Antibacterial and Antiyeast Compounds from Marine-Derived Bacteria

Abstract
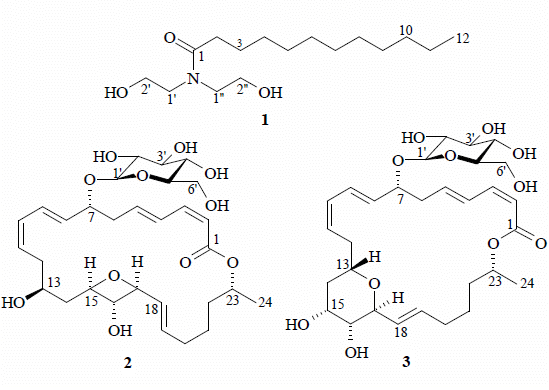
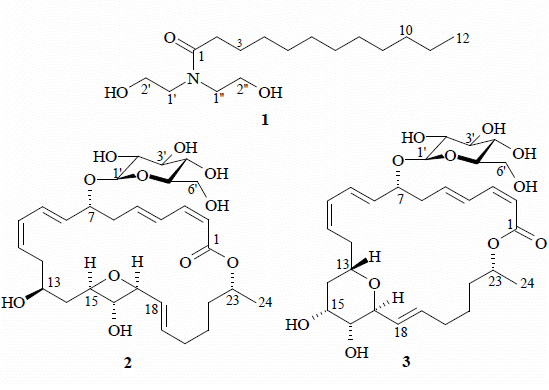
:
1. Introduction
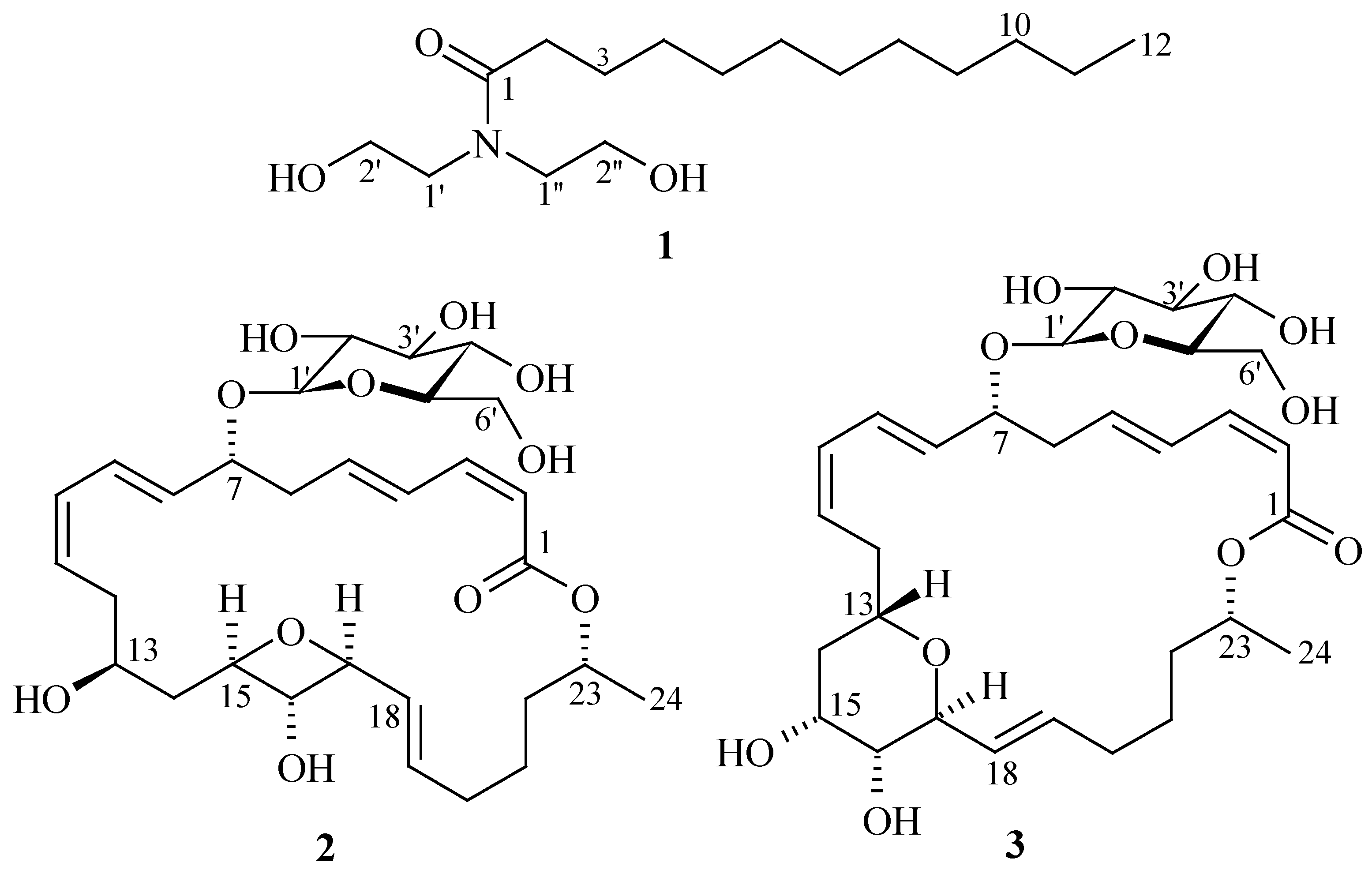
2. Results and Discussion
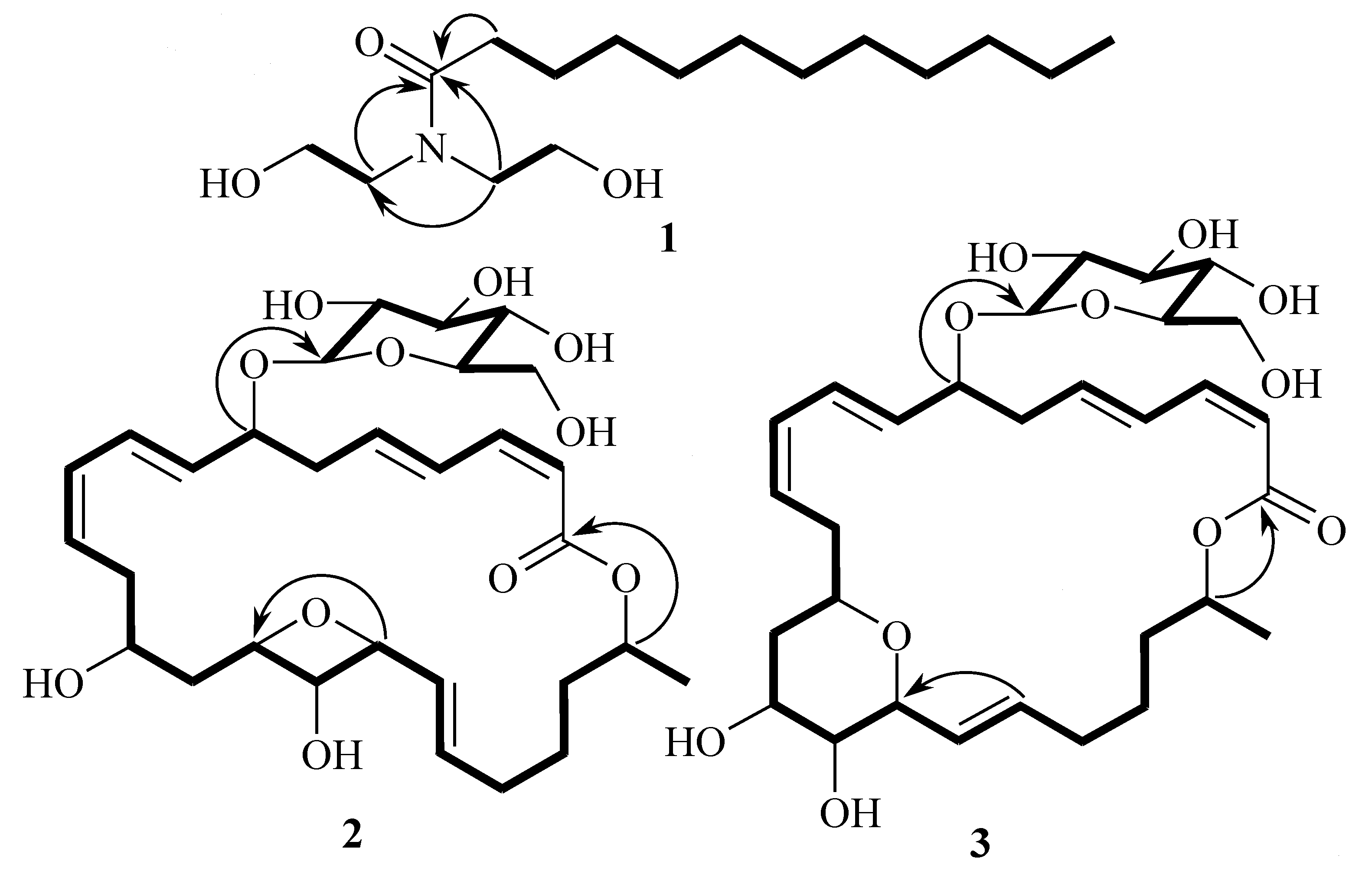

{kind=link}
{kind=link}
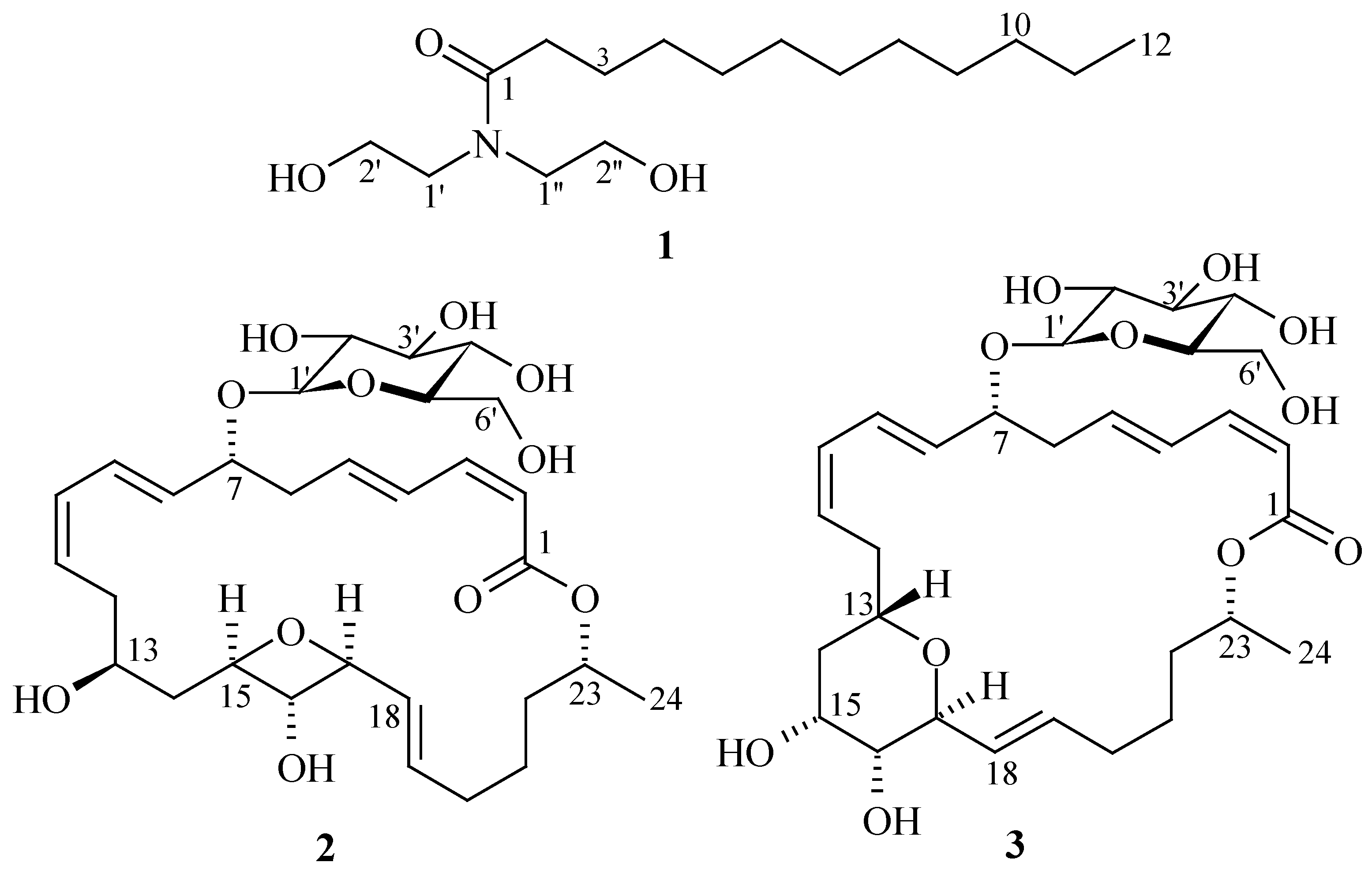{kind=link}
{kind=link}
| No. | 1 a | 2 a | 3 a | |||
|---|---|---|---|---|---|---|
| δC | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) | |
| 1 | 176.8 | 168.3 | 168.1 | |||
| 2 | 34.3 | 2.43, t (7.5) | 118.1 | 5.50, d (11.5) | 118.1 | 5.51, d (11.5) |
| 3 | 26.7 | 1.60, m | 144.6 | 6.65, t (11.5) | 144.6 | 6.30, t (11.5) |
| 4–11/4 | 23.8–33.2 | 1.32–1.29, m | 130.6 | 7.17, dd (15.5, 11.5) | 130.6 b | 7.17, dd (15.0, 11.5) |
| 12/5 | 14.5 | 0.89, t (7.0) | 141.6 | 6.35, dt (15.5, 7.0) | 141.3 | 6.27, dt (15.0, 7.5) |
| 1′/6 | 50.3 | 3.49, t (6.0) | 41.6 | 2.46, m; 2.57, m | 41.5 | 2.54, m |
| 2′/7 | 61.0 | 3.67, t (5.5) | 78.3 | 4.51, m | 77.9 b | 4.54, m |
| 1″/8 | 52.7 | 3.54, t (6.0) | 132.8 | 5.52, dd (15.5, 8.5) | 135.6 | 5.60, dd (15.1, 8.0) |
| 2″/9 | 60.9 | 3.69, t (5.5) | 131.0 | 6.63, dd (15.5, 11.5) | 129.8 | 6.67, dd (15.1, 11.0) |
| 10 | 131.9 | 6.07, t (11.5) | 130.6 b | 6.06, t (11.0) | ||
| 11 | 130.6 | 5.46, dt (11.5, 5.0) | 130.3 | 5.52, dt (11.0, 6.5) | ||
| 12 | 35.4 | 2.60, m | 34.3 | 2.06, m; 2.93, m | ||
| 13 | 75.8 | 3.45, m | 72.1 | 3.72, m | ||
| 14 | 41.1 | 1.38, q (11.5); 1.95, m | 35.7 | 1.77, m; 2.06, m | ||
| 15 | 73.9 | 3.52, ddd (11.4, 9.0, 5.0) | 67.7 | 3.86, m | ||
| 16 | 77.6 | 2.90, dd (9.0, 9.0) | 72.0 | 3.54, dd (4.3, 4.3) | ||
| 17 | 80.0 | 3.42, dd (9.0, 5.0) | 76.6 | 4.28, dd (4.3, 4.3) | ||
| 18 | 129.0 | 5.64 b [5.62, dd (15.3, 5.0)] c | 128.4 | 5.42 dd (15.6, 4.3) | ||
| 19 | 132.0 | 5.64 b [5.46, dt (15.3, 8.5)] c | 135.6 | 5.65, dt (15.6, 8.0) | ||
| 20 | 34.6 | 2.01, m; 2.07, m | 33.7 | 2.06, m; 2.14, m | ||
| 21 | 26.9 | 1.44, m; 1.52, m | 26.0 | 1.48, m; 1.57, m | ||
| 22 | 37.0 | 1.64, m | 36.5 | 1.60, m; 1.67, m | ||
| 23 | 73.0 | 4.93, m | 72.4 | 4.97, m | ||
| 24 | 20.0 | 1.25, d (6.5) | 20.1 | 1.25, d (6.0) | ||
| 1′ | 100.6 | 4.29, d (8.0) | 101.2 | 4.31, d (8.0) | ||
| 2′ | 75.1 | 3.22, dd (9.0, 8.0) | 75.1 | 3.20, dd (8.7, 8.0) | ||
| 3′ | 71.8 | 3.23, dd (9.0, 8.4) | 71.7 | 3.27, dd (8.7, 8.5) | ||
| 4′ | 78.1 | 3.32, dd (9.5, 8.4) | 78.1 | 3.32, dd (9.0, 8.5) | ||
| 5′ | 78.0 | 3.18, m | 77.9 b | 3.18, m | ||
| 6′ | 62.8 | 3.64, dd (12.0, 6.0) 3.85, dd (12.0, 2.0) | 62.7 | 3.65, dd (12.0, 5.5) 3.86, dd (12.0, 2.0) | ||
| Test Organisms | MICs (μM/mL) | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | AZ | AmpB | |
| Bacillus subtilis | 0.055 | 0.027 | 0.055 | 0.005 | - |
| Escherichia coli | 0.055 | 0.220 | 0.220 | 0.005 | - |
| Pseudomonas aeruginosa | 0.110 | 0.055 | 0.055 | 0.005 | - |
| Staphylococcus aureus | 0.110 | 0.055 | 0.055 | 0.005 | - |
| Saccharomyces cerevisiae | 0.220 | 0.220 | 0.220 | - | 0.002 |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Isolation and Taxonomy of the Strain O6CH80 and 09ID194
3.3. Seed and Large-Scale Cultures of the Strains O6CH80 and 09ID194
3.4. Extraction and Isolation
 −97 (c 0.05, MeOH); UV (MeOH) λmax (log ε) 237 (4.18) and 260 (4.15) nm; IR (MeOH) νmax 3297 (br), 2941, 2830, 1600, 1022 cm−1; 1H and 13C NMR data (CD3OD), see Table 1; HRESIMS m/z 603.2775 [M + Na]+ (calcd for C30H44O11Na, 603.6535). −88 (c 0.05, MeOH); UV (MeOH) λmax (log ε) 236 (4.04) and 256 (4.20) nm; IR (MeOH) νmax 3312 (br), 2941, 2830, 1600, 1022 cm−1; 1H and 13C NMR data (CD3OD), see Table 1; HRESIMS m/z 603.2776 [M + Na]+ (calcd for C30H44O11Na, 603.6535).
−97 (c 0.05, MeOH); UV (MeOH) λmax (log ε) 237 (4.18) and 260 (4.15) nm; IR (MeOH) νmax 3297 (br), 2941, 2830, 1600, 1022 cm−1; 1H and 13C NMR data (CD3OD), see Table 1; HRESIMS m/z 603.2775 [M + Na]+ (calcd for C30H44O11Na, 603.6535). −88 (c 0.05, MeOH); UV (MeOH) λmax (log ε) 236 (4.04) and 256 (4.20) nm; IR (MeOH) νmax 3312 (br), 2941, 2830, 1600, 1022 cm−1; 1H and 13C NMR data (CD3OD), see Table 1; HRESIMS m/z 603.2776 [M + Na]+ (calcd for C30H44O11Na, 603.6535).3.5. Acid Hydrolysis of Glycosylated Macrolactins 2 and 3
+41 (c 0.04, H2O) and from 3 was +44 (c 0.04, H2O).3.6. Antimicrobial Activity Study
4. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Edmond, M.B.; Ober, J.F.; Weinbaum, D.L.; Pfaller, M.A.; Hwang, T.; Sanford, M.D.; Wenzel, R.P. Vancomycin-resistant Enterococcus faecium bacteremia: Risk factors for infection. Clin. Infect. Dis. 1995, 20, 1126–1133. [Google Scholar] [CrossRef]
- Leclercq, R. Epidemiological and resistance issues in multidrug-resistant Staphylococci and Enterococci. Clin. Microbiol. Infect. 2009, 15, 224–231. [Google Scholar] [CrossRef]
- Demain, A.L.; Sanchez, S. Microbial drug discovery: 80 Years of progress. J. Antibiot. 2009, 62, 5–6. [Google Scholar] [CrossRef]
- Katz, M.L.; Mueller, L.V.; Polyakov, M.; Weinstock, S.F. Where have all the antibiotic patents gone. Nat. Biotechnol. 2006, 24, 1529–1531. [Google Scholar] [CrossRef]
- Kong, D.X.; Jiang, Y.Y.; Zhang, H.Y. Marine natural products as sources of novel scaffolds: Achievement and concern. Drug Discov. Today 2010, 15, 884–886. [Google Scholar] [CrossRef]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef]
- Bhatnagar, I.; Kim, S.K. Immense essence of excellence: Marine microbial bioactive compounds. Mar. Drugs 2010, 8, 2673–2701. [Google Scholar] [CrossRef]
- Liu, Y. Renaissance of marine natural product drug discovery and development. J. Mari. Sci. Res. Dev. 2012. [Google Scholar] [CrossRef]
- Trowbridge, J.R.; Falk, R.A.; Krems, I.J. Fatty acid derivatives of diethanolamine. J. Org. Chem. 1955, 20, 990–995. [Google Scholar] [CrossRef]
- Mondol, M.A.M.; Tareq, F.S.; Kim, J.-H.; Lee, M.A.; Lee, H.-S.; Lee, Y.-J.; Lee, J.-S.; Shin, H.J. Cyclic ether-containing macrolactins, antimicrobial 24-membered isomeric macrolactones from a marine Bacillus sp. J. Nat. Prod. 2011, 74, 2582–2587. [Google Scholar] [CrossRef]
- Schneider, K.; Chen, X.-H.; Vater, J.; Franke, P.; Nicholson, G.; Borriss, R.; Süssmuth, R.D. Macrolactin is the polyketide biosynthesis product of the pks2 cluster of Bacillus amyloliquefaciens FZB42. J. Nat. Prod. 2007, 70, 1417–1423. [Google Scholar] [CrossRef]
- Castillo, J.A.; Clapes, P.; Infante, M.R.; Jaume, C.; Manresa, A. Comparative study of the antimicrobial activity of bis(Nα-caproyl-l-arginine)-1,3-propanediamine dihydrochloride and chlorhexidine dihydrochloride against Staphylococcus aureus and Escherichia coli. J. Antimicrob. Chemother. 2006, 57, 691–698. [Google Scholar] [CrossRef]
- Zheng, C.-J.; Lee, S.; Lee, C.-H.; Kim, W.-G. Macrolactins O–R, glycosylated 24-membered lactones from Bacillus sp. AH159-1. J. Nat. Prod. 2007, 70, 1632–1635. [Google Scholar] [CrossRef]
- Kabara, J.J. Structure-function relationships of surfactants as antimicrobial. J. Soc. Cosmet. Chem. 1978, 29, 733–741. [Google Scholar]
- Mondol, M.A.M.; Shin, H.J.; Islam, M.T. Diversity of secondary metabolites from marine Bacillus species: Chemistry and biological activity. Mar. Drugs 2013, 11, 2846–2872. [Google Scholar] [CrossRef]
- Nagao, T.; Adachi, K.; Sakai, M.; Nishijima, M.; Sano, H. Novel macrolactins as antibiotic lactones from a marine bacterium. J. Antibiot. 2001, 54, 333–339. [Google Scholar] [CrossRef]
- Romero-Tabarez, M.; Jansen, R.; Sylla, M.; Lunsdorf, H.; Haubler, S.; Santosa, D.A.; Timmis, K.N.; Molinari, G. 7-O-malonyl macrolactin A, a new macrolactin antibiotic from Bacillus subtilis active against methicillin-resistant Staphylococcus aureus, vancomycin-resistant enterococci, and a small-colony variant of Burkholder. Antimicrob. Agents Chemother. 2006, 50, 1701–1709. [Google Scholar] [CrossRef]
- Jaruchoktaweechai, C.; Suwanborirux, K.; Tanasupawatt, S.; Kittakoop, P.; Menasveta, P. New macrolactins from a marine Bacillus sp. Sc026. J. Nat. Prod. 2000, 63, 984–986. [Google Scholar] [CrossRef]
- Mondol, M.A.M.; Kim, J.-H.; Lee, M.A.; Lee, H.-S.; Lee, Y.-J.; Shin, H.J. Macrolactin W, a new antibacterial macrolide from a marine Bacillus sp. Bioorg. Med. Chem. Lett. 2011, 21, 3832–3835. [Google Scholar] [CrossRef]
- Lee, Y.B.; Park, H.J.; Kwon, M.J.; Jeong, S.K.; Cho, S.H. Beneficial effects of pseudoceramide-containing physiologic lipid mixture as a vehicle for topical steroids. Eur. J. Dermatol. 2011, 21, 710–716. [Google Scholar]
- Uchida, Y.; Holleran, W.M.; Elias, P.M. On the effects of topical synthetic pseudoceramides: comparison of possible keratinocyte toxicities provoked by the pseudoceramides, PC104 and BIO391, and natural ceramides. J. Dermatol. Sci. 2008, 51, 37–43. [Google Scholar] [CrossRef]
- Mizushima, H.; Fukasawa, J.-I.; Suzuki, T. Thermotropic behavior of stratum corneum lipids containing a pseudo-ceramide. Lipids 1995, 30, 327–332. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mondol, M.A.M.; Shin, H.J. Antibacterial and Antiyeast Compounds from Marine-Derived Bacteria. Mar. Drugs 2014, 12, 2913-2921. https://doi.org/10.3390/md12052913
Mondol MAM, Shin HJ. Antibacterial and Antiyeast Compounds from Marine-Derived Bacteria. Marine Drugs. 2014; 12(5):2913-2921. https://doi.org/10.3390/md12052913
Chicago/Turabian StyleMondol, Muhammad Abdul Mojid, and Hee Jae Shin. 2014. "Antibacterial and Antiyeast Compounds from Marine-Derived Bacteria" Marine Drugs 12, no. 5: 2913-2921. https://doi.org/10.3390/md12052913
APA StyleMondol, M. A. M., & Shin, H. J. (2014). Antibacterial and Antiyeast Compounds from Marine-Derived Bacteria. Marine Drugs, 12(5), 2913-2921. https://doi.org/10.3390/md12052913