B (
42) in their structure [
17]. On the other hand, the signals related to C-6 to C-10 in
13C-NMR spectra, in addition to the signal of the methoxyl group (C-13), the methyl protons C-12, C-14 and C-15 and the signal of hydroxy group positioned at C-10 in the
1H-NMR spectra, establish the presence of C and D ring fused at 1,4-benzoquinone/1,1-dioxo-1,4-thiazide bicyclic system (A and B rings) of conithiaquinone skeleton [
58].







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}